

Abstract
Incorporation of 2,3,6-trifluorotyrosine (F3Y) and a rhenium bipyridine ([Re]) photooxidant into a peptide corresponding to the C-terminus of the β protein (βC19) of Escherichia coli ribonucleotide reductase (RNR) allows for the temporal monitoring of radical transport into the α2 subunit of RNR. Injection of the photogenerated F3Y radical from the [Re]–F3Y–βC19 peptide into the surface accessible Y731 of the α2 subunit is only possible when the second Y730 is present. With the Y–Y established, radical transport occurs with a rate constant of 3 × 105 s−1. Point mutations that disrupt the Y–Y dyad shut down radical transport. The ability to obviate radical transport by disrupting the hydrogen bonding network of the amino acids composing the co-linear proton-coupled electron transfer pathway in α2 suggests a finely tuned evolutionary adaptation of RNR to control the transport of radicals in this enzyme.
Keywords: Ribonucleotide Reductase, Proton-Coupled Electron Transfer, Radical Transport
Introduction
Ribonucleotide reductases (RNR) are the keystone of DNA biosynthesis in all organisms. They remove the 2′ hydroxyl from nucleoside diphosphates (NDPs) to generate deoxynucleoside diphosphates (dNDPs).1 The Escherichia coli class Ia RNR is composed of two subunits—α2 and β2—that form an active α2: β2 complex when in the presence of substrate (S) and effectors (E). The α2 subunit houses the catalytic cysteine (C439) as well as two allosteric sites that control both substrate specificity and turnover rate. β2 stores a diiron-centered tyrosyl radical cofactor (Fe2–•Y122) that is essential for catalysis in α2. The binding of substrate and effector enhances inter-subunit interactions and triggers Fe2–•Y122 mediated C439 oxidation in α2 from a distance of 35 Å. Whereas the mechanism of catalysis2 by the enzyme is understood, the basis for S/E mediated conformational change3 and the attendant dynamic transport of the radical through the protein to the active site is yet to be unraveled. This report focuses on the use of a phototriggered β2 surrogate to monitor, for the first time, the kinetics of proton-coupled electron transfer (PCET) within α2.
At 35 Å separation, the vanishingly small overlap of the amino acid wave functions precludes a single-step superexchange mechanism.4 Accordingly, current models4–7 posit a stepwise translation of the radical across the two proteins:
This proposed pathway is an outgrowth of a docking model between α2 and β2 whose structures were determined crystallographically.8 It is bolstered by observations that the enzyme is inactive when point mutations into residues that cannot be oxidized are made on the pathway.9–12 Measurement of radical transport along the pathway is masked by rate-limiting conformational changes caused by S/E binding to α2 prior to rapid radical propagation and nucleotide reduction.
To permit investigation of this radical transport pathway, methods have been developed to site-specifically incorporate unnatural amino acids in place of each proposed tyrosine in the pathway. This allows for enzyme turnover to be monitored as a function of modulated phenolic pKa and reduction potential.13,14 Studies using the more easily oxidized 3-amino-tyrosine (NH2Y) in place of Y356 (β2), Y730 (α2), and Y731 (α2) establish the conformational gating and its complexity,13–16 and PELDOR spectroscopy of these protein mutants establish for long-range radical initiation and more generally the validity of the docking model.17 Incorporation of 3-NO2Y, a strong oxidant, at position 122 in β2 has fortuitously resulted in uncoupling of conformational gating.18 Millisecond time-scale kinetics experiments have revealed that 3-NO2Y122• in the presence of α2, CDP (S) and ATP (E) is rapidly reduced to the 3-NO2Y phenolate concomitant with dCDP formation and generation of a mixture of three tyrosyl radicals at 356, 731 and 730. The baseline catalytic turnover of the wild type α2: β2 complex is 2–10 s−1.4 The observed rate constant in these mutants is 100–300 s−1, or 10–30 times faster than wild-type turnover.18 These studies provide the direct observation of pathway radicals and start to define their relative redox potentials.
The PCET kinetics among these pathway radicals may be isolated with photoRNRs. In this construct, a short peptide (Y–βC19) has been employed in place of full-length β2.19,20 This peptide contains the C-terminal 20 amino acids of the β protein (βC20), including both the determinant for binding β2 to α2 and Y356 in β2, which facilitates radical transport at the α2: β2 interface.21 By appending a photooxidant (PO) to this peptide (PO–Y–βC19), the equivalent of •Y356 (•Y–βC19) can be photochemically generated. Photoinitiated substrate turnover can be observed for the PO–Y–βC19: α2 construct upon illumination.20,22 For instance, installation of 3,5-difluorotyrosine in PO–Y–βC19 can generate a transient 3,5-•F2Y that is able to generate dNDPs in the absence of β2.20 However, to date, a kinetic measurement of the radical on pathway has not been achieved and more generally, the physiologically relevant dynamic transport of a radical in any enzyme has yet to be observed.
In this work, we transiently generate an unnatural fluorotyrosyl radical with the rhenium PO in the presence of four mutants of the catalytic subunit of RNR shown in Figure 1. We temporally observe the radical as it propagates into α2 and measure the radical injection rate into the Y731 ⇆ Y730 ⇆ C439 pathway of α2.
Figure 1.

A photoactive peptide [Re]–F3Y–βC19 displays a fluorotyrosyl radical at the equivalent position to Y356 in β2. The method of radical transport into α2 from Y356→Y731→Y730→ C439 is deciphered by monitoring the radical in the presence of the stop mutants Y731F-α2, Y730F-α2, C439S-α2 as well as the wt-α2.
Experimental
Materials
The βC19 peptide was synthesized on resin by Pi Proteomics (Huntsville, AL; piproteomics.com) by starting with Fmoc-L-Leu-PEG-PS resin (Applied Biosystems, 180 μmol/g), and using our previously described protocol.23 4-methyl-4′-carboxyl-2,2′-bipyridine, RNR subunit β2 (1.2 •Y122/β2, 5,400 nmol/min/mg), E. coli thioredoxin (TR, 40 U/mg), E. coli thioredoxin reductase (TRR, 1400 U/mg), [Re]–Y–βC19, and hydroxybenzotriazole (HOBT) were available from previous studies. Protected Fmoc-2,3,6-trifluorotyrosine was prepared as previously reported.13,20 [5-3H]-CDP was purchased from ViTrax (Placentia, CA). Other chemicals were of reagent grade or higher, sourced commercially, and used as received. These compounds, and their abbreviations, are listed in the Supporting Information.
Cell stocks, plasmids and primers
E. coli BL21(DE3) cells were purchased from Novagen. E. coli XL-10 Gold cells were purchased from Agilent (formerly Stratagene). The pET-nrdA(wt) plasmid encoding for N-terminally (His)6-tagged wt RNR protein α was available from a previous study.16 Primers used in site-directed mutagenesis were purchased in purified form as a custom synthesis from Invitrogen.
pET-nrdA(wt) site-directed mutagenesis and transformation
Site-directed mutagenesis (SDM) was carried out with the Quickchange Kit from Stratagene. Each mutant was generated by amplifying the template, pET-nrdA(wt), with Pfu Ultra II polymerase in the presence of the forward and reverse primers whose sequences are listed in the supporting information. Amplification of pET-nrdA mutant plasmids was accomplished by transformation of SDM reaction mixtures into XL-10 gold cells by following the manufacturer’s instructions. Plasmids were isolated using a Miniprep kit from Qiagen, eluting the final plasmid with di-H2O. DNA sequencing was performed by the MIT Biopolymers Lab. pET-nrdA transformation into BL21(DE3) cells was completed by following the manufacturer’s instructions.
NrdA mutant expression
A solution of 75 μL of the SOC media BL21(DE3) transformant was spread aseptically onto an LB-agar plate containing kanamycin (Km) at 50 μg/mL. The plate was then incubated overnight at 37 °C. After 14 h of growth, the plate showed well-dispersed individual colonies. One colony from the plate was picked, which was incubated at 37 °C in 5 mL of LB media containing 50 μg/mL Km on a rotating tumbler until saturated (10–20 h). One mL of this culture was then diluted into 100 mL of LB-Km media in a 500 mL Erlenmeyer flask and incubated at 37 °C while shaking at 220 rpm for 12 h. 50 mL of this saturated culture was then diluted into 10 L of LB-Km media in a Beckman Scientific fermentor. The temperature was set to 37 °C with air sparging at 10 L/h and stirring at 500 RPM. After 2.5 h of growth (OD600 = 0.67), protein production was induced by adding 10 mL of 1 M IPTG, giving 1 mM in solution. Growth continued for 4 h, at which point the cells were harvested by centrifugation (10 min, 7,000 × g), flash frozen at 77 K, and stored at −80 °C. Typical yield was 3–5 g/L wet cell paste.
(His)6-α2 purifications
“Lysis buffer” consisted of 50 mM Tris (pH 7.6 at 4 °C) containing 5% glycerol, 1 mM PMSF, 10 mM imidazole and 10 mM DTT. The solid DTT and PMSF (0.2 M in EtOH) were added just before use. The details for a single purification of the Y730F mutant follow, and the other mutants were purified in the same manner. Frozen cell pellet (7.4 g) was thawed on ice with 5 mL of lysis buffer per gram of pellet (35 mL total). The suspension was then prepared for lysis with several passes through a Teflon/glass homogenizer. The cells were then lysed with a single pass through a SLM-Aminco French pressure cell (with the cell and its fittings pre-cooled on ice) at between 16,000 and 18,000 psi. The resulting suspension was then centrifuged (25,000 × g, 30 min). The pellet was discarded and the supernatant was poured into a stirring solution of 0.2 vol equiv (16 mL) of 6% w/v streptomycin sulfate at 4 °C. The solution was stirred for 30 min at 4 °C and centrifuged (25,000 × g, 30 min). The supernatant was then loaded onto a Ni-NTA superflow (Qiagen) column (25 mL), which had been equilibrated with lysis buffer containing 500 mM NaCl. The column was then washed with 10 CV (250 mL) of lysis buffer to remove cellular proteins, and collected in five 50 mL fractions. The protein was then eluted with a 500 mL linear gradient of 10–300 mM imidazole in lysis buffer and collected in 1.5 min (12 mL) fractions. The fractions containing only α protein (by SDS-PAGE) were pooled and concentrated to 30 mL while stirring in an Amicon pressure concentrator fitted with a 30 kD MWCO membrane at 50 psi N2. The solution was then loaded onto a 200 mL G-25 column and eluted with “spectroscopy buffer” (50 mM sodium borate (pH 8.3 at 23 °C), containing 15 mM MgSO4 and 5% v/v glycerol). The eluent was collected in 3 min (10 mL) fractions. Those fractions containing protein, as judged by the Bradford assay, were pooled and concentrated in 30 kD MWCO centrifugal concentrators. Final purity was determined by SDS-PAGE (Figure 2a,b). Final concentration was determined with the known ε278 of 0.189 μM−1 cm−1.24 Activity measurements were performed as previously described (Figure 2c).16
Figure 2.

Purification and activity of α2 mutants. (a) SDS-PAGE of each mutant after purification indicates a monomer molecular weight at the expected retention (85 kD). (b) Quantification of the gel lanes in (a) by integrating the band density indicates that >98% of the protein in solution is α2 for each mutant prepared. (c) Activity of each enzyme quantified by counting turnover of [3H]-labeled CDP.
Synthesis of Re(I)(CO)3(CN)(Mebpy-COO-PFP) ([Re]-OPFP)
The synthesis of Re(I)(CO)3(CN)(Mebpy-COOH) ([Re]-COOH) has been previously described.20 It was determined that the coupling yields of the rhenium carboxylate to the N-terminus of the peptide were low under previously reported HATU coupling conditions. As such, the [Re]-COOH was pre-activated as a pentafluorophenyl ester. To do so, [Re]-COOH (235 mg, 0.46 mmol, 1 equiv.) was diluted with 2.6 mL DCM and 75 μL DIPEA. The solution was transferred to a 25 mL round-bottom flask under a nitrogen atmosphere, and the vial used to dissolve the carboxylate was washed with an additional 2 mL DCM and 50 μL DIPEA and transferred to the reaction flask. The flask headspace was purged with N2 and then PFP-TFA (258 mg, 0.92 mmol, 158 μL, 2 equiv) was added dropwise via syringe. The solution was stirred magnetically for 2 h, at which point an additional 1 equiv of PFP-TFA was added and the reaction stirred 90 min more. The reaction was monitored by TLC viewed in 4:1 DCM:EtOAc. Upon consumption of the baseline starting material determined by TLC, the reaction solution was condensed under reduced pressure. The product was purified by flash chromatography on silica gel (2 × 15 cm). The product was loaded onto the column in DCM, and washed with DCM to elute a pale yellow band (Rf = 0.9) corresponding to hydrolyzed pentafluorophenol. This band was discarded. The bright orange product band was then eluted with 3:1 DCM:EtOAc and collected in 10 mL fractions. Those showing product by UV illumination of TLC spots were pooled, condensed under reduced pressure, and dried in vacuo. Yield 221 mg (71%, bright red-orange solid). 1H NMR (400 MHz, CDCl3, 7.26): δ 2.65 (s, 3H, bpy-CH3), 7.43 (dd, 1H, J = 5.6, 0.8, bpy-H), 8.17 (dd, 1H, J = 1.6, 6, bpy-H), 8.23 (s, 1H, bpy-H), 8.85 (d, 1H, J = 0.8, bpy-H), 8.89 (d, 1H, J = 5.6, bpy-H), 9.31 (d, 1H, J = 6, bpy-H). 13C NMR (400 MHz, CDCl3, 77.00): 21.6 (bpy-CH3), 122.99 (bpy), 125.00 (bpy), 126.50 (bpy), 129.09 (bpy), 136.51 (bpy), 143.61(bpy), 151.98 (bpy), 153.08 (bpy), 154.21 (bpy), 154.72 (PFP C-F), 157.73 (PFP C-F), 159.59 (PFP C-F), 190.07 (-O-(C=O)-Ar, ester), 194.59 (Re-CO), 194.92 (Re-CN). HPLC: 99%, 6.6 min (isocratic 1:1 MeCN:H2O w/0.1% TFA).
Synthesis of (N-Fmoc)-((2,3,6-trifluoro)-tyrosyl)-βC19; βC19 = LVGQIDSEVDTDDLSNFQL
The as-received βC19 resin (126 μmol/g for the protected peptide, 1 equiv, 992 mg resin) was added to a 20 mL econopac column (Bio-Rad). To this was added 10 mL of deblocking solution (20% v/v piperidine/DMF containing 0.1 M HOBT). The column was capped and vortexed for 10 min at room temperature. The solution was drained and the deblocking was repeated twice more. The resin was then washed with DMF (5 × 10 mL for 60 s each) and DCM (3 × 10 mL for 60 s each). To the column was then added the reaction solution, including (N-Fmoc)-2,3,6-trifluorotyrosine (343 mg, 75 mM, 6 equiv), DIPEA (250 μL, 194 mg, 0.15 M, 12 equiv), and DMF (9.75 mL). The reaction was initiated by the addition of HCTU (280 mg, 67.5 mM, 5.4 equiv). The column was vortexed for 2 h at room temperature. The solution was then drained, and the resin was again washed five times with DMF and three times with DCM, as described above. The resin was then dried in the econopac column by pulling on the outlet with vacuum for 2 h. The product was characterized by HPLC and MALDI-MS of a test cleavage of 10 mg of the resin, which confirmed the conjugation. The dry material was stored at 4 °C.
Synthesis of [Re]–F3Y–βC19
The Fmoc-F3Y–βC19 resin (122 μmol/g for the protected peptide, 1 equiv, 1.02 g resin) was added to a 20 mL econopac column (Bio-Rad). To this was added 10 mL of deblocking solution (20% v/v piperidine/DMF containing 0.1 M HOBT). The column was capped and vortexed for 10 min at room temperature. The solution was drained and the deblocking was repeated twice more. The resin was then washed with DMF (5 × 10 mL for 60 s each) and DCM (3 × 10 mL for 60 s each). The reaction solution was then added to the column, including [Re]-OPFP (224 mg, 33 mM, 2.6 equiv), HOBT (110 mg, 80 mM, 6.5 equiv), and DMF. The column was vortexed for 90 min at room temperature. The solution was then drained, and the resin was again washed five times with DMF and three times with DCM, as described above. To isolate the peptide from the resin, the econopac was then filled with 10 mL of 95/2.5/2.5 TFA/TIPS/H2O, capped, and vortexed for 4 h at room temperature. After, the solution was bright yellow and it was drained into a 20 mL scintillation vial and condensed by hand under a stream of N2 to ~2 mL. We note that TFA vapors are noxious and toxic and proper personal protective equipment is imperative. Condensation of TFA by rotary evaporation was avoided. After condensation, the solution was dripped into 45 mL of Et2O stirring in a 50 mL falcon tube, which caused immediate precipitation of a bright yellow flocculent solid. The falcon tube was then capped and incubated at 4 °C for 12 h to encourage further precipitation. The solution was then centrifuged (8,000 × g, 30 min) and the supernatant discarded. The yellow pellet was then resuspended in 25 mL of Et2O with vigorous vortexing, and recentrifuged, as before. This wash step was necessary to remove residual TFA, which makes the final peptide difficult to dissolve in dilute basic solutions. The final product was then dried for 12 h under a stream of N2. The solid was stored at 4 °C.
Purification of [Re]–F3Y–βC19
HPLC analysis of received βC19 peptide established that further purification was necessary. As such, the final peptide was purified by RP-HPLC. A separation method was first developed on analytical scale. It was found that a 2–17.5% gradient of MeCN in water with 0.1% v/v Et3N gave adequate separation in 20 min. For semi-preparative scale, this same gradient was applied to a 40 min separation time. For each semi-preparative run, fractions were collected in 0.5 min increments across the peak of interest. Injections consisted of a 500 μL solution of ~10 μmol of crude material dissolved in 5% v/v Et3N. Fractions containing the desired product, as determined by inline UV and fluorescence, were pooled and condensed in vacuo (50 mTorr). From 16 serial semi-preparative runs 30 mg of purified peptide was recovered (8% isolated yield) (Figure 3). HPLC: 98%, 9.5 min (5–15% MeCN in H2O w/0.1% Et3N over 15 min). MALDI-MS: [M–CN]+ e.m. expected 2791.03; found 2790.81.
Figure 3.

Purification, identification, and pKa of [Re]–F3Y–βC19. (a) HPLC of purified peptide on C-18 resin with a gradient of 5–15% MeCN in 0.1% aqueous Et3N. (b) UV–vis absorption spectrum of the peak at 9.5 min in (a). (c) HRMS of the peptide in (a). (d) Determination of the pKa of the phenolic proton by fluorometric titration. Each data point is the intensity at 600 nm for a 5 μM solution in aqueous buffer.
pKa of [Re]–F3Y–βC19
The pKa of the peptide was determined by fluorometric titration (Figure 3). To prepare samples at 5 μM for spectroscopy, 13 μL of a 73 μM stock solution of peptide was diluted into 187 μL of buffer at the following pH values: 5.99, 6.19, 6.41, 6.62, 6.80, 7.00, 7.20, 7.38, 7.55, 7.79, 8.00, 8.20, 8.40 and 8.58. From 5.99 to 7.00 the buffer was 100 mM potassium phosphate. From 7.20 to 8.58 the buffer was 100 mM tris. Each spectrum was recorded by exciting at 315 nm and monitoring from 475–800 nm, in 1 nm increments with 1 s integration per nm. The intensity at 600 nm was then plotted versus pH and fit using origin software according to a method previously outlined,25 with the assumption that protonation and deprotonation does not occur in the excited state.
KD of [Re]–F3Y–βC19 and wt-α2
To measure the dissociation of the photopeptide from α2, we used a previously developed26 competitive inhibition assay. The inhibitor was titrated against a solution of wt RNR. In a total volume of 150 μL, each solution contained 100 nM α2, 200 nM wt β2, 200 μM NADPH, 30 μM TR 500 nM TRR, 1 mM CDP, 3 mM ATP, inhibitor peptide, and buffer. The volume of inhibitor peptide (250 μM stock) and buffer (50 mM borate, pH 8.3 with 15 mM MgSO4 and 5% glycerol) were adjusted so that the peptide was present in the total volume at the following concentrations: 5, 15, 25, 35, and 50 μM.
Transient spectroscopy and data analysis
Solutions for time-resolved spectroscopy were prepared in 1.5 mL eppen-dorf tubes at room temperature. Reagents were added to the vial in the following order: 1st half of buffer, flash quencher (if used), ATP, CDP, peptide, 2nd half of buffer (to encourage mixing), and then protein (if used). Care was taken to avoid using pipette tips for more than one draw of a protein solution. It was found that the second draw of these very concentrated protein solutions in the same pipette tip can lead to small amounts of precipitation on the plastic sidewall. All samples were equilibrated in a water bath at 23 °C for 3 min before analysis. Unless otherwise noted, “buffer” for all transient spectroscopy samples consisted of 50 mM sodium borate (pH 8.3 at 23 °C) with 15 mM MgSO4 and 5% v/v glycerol.
Each solution was analyzed while flowing at 10 mL/min through the cuvette. Great care was taken to ensure that no bubbles were in the path length of the cuvette during measurement. In addition, we found that in alkaline solutions, the flash quencher decomposed over time into RuO2. At low concentration this side product can slowly deposit on the cell glass, and at high concentrations it rapidly clouds the solution. To alleviate these problems, during the experiment the samples were run continuously through a 13 mm 0.2 μm Supor inline filter (Pall Corp., Port Washington, NY) to remove the byproduct as it is generated. We found that 650 μL was the minimum volume required to flow a sample through the cuvette, pump lines, and inline filter.
Each buffered solution used for time-resolved emission measurements of [Re]–F3Y–βC19 contained in final concentration: 10 μM [Re]–F3Y–βC19, 200 μM Ru(NH3)6Cl3, 1 mM CDP, and 3 mM ATP. Each 650 μL buffered solution used for time-resolved emission measurements of [Re]–F3Y–βC19 with α2 variants contained in final concentration: 10 μM [Re]–F3Y–βC19, 20 μM α2 mutant, 200 μM Ru(NH3)6Cl3, 1 mM CDP, 3 mM ATP. Each buffered solution used for transient absorption measurements of [Re]–F3Y–βC19 contained in final concentration: 50 μM [Re]–F3Y–βC19, 1 mM Ru(NH3)6Cl3, 1 mM CDP, 3 mM ATP. Each 650 μL buffered solution used for transient absorption measurements of [Re]–F3Y–βC19 with α2 variants contained in final concentration: 50 μM [Re]–F3Y–βC19, 100 μM α2 mutant, 1 mM Ru(NH3)6Cl3, 1 mM CDP, 3 mM ATP.
The calculation of rate constants for the oxidation of Y by •F3Y was performed using the equation
| (1) |
where kon (τon) is the rate constant (time constant) for •F3Y decay when the tyrosyl is in the “on” conformation, bound to the protein, and koff (τoff) is the rate constant (time constant) for •F3Y decay when the tyrosyl is in the “off” solvated, conformation. The error of each measurement was propagated using standard methods.27 Transient absorption spectra were an average of three independently collected data sets, corrected for noise inherent to the instrument by fast Fourier transform (FFT) filtering of high frequency noise across the data set. To determine the filter level, a FFT was first performed on the raw data to determine the frequency of signal/noise cutoff. That cutoff filter was then applied to the data set using Origin (see software, SI). Kinetic decay traces were collected from the PMT as intensity values corresponding to a change in voltage. Transient optical density was calculated using,
| (2) |
V0 was determined by averaging the first 40 data points collected before the 0 time point.
Each decay trace from which a lifetime was calculated was an average of 3–6 individually collected data sets. The standard deviation at each x,y pair was used as the weight term in the fitting. All rate constants were calculated using weighted least-squares regression analysis of Cartesian data pairs in Origin by modulation of variables until the reduced ξ2 ceased changing. The goodness-of-fit parameter (R2) was used as a starting point for determining the accuracy of fit; all fits reported are 0.98 or greater R2. Subsequently, graphical residual analysis was employed. Those residuals demonstrating significant asymmetry or periodicity with respect to the independent variable were fit again with an additional phase. Error bars were calculated as 67% confidence intervals (one standard deviation). Decays of short (ns) lifetimes were fit using all data points appearing after the time point with maximum amplitude. Decays of radical lifetimes were fit by excluding those data points corresponding to the residual charge-separated state of [Re] by starting at the 1 μs timepoint.
Results
Synthesis, purification and characterization of materials
The synthesis of [Re]–F3Y–βC19 was accomplished on solid phase using established methods. The identity of the modified peptide was confirmed by high-resolution MALDI-TOF MS, its purity by HPLC, and the pKa of the F3Y phenol when incorporated within the peptide was found to be 7.1 ± 0.1; these data are shown in Figure 3. The observed pKa of F3Y in [Re]–F3Y–βC19 is in accordance with previous measurements of model dipeptides of the FnY residue.17 We determined the KD between the α2 subunit and the [Re]–F3Y–βC19—under the conditions of spectroscopy—to be 9 ± 1 μM using a competitive inhibition assay (Figure S1), which agrees with measurements on our previous photoRNR systems.18 Due to the modest affinity of the peptide for the subunit, the spectroscopic measurements require large amounts of α, thus the expression and purification of each α2 (Figure 2) was accomplished on g scale with hexahistidine affinity chromatography.
[Re]–F3Y–βC19 charge-separated state
Spectroscopic observation of F3Y radical injection into α2 requires a detailed understanding of all transient spectroscopic features present. The generation of the F3Y radical was confirmed by comparing the excitation of a peptide that cannot be oxidized, [Re]–F–βC19, to the peptide containing the fluorotyrosine. Photolysis of [Re]–F–βC19 at pH 8.3 generates a [ReI]* (3MLCT) excited state, which cannot oxidize the adjacent phenylalanine. The [ReI]* can be monitored by recording the decay of the emission intensity at λmax = 610 nm; a monoexponential lifetime of 59 ns is observed.28 In contrast to [Re]–F–βC19, excitation of [Re]–F3Y–βC19 at pH 8.3 generates [ReI]* that decays with biphasic kinetics (Figure S2). The long component (58 ns, 19%) corresponds to [ReI]* that is unchanged by the proximal F3Y, while the short component (22 ns, 81%), represents the quenching of [ReI]* by the deprotonated F3Y−. The decay is biexponential as a result of measuring two chemical species in solution—protonated and deprotonated tyrosine.
Whereas the emission decay of the [ReI]* can be used to reveal the quenching of the 3MLCT, the photoproducts of the quenching reaction may be captured by transient absorption (TA) spectroscopy. Figure 4 shows the time-resolved spectral features upon excitation of [Re]–F3Y–βC19 at three different time points. The absorption features for [ReI]* appear as broad bands at 380 and 480 nm;28 the decay of the transient signal at these two TA wavelengths yields decay time constants of 60 ± 2 and 61 ± 2 ns, respectively. In addition, new spectral features corresponding to the bpy•− and the •F3Y appear in the spectrum at λmax = 525 and 425 nm, respectively.29 The •F3Y signal decays monoexponentially with a time constant of 68 ns. In contrast, the signal for the bpy•− is best fit to a biexponential as a combination of growth and decay. This complication in the measured TA kinetics arises from the spectral congestion between the emission of [ReI]* excited state (τ = 22 ns) and the decay of bpy•− anion over 76 ns. The appearance of signals for both the •F3Y and the bpy•− anion with the concomitant disappearance of [ReI]* establishes that the excited state is quenched by deprotonated tyrosine in an intramolecular charge transfer event to result in a charge-separated [Re(bpy•−)]− •F3Y–βC19 intermediate. The duration of this charge-separated state agrees well with previously reported lifetimes for model compounds.19
Figure 4.
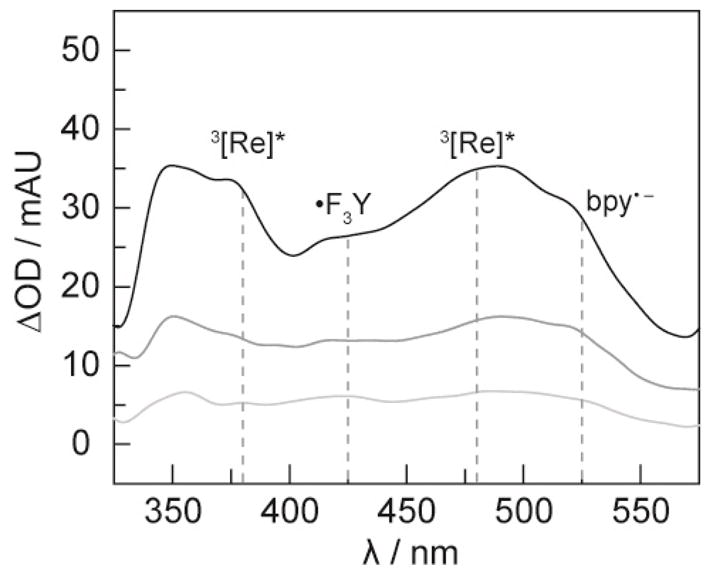
TA spectra of the charge-separated state [Re(bpy•−)]–•F3Y–βC19 collected 25 ns (top), 75 ns (middle) and 125 ns (bottom) after a 7 ns excitation pulse of laser light, λexc = 355 nm. Lifetime decays of the transient spectrum were measured at the four wavelengths indicated by the dashed lines: λdet at 380 and 480 nm correspond to the 3MLCT of [ReI]; 28 λdet = 525 nm is that of bpy•−;29 and, λdet = 425 nm is that of •F3Y. The solution contained 50 μM [Re]–F3Y–βC19 with 1 mM CDP, 3 mM ATP, 15 mM MgSO4, and 5% glycerol in 50 mM sodium borate (pH 8.3).
Generation of a long-lived •F3Y by the flash-quench method
The rapid decay of the charge-separated state (discussed above) on a sub 100 ns timescale precludes faithful measurements of radical injection rates slower than 107 s−1. The likelihood for oxidative injection from the •F3Y radical into α2 increases as the lifetime of the radical increases. The •F3Y lifetime can be increased by employing the flash-quench method to irreversibly remove the electron from the quenched [ReI]* excited state by electron transfer to a flash quencher (FQ), Ru(NH3)6Cl3. By removing the electron from the system with the FQ, back electron transfer is averted and the flash-quenched ReII is able to oxidize F3Y. Spectra of •Y and the •F3Y were collected 50 ns after irradiating a solution of either [Re]–Y–βC19 (at pH 12) or [Re]–F3Y–βC19 (at pH 8.3) in borate buffer and in the presence of 200 mol equiv of FQ. The spectra, which are reproduced in Figure S3, exhibit the expected isolated peaks for the •F3Y radical at λmax = 425 nm and the •Y radical at λmax = 412 nm. We also observe that the •F3Y radical in tris buffer shifts to λmax = 418 nm.
The optimal FQ concentration was determined by titrating solutions of 50 μM [Re]–F3Y–βC19 with 5–1000 mol equiv of FQ. As Figure S4 shows, the best concentration of FQ was determined to be 20 mol equiv, which was the concentration used to perform all subsequent kinetic analysis involving the •F3Y radical. At 20 mol equiv of FQ, not all of the [ReI]* is quenched, and thus the flash quench TA spectrum of [Re]–•F3Y–βC19 taken 25 ns after laser excitation (λexc = 355 nm) appears as a sum of the •F3Y and the remaining charge-separated state. Figure S5 shows the decay profile of the TA spectrum at λdet = 425 nm. A fast time component of 100 ns corresponding to the recombination of the charge separated state is followed by a slower time component corresponding to the decay of the •F3Y radical produced by flash quenching. The TA spectral profile of the flash-quenched radical may be spectrally isolated by delaying its detection (typically by 0.5–1 μs) until after the decay of the charge-separated state. Figure 5a shows the TA spectrum of a flash-quenched peptide 500 ns after laser excitation; the λmax = 425 nm of •F3Y is clearly observed. The radical, monitored at 395, 410, and 425 nm, decays monoexponentially with lifetimes of τ395 = 14.7 ± 0.2 μs, τ410 = 13.5 ± 0.2 μs and τ425 = 14.5 ± 0.4 μs, Figure 5b. In this optimized system, the photochemical yield of •F3Y formation is calculated to be 4.9% (see SI).
Figure 5.

(a) TA spectra and of [Re]–•F3Y–βC19 collected 500 ns (top), 5 μs (middle) and 30 μs (bottom) after a 7 ns 355 nm excitation pulse of laser light. (b) Lifetime decay of TA signal at 395 (
 ), 410 (
), 410 (
 ), and 425 (
), and 425 (
 ) nm. The monoexponential fit is shown by the solid lines. The solution contained 50 μM [Re]–F3Y–βC19 with 20 equiv (1 mM) Ru(NH3)6Cl3, 1 mM CDP, and 3 mM ATP, 15 mM MgSO4 and 5% glycerol in 50 mM borate buffer (pH 8.3). Colored vertical bands (a) indicate the portion of the spectrum polled for recording the kinetic decays in (b).
) nm. The monoexponential fit is shown by the solid lines. The solution contained 50 μM [Re]–F3Y–βC19 with 20 equiv (1 mM) Ru(NH3)6Cl3, 1 mM CDP, and 3 mM ATP, 15 mM MgSO4 and 5% glycerol in 50 mM borate buffer (pH 8.3). Colored vertical bands (a) indicate the portion of the spectrum polled for recording the kinetic decays in (b).
Two conformations of [Re]–F3Y–βC19 bound to α2
The relatively weak binding between [Re]-X-βC20 and α2 manifests in the dynamics of the peptide N-terminus, as we have shown previously.28 Crystallographic measurements of βC20 and α2 reveal the C-terminal 16 amino acids of the peptide, but the N-terminal four are not visible, suggesting that they are not held tightly to the protein surface. Both the [Re] and the Y356 are located in this flexible region of the peptide, and when [Re]–X–βC19 is in the presence of α2, the lifetime of the [ReI]* reports on the local environment of both the [Re] complex as well as the proximal fluorotyrosine. The decay signal from the [ReI]*—in the presence of an amino acid that it cannot oxidize—is biexponential when the peptide is bound to the protein.28 The fast time decay, at 60 ns, is similar to [ReI]* in solution (59 ns), and was assigned to an “off” state where the chromophore is largely solvated. A longer time decay, at 155 ns, was ascribed to the N-terminus binding closely to the surface of α2 in an “on” state, owing to occluding solvent from [Re].
The [ReI]* lifetime measurements were repeated for the [Re]–X–βC19 peptide with non-oxidizable (X = F) and oxidizable (X = F3Y) amino acids associated to each of the α2 mutants prepared here. All decays were biexponential; the lifetimes obtained from a fit of the lifetime decay of [Re]–F–βC19 are shown in Figure 6a. The “off” lifetime increases marginally from 52 to 60 ns, and the “on” lifetime of 180 ns is the same for all four mutant proteins. A biphasic decay for the [Re]–F3Y–βC19 peptide bound to α2 is also obtained (Figure 6b), but with significantly attenuated lifetimes (“on” lifetime of 29 ns, “off” lifetime of 100 ns) owing to the quenching of [ReI]* by F3Y− as described above. The similarity of the percentage of the short and long lifetime components to the overall decay suggests similar binding conformations for both peptides. It is likely that all four states of the [ReI]* (on/quenched, off/quenched, on/unquenched, off/unquenched) are present in solution.
Figure 6.
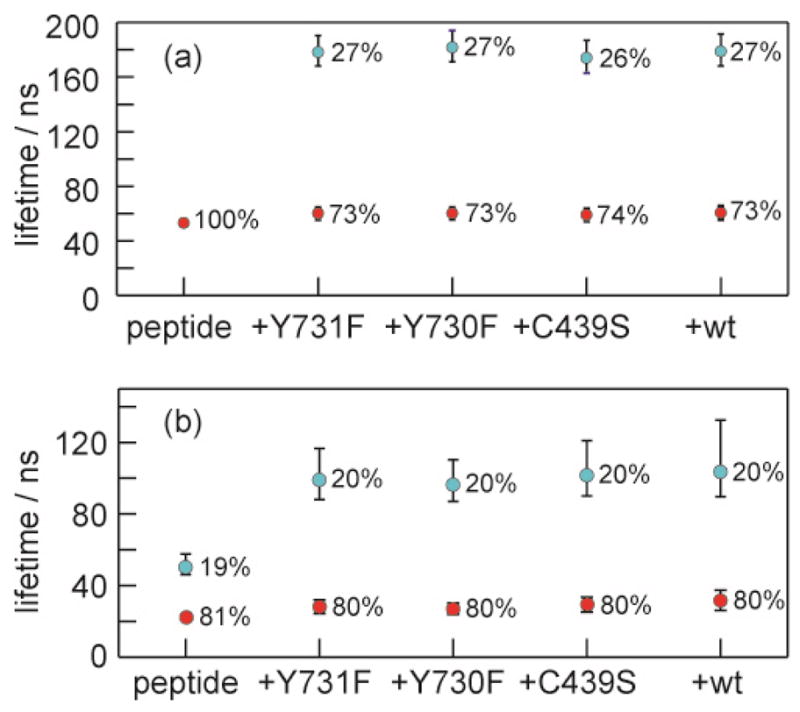
Lifetime of [ReI]* excited state of 10 μM [Re]–X–βC19 for (a) X = F and (b) X = F3Y peptide in the presence of α2 mutants at 20 μM, in a solution of 1 mM CDP, 3 mM ATP, 15 mM MgSO4, and 5% glycerol, in 50 mM borate (pH 8.3). For biexponential decays, the percentage contribution of short and long components to the decay is listed.
[Re]–•F3Y–βC19 lifetimes in the presence of α2 mutants
The emission lifetimes permit the formation of the •F3Y to be monitored. Radical injection, however, requires that the •F3Y on the photopeptide in the presence of each α2 mutant be directly observed by TA spectroscopy. Figure S6a shows the spectra recorded at 1 μs for the peptide alone, and in the presence of each protein. For the free peptide and for the Y731F mutant, for which radical injection is blocked, the typical spectrum of the •F3Y with λmax = 425 nm is observed, with a small shoulder at 410 nm; this spectral profile is typical of fluorotyrosyl radicals.13 Figure S6b displays the TA spectrum of •F3Y in the presence of mutants that are theoretically capable of radical injection. The radical profile changes significantly. The λmax = 425 nm broadens and a pronounced shoulder at λmax = 400 nm appears; the spectrum of the C439S mutant shown in Figure 7a is representative. We chose to monitor kinetics at three wavelengths, 425, 410 and 395 nm, as these correspond to the λmax of •F3Y, its typically observed shoulder, and the new, shifted shoulder that appears in the presence of the protein. For Y731F and Y730F, the TA signal decays monoexponentially at all wavelengths. In contrast, the signals for [Re]–•F3Y–βC19 in the presence of C439S and wt mutants decay biexponentially with a significantly shorter lifetime component. Figure 7b shows the decay of •F3Y for the C439S mutant; the residuals for the mono- and biexponential fits are shown in Figure S7. A summary of the lifetimes of the TA signal of •F3Y at the three different wavelengths for the free peptide and the peptide in the presence of Y731F-α2, Y730F-α2, C439S-α2 as well as the wt-α2 is plotted in Figure 8; a tabulation of these lifetimes and their amplitude components for the four systems is given in Table S1.
Figure 7.

(a) TA spectra of [Re]–•F3Y–βC19 in the presence of C439S-α2 collected 1 μs (top), 15 μs (middle) and 30 μs (bottom) after a 7 ns 355 nm excitation pulse of laser light. (b) Lifetime decay of TA signal at 395 (
), 410 (
), and 425 (
) nm. The biexponential fit is shown by the solid lines. The solution contained 50 μM [Re]–F3Y–βC19, 100 μM C439S-α2, 20 equiv (1 mM) Ru(NH3)6Cl3, 1 mM CDP, and 3 mM ATP, 15 mM MgSO4 and 5% glycerol in 50 mM borate buffer (pH 8.3). Colored vertical bands (a) indicate the portion of the spectrum polled for recording the kinetic decays in (b).
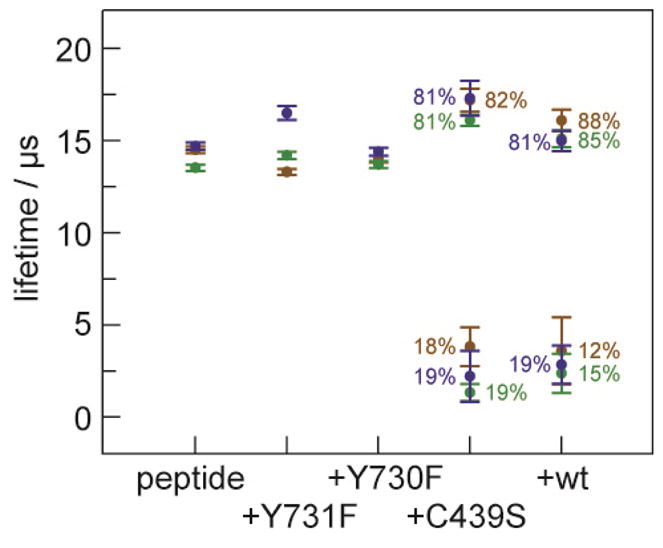
Figure 8.
Lifetime of [Re]–•F3Y–βC19 peptide, and it in the presence of the four α2 variants. Decay lifetimes of TA signal of •F3Y were observed at 395 (
 ), 410 (
), 410 (
 ) and 425 (
) and 425 (
 ) nm. For the peptide in solution, in the presence of Y731F-α2, or Y730F-α2, there is no significant change in radical lifetime. With C439S-α2 and the wt-α2 subunit, the TA signal of •F3Y exhibits a biexponential decay owing to the contribution of a significantly shorter lifetime component arising from radical injection into the protein.
) nm. For the peptide in solution, in the presence of Y731F-α2, or Y730F-α2, there is no significant change in radical lifetime. With C439S-α2 and the wt-α2 subunit, the TA signal of •F3Y exhibits a biexponential decay owing to the contribution of a significantly shorter lifetime component arising from radical injection into the protein.
Discussion
The unnatural tyrosine—2,3,6-trifluorotyrosine (F3Y)—permits a photoRNRα2 to be constructed by binding [Re]–F3Y–βC19 to α2. The F3Y-photoRNRα2 construct provides three important features to enable the kinetics of radical initiation and propagation to be uncovered. First, Figure 3d establishes the pKa of F3Y, when incorporated into the full-length peptide, to be 7.1 ± 0.1. In order to generate proton-independent oxidation of the F3Y by the [ReI]* (electron transfer only), the F3Y must be in its deprotonated state. Thus the proton-independent oxidation of F3Y by [ReI]* may occur at a mild pH; we chose to perform spectroscopic experiments at pH 8.3 where F3Y is ~94% deprotonated. Second, the reduction potential of F3Y is ~180 mV above tyrosine at pH 8.3,13 thus providing a driving force for oxidation of the tyrosines of α2. Third, the absorption maximum of the •F3Y is sensitive to its local environment, shifting 7 nm by changing buffer identity in solution (Figure S6). This spectral shift allows •F3Y to be spectrally isolated and the radical can be time resolved at multiple wavelengths.
F3Y radical photoinitiation
The F3Y radical is generated efficiently by laser flash photolysis of [ReI] incorporated within the [Re]–F3Y–βC19 peptide. The TA spectrum of the photolyzed peptide in the presence of substrate, effector, and buffer establishes that the 3MLCT excited state of [Re], [ReI]*, is readily quenched by F3Y− to generate the intramolecular charge-separated state [Re(bpy•−)]− •F3Y–βC19. The absorption features characteristic of the charge transfer photoproducts, •F3Y and bpy•−, are observed in the TA spectrum of Figure 4. These features decay concomitantly with one another, and they persist for a time longer than the [ReI]* excited state. Identical lifetimes for the charge-separated state were observed for each of the protein mutants prepared here.
In order to extend the lifetime of •F3Y and isolate its spectral signatures, radical photoinitiation was performed in the presence of the reversible flash-quench reagent Ru(NH3)6Cl3. The RuIII complex oxidatively quenches the [ReI]* within the laser pulse to furnish the [ReII] ground state.28 [ReII] is also thermodynamically competent for tyrosinate oxidation,29 subsequently regenerating the [ReI] ground state and a free •F3Y. Since neither the [ReI]* nor the intramolecular charge-separated state are present upon flash quenching, the resulting TA features are solely representative of the radical. Spectra of •F3Y and •Y under flash quench conditions clearly show disparate absorption maxima (Figure S3), which concur with those observed for tyrosyl radicals in the βC19 construct that were generated stoichiometrically with a benzophenone photooxidant.29 In addition, we found that the λmax of the •F3Y changes as a function of buffer composition, highlighting the sensitivity of the radical to its environment (Figure S6). There is an optimum concentration of the FQ reagent. High concentrations of FQ decrease the observed yield of radical because contaminating RuII (resulting from the flash quench process) is present at higher concentration in solution, which is able to reduce the photogenerated radical. Figure S4 shows that 20 mol equiv engenders the highest yield of radical with the longest observable lifetime.
Spectroscopy of [Re]–F3Y–βC19 bound to α2 mutants
The equilibrium binding of [Re]-labeled peptides with the α2 subunit can be measured by fluorometric titration.28 However, in this case, the quenching of [Re] by the deprotonated F3Y− was so efficient that the emission intensity of [ReI]* was too low to provide a reliable measurement of the binding between the peptide and α2. Thus we reverted to competitive inhibition assay measurements to reveal that KD is 9 ± 1 μM. With this KD, spectroscopic measurements employed a mixture of 50 μM peptide and 100 μM α2 to ensure that 86% of the peptide was bound to α2.
Even in this bound state, the N-terminus of the photopeptide resides in two states, one in which it is closely associated with the α2 subunit (“on”) and one in which it is largely solvent exposed (“off”).28 This on/off dynamic was measured by monitoring the emission lifetime of the [ReI]* for a control [Re]–F–βC19 peptide, Figure 6a. The lifetime of the solvent-exposed excited state is much shorter than when it is adsorbed to the protein surface. For the experiments reported here, we also wished to define this two state dynamic for the fluorotyrosine-containing peptide bound to each of the point mutated α2 proteins. All constructs displayed the two-state behavior, as shown by Figure 6b. We note that the [ReI]* lifetime of the F3Y–βC19:Y731F-α2 construct corresponding to the “on” state (τ = 29 ns) is unquenched relative to its lifetime in buffered solution (τ = 22 ns). This observation confirms that the highly oxidizing [ReI]* is unable to extract electrons from protein sidechains via direct oxidation of tyrosine within the protein. Consistent with this observation, the “on” lifetime of the [ReI]* is the same for all constructs (see Figure 6). Thus the primary quenching pathway of the [ReI]* is electron transfer solely from the adjacent F3Y on the peptide.
The decay of [ReI]* is accompanied by the appearance of absorption features characteristic of •F3Y at λmax = 425 nm in borate buffer and λmax = 418 nm in an otherwise identical solution of the mildly more hydrophobic Tris buffer (Figure S3). The lifetime of the photogenerated •F3Y is provided, independent of protein oxidation, by the [Re]–F3Y–βC19:Y731F-α2 construct (Figure 1a). As summarized in Figure 8, the lifetime of the •F3Y marginally depends upon the wavelength of observation (τ395 = 13.3 ± 0.2 μs, τ410 = 14.2 ± 0.2 μs, and τ425 = 16.5 ± 0.4 μs). At λdet = 425 nm, this lifetime is slightly shorter than that observed for the free peptide in solution. The lifetime at λdet = 410 nm matches the solution lifetime, and the lifetime at λdet = 395 nm is longer than the lifetime when protein is not present. These lifetimes correspond to k0 denoted in Figure 1. This heterogeneity suggests that the radical, whose spectral features are sensitive to local environment, may experience multiple conformations when in the presence of the unnatural phenylalanine on the surface of the protein. The local environment at Y356 does depend on the residue at 731 in the intact α2: β2 system. The •Y356 radical may be trapped when •NO2Y122 is introduced into β2.30 The g-value for trapped •Y356 in Y731F-α2 (2.0073) is shifted relative to wt-α2 (2.0063). The g-value for •Y in proteins is known to vary between 2.006 and 2.009 as a function of local environment, with the lower values corresponding to tyrosyl radicals involved in hydrogen-bonding. This result implies that •Y356 is involved in hydrogen-bonded stabilization in the wt enzyme, but not Y731F-α2. Together, these results suggest that the radical in the presence of Y731F experiences multiple solution/surface conformations. Of these different conformations, the amplitude of the monoexponential lifetime of •F3Y conveys that a significant portion of the peptide N-terminus (20–30%) is in the conformer of the “on” state. However, even in this conformation, •F3Y cannot competently inject the radical into α2 owing to the phenylalanine block.
Radical propagation into α2
With the ability to isolate the •F3Y radical and temporally profile its lifetime in the absence of protein oxidation, the kinetics for radical transfer along the RNR pathway are revealed for the first time. The F point mutation was moved from position 731 to position 730 (Figure 1b), which would allow for the oxidation of Y731 but block any further transport of the radical into the α2 subunit. Photogeneration of [Re]–•F3Y–βC19 in the presence of Y730F-α2 again reveals monoexponential decays at the three wavelengths: 425 nm, 14.0 ± 0.2 μs; 410 nm, 13.7 ± 0.2 μs; 395 nm, 14.4 ± 0.2 μs. The lifetimes are all within error of one another, suggesting that the radical is in a homogenous environment in the presence of the native Y731, to which it can hydrogen bond. A new spectral feature at 400 nm is a signature of this hydrogen-bonded environment. This result suggests that the radical residing in the “on” conformations has a spectral signature that is shifted to higher energy relative to the free peptide or the dynamic Y731F radical. Though both “on” and “off” conformations are present, the observed lifetime of •F3Y should be monoexponential as it corresponds to the sum of k0 and k1 (Figure 1b). Inasmuch as (k0 + k1) of [Re]–F3Y–βC19:Y730F-α2 is similar to k0 of [Re]–F3Y–βC19:Y731F-α2Y731F, we conclude that radical injection into the surface of the protein is blocked when the radical cannot further propagate along the pathway.
Consistent with this contention, dramatic changes occur when Y730 is present to propagate the radical into the subunit. As shown by the data in Figure 8, the rate of [Re]–•F3Y–βC19 decay increases significantly for both the C439S-α2 mutant and the wt enzyme. In each case, both Y730 and Y731 are present, and in each case the lifetimes at all wavelengths become biexponential. For C439S, in which a serine substitutes for the active site cysteine (Figure 1c) the TA signal at 395 nm consists of a long phase (17.3 ± 0.9 μs, 81%) and a short phase (2.2 ± 1 μs, 19%) (see Figure 7b). Similar pairs of lifetimes were observed for 410 and 425 nm, as shown in Table S1. The amplitudes of each signal can be used to assign each phase of decay to a conformation on the protein surface. As in the measurement of emission decay of the [ReI]*, the large amplitude component (80%) of the radical decay is attributed to the portion of the peptide that is largely solvated, in the “off” state. The small amplitude (20%) matches the “on” state. In this conformation, the •F3Y lifetime is much shorter, indicating that it is able to oxidize Y731. Previous analysis of the crystal structure of wt-α2 concluded that there is hydrogen-bonding between Y731 and Y730.8 Since the installation of Y730F—a change of only one hydroxyl group from Y730—prevents oxidation of Y731 from occurring, we postulate that the enzyme has evolved to employ the hydrogen bonding between Y730 and Y731 (a Y–Y dyad) in proton-coupled tyrosine oxidation.
The “off” lifetime then serves as a baseline (k0, Figure 1c) for the rate of oxidation of Y–Y by •F3Y. Substituting the averages across the three wavelengths for the lifetime of the radical in the “on” (2.5 ± 1.0 μs) and “off” (16.9 ± 0.7 μs) states into Eq. 1 yields a rate constant for radical injection of C439S-α2 to be (4 ± 2) × 105 s−1. This injection event is rate limited by the slower of k1 and k2, Figure 1c. Since oxidation of Y731 is not possible without pre-arrangement with Y730, as shown in the Y730F-α2 mutant, we believe k1 is the limiting step. A similar oxidation event is observed for the wt enzyme, which contains the active site cysteine. The averaged lifetimes for the long phase (15.4 ± 0.5 μs) and the short phase (3.0 ± 1.4 μs) of •F3Y in [Re]–F3Y–βC19:wt-α2 correlate to an injection rate of (3 ± 2) × 105 s−1. As in the C439S-α2 experiment, this rate constant corresponds to the rate-limiting step among k1, k2 and k3, Figure 1d, and it is within error limit of the injection kinetics observed in [Re]–F3Y–βC19:C439S-α2. The requirement of hydrogen-bonding between Y731 and Y730 again lead us to conclude that the rate-limiting oxidation for [Re]–F3Y–βC19:wt-α2 is k1, Figure 1d.
The need for the Y–Y dyad to promote radical injection into α2 is consistent with the PCET reactivity of tyrosine in model compounds. The rate constant for oxidation of tyrosine depends dramatically on the proximity of a hydrogen-bonding partner to the phenol.31–34 Specifically, in a series of phototriggered RuII(bpy)3–Y model dyads, the addition of a carboxylate at the ortho position of the tyrosine phenol induces a hydrogen bond and increases the rate for PCET at pH 8 from 104 s−1 to 105–106 s−1.31 The criticality of coupling oxidation to a hydrogen bond is further demonstrated by its ability to drive less favored reactions. For instance, a pair of RuII(bpy)3–Y models containing benzimidazole hydrogen-bond partners were synthesized in which one congener contained,35 a 0.2 eV lower driving force but a 0.2 Å shorter hydrogen-bond distance. The shorter hydrogen bond drives the rate of oxidation by nearly one order of magnitude greater than for the congener with the greater thermodynamic driving force but longer hydrogen bond.35 These observations for model tyrosyl radical systems are in line with the similarity of the observed radical kinetics of [Re]–F3Y–βC19:Y731F-α2 and [Re]–F3Y–βC19:Y730F-α2. In [Re]–F3Y–βC19:Y730F-α2, a hydrogen bond to Y730 is absent thus obviating PCET.
Conclusion
These results reported herein show that radical injection and propagation in RNR with any appreciable rate requires that both Y731 and Y730 be in place to establish the hydrogen-bonded network needed for PCET. With the Y–Y dyad present, we are able to transiently monitor a radical on the RNR pathway for the first time. The rate for radical injection and transport in α2 is fast, on the order of 3 × 105 s−1. The ability to shut down this efficient PCET pathway for radical propagation by modifying the hydrogen-bonding network of the amino acids composing a co-linear PCET pathway in α2 suggests a finely tuned evolutionary adaptation of RNR to control radical transport in this enzyme.
Supplementary Material
Acknowledgments
Funding Sources
The authors gratefully acknowledge the National Institutes of Health (GM 47274, D.G.N; GM 29595 J.S.) for provided funding to enable this research. P.G.H acknowledges a fellowship from the National Institutes of Health (GM 087034).
We are grateful to Ellen C. Minnihan for her expertise and time.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Material vendors and abbreviations used; sequence of primers used in mutagenesis; full description of all tools and instruments; calculations of uncertainty; [Re]–F3Y–βC19 emission quenching at pH 8.3; residuals of C439S and wt radical decays; numerical table of radical lifetimes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nordlund P, Reichard P. Annu Rev Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 2.Kolberg M, Strand KR, Graff P, Kristoffer Andersson K. Biochim Biophys Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Kashlan OB, Scott CP, Lear JD, Cooperman BS. Biochemistry. 2002;41:462–474. doi: 10.1021/bi011653a. [DOI] [PubMed] [Google Scholar]
- 4.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 5.Reece SY, Nocera DG. In: Quantum Tunneling in Enzyme Catalyzed Reactions. Scrutton N, Allemann R, editors. Royal Society of Chemistry Press; London: 2009. [Google Scholar]
- 6.Reece SY, Hodgkiss JM, Stubbe J, Nocera DG. Phil Trans Royal Soc B. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reece SY, Nocera DG. Annu Rev Biochem. 2009;78:673–699. doi: 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uhlin U, Eklund H. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- 9.Ekberg M, Sahlin M, Eriksson M, Sjoberg BM. J Biol Chem. 1996;271:20655–20659. doi: 10.1074/jbc.271.34.20655. [DOI] [PubMed] [Google Scholar]
- 10.Aberg A, Hahne S, Karlsson M, Larsson A, Ormö M, Ahgren A, Sjöberg BM. J Biol Chem. 1989;264:12249–12252. [PubMed] [Google Scholar]
- 11.Larsson A, Sjöberg BM. EMBO J. 1986;5:2037–2040. doi: 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkin SE, Chen S, Ley BA, Mangravite L, Edmondson DE, Huynh BH, Bollinger JM. Biochemistry. 1998;37:1124–1130. doi: 10.1021/bi9723717. [DOI] [PubMed] [Google Scholar]
- 13.Seyedsayamdost MR, Reece SY, Nocera DG, Stubbe J. J Am Chem Soc. 2006;128:1569–1579. doi: 10.1021/ja055926r. [DOI] [PubMed] [Google Scholar]
- 14.Seyedsayamdost MR, Argirevi T, Minnihan EC, Stubbe J, Bennati M. J Am Chem Soc. 2009;131:15729–15738. doi: 10.1021/ja903879w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minnihan EC, Seyedsayamdost MR, Stubbe J. Biochemistry. 2009;48:12125–12132. doi: 10.1021/bi901439w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minnihan EC, Seyedsayamdost MR, Uhlin U, Stubbe J. J Am Chem Soc. 2011;133:9430–9440. doi: 10.1021/ja201640n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennati M, Robblee JH, Mugnaini V, Stubbe J, Freed JH, Borbat J. Am Chem Soc. 2005;127:15014–15015. doi: 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- 18.Yokoyama K, Uhlin U, Stubbe J. J Am Chem Soc. 2010;132:15368–15379. doi: 10.1021/ja1069344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang MCY, Yee CS, Stubbe J, Nocera DG. Proc Natl Acad Sci USA. 2004;101:6882–6887. doi: 10.1073/pnas.0401718101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2007;129:13828–13830. doi: 10.1021/ja074452o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seyedsayamdost MR, Stubbe J. J Am Chem Soc. 2006;128:2522–2523. doi: 10.1021/ja057776q. [DOI] [PubMed] [Google Scholar]
- 22.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2007;129:8500–8509. doi: 10.1021/ja0704434. [DOI] [PubMed] [Google Scholar]
- 23.Seyedsayamdost MR, Yee CS, Stubbe J. Nat Protoc. 2007;2:1225–1235. doi: 10.1038/nprot.2007.159. [DOI] [PubMed] [Google Scholar]
- 24.Thelander L. J Biol Chem. 1973;248:4591–4601. [PubMed] [Google Scholar]
- 25.Novikov E, Stobiecka A, Boens N. J Phys Chem A. 2000;104:5388–5395. [Google Scholar]
- 26.Climent I, Sjoeberg BM, Huang CY. Biochemistry. 1992;31:4801–4807. doi: 10.1021/bi00135a009. [DOI] [PubMed] [Google Scholar]
- 27.Harris D. Quantitative Chemical Analysis. 6. W. H. Freeman; New York: 2003. [Google Scholar]
- 28.Reece SY, Lutterman DA, Seyedsayamdost MR, Stubbe J, Nocera DG. Biochemistry. 2009;48:5832–5838. doi: 10.1021/bi9005804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reece SY, Seyedsayamdost MR, Stubbe J, Nocera DG. J Am Chem Soc. 2006;128:13654–13655. doi: 10.1021/ja0636688. [DOI] [PubMed] [Google Scholar]
- 30.Yokoyama K, Smith AA, Corzilius BRG, Griffin RG, Stubbe J. J Am Chem Soc. 2011;50 doi: 10.1021/ja207455k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irebo T, Johansson O, Hammarström L. J Am Chem Soc. 2008;130:9194–9195. doi: 10.1021/ja802076v. [DOI] [PubMed] [Google Scholar]
- 32.Irebo T, Reece SY, Sjödin M, Nocera DG, Hammarström L. J Am Chem Soc. 2007;129:15462–15464. doi: 10.1021/ja073012u. [DOI] [PubMed] [Google Scholar]
- 33.Rhile IJ, Mayer JM. J Am Chem Soc. 2004;126:12718–12719. doi: 10.1021/ja031583q. [DOI] [PubMed] [Google Scholar]
- 34.Markle TF, Mayer JM. Angew Chem Int Ed. 2008;47:738–740. doi: 10.1002/anie.200702486. [DOI] [PubMed] [Google Scholar]
- 35.Zhang MT, Irebo T, Johansson O, Hammarström L. J Am Chem Soc. 2011;133:13224–13227. doi: 10.1021/ja203483j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.