

Abstract
Traumatic brain injury (TBI) is a major environmental risk factor for subsequent development of Alzheimer disease (AD). Pathological features that are common to AD and many tauopathies are neurofibrillary tangles (NFTs) and neuropil threads composed of hyperphosphorylated tau. Axonal accumulations of total and phospho-tau have been observed within hours to weeks and intracytoplasmic NFTs have been documented years following severe TBI in humans. We previously reported that controlled cortical impact TBI accelerated tau pathology in young 3×Tg-AD mice. Here, we used this TBI mouse model to investigate mechanisms responsible for increased tau phosphorylation and accumulation following brain trauma. We found that TBI resulted in abnormal axonal accumulation of several kinases that phosphorylate tau. Notably, c-Jun N-terminal kinase (JNK) was markedly activated in injured axons and colocalized with phospho-tau. We found that moderate reduction of JNK activity (40%) by a peptide inhibitor, DJNKi1, was sufficient to reduce total and phospho-tau accumulations in axons of these mice with TBI. Longer-term studies will be required to determine whether reducing acute tau pathology proves beneficial in brain trauma.
Keywords: c-Jun N-terminal kinase, Controlled cortical impact, D-JNKi1, Kinase, Phosphorylation, tau, Traumatic brain injury
INTRODUCTION
Progressive accumulation of hyperphosphorylated microtubule-associated protein tau into neurofibrillary tangles (NFTs) and neuropil threads is a common feature of many neurodegenerative tauopathies, including Alzheimer disease (AD), Pick disease, progressive supranuclear palsy, and frontotemporal dementias (1). Tau pathology has also been documented in individuals who suffered from a single severe traumatic brain injury (TBI) or multiple mild, concussive injuries. In particular, acute axonal accumulations of total and phospho-tau have been documented within hours to weeks (2, 3), whereas NFTs have been detected years following single severe TBI in humans (4). Furthermore, NFT pathology is widespread in patients with lifetime histories of multiple concussive injuries (5–11). Tau pathologies in AD and TBI share similar immunohistochemical and biochemical features (10, 11). In both conditions, somatodendritic tau immunoreactivity is prominent; however, tau-immunoreactive neurites observed in TBI have been suggested to have an axonal origin, which may be distinct from the threadlike forms in AD suggested to be dendritic in origin (2, 3, 8, 11–13). Furthermore, the anatomical distribution of NFTs may be different following TBI than is typically seen in AD (8). Thus, the mechanisms leading to tau hyperphosphorylation in TBI may differ from those in AD.
The physiological function of tau is to stabilize microtubules (MTs) (14). Tau binding to MTs is regulated by serine/threonine phosphorylation. Abnormally phosphorylated tau has reduced MT binding, which results in MT destabilization. This in turn may compromise normal cytoskeletal function, ultimately leading to axonal and neuronal degeneration (15–17). This is the basis for the hypothesis that tau hyperphosphorylation leads to neurodegeneration in tauopathies. Identification of many mutations in the tau gene, which cause frontotemporal dementia with parkinsonism linked to chromosome-17 and result in tau hyperphosphorylation, supports this hypothesis (18). Findings from experimental models in which human mutant tau is expressed provide further support for this hypothesis. In these models, hyperphosphorylation of tau often precedes axonopathy and degeneration (1). Consequently, targeting tau either by reducing its phosphorylation state or aggregation has been a focus of preclinical therapeutic development for AD and related dementias (19, 20).
Two major mechanisms proposed to underlie tau hyperphosphorylation are aberrant activation of kinases and downregulation of protein phosphatases. Cyclin-dependent kinase-5 (CDK5) and its co-activator p25 (21–23), glycogen synthase kinase-3β (GSK-3β) (24, 25), and protein phosphatase 2A (26–28) have been implicated in hyperphosphorylation of tau in vivo. Others such as protein kinase A (PKA)(29, 30), extracellular signal-regulated kinase 1/2 (ERK1/2) (31, 32), and c-Jun N-terminal kinase (JNK) (33–36) have only been shown to regulate tau phosphorylation in vitro. It is not known whether these kinases and phosphatase contribute to TBI-induced tau pathology.
We previously reported that controlled cortical impact TBI accelerated tau pathology in young 3×Tg-AD mice (37). Importantly, the post-traumatic tau pathology appeared to be independent of β-amyloid (Aβ). Furthermore, TBI-induced tauopathy in these mice resembled tau pathology observed in humans in that tau immunoreactivity was evident in both axonal and somatodendritic compartments. In this study, we used this experimental TBI mouse model to investigate mechanisms responsible for increased tau phosphorylation following moderately severe brain trauma. We found JNK to be critically involved in this process.
MATERIALS AND METHODS
Animals
Five-to 7-month-old homozygous 3×Tg-AD mice were used. 3×Tg-AD mice express 3 mutant human genes: PS1M146V knockin, APPswe, and TauP301L mutations (38). 3×Tg-AD mice were derived from the founders received from the Laferla lab (Irvine, CA) since 2007. There was no evidence of genetic drift. Mice were housed in standard cages in 12-hour light, 12-hour dark cycle and given food and water ad libitum. Mice of both sexes were randomly assigned to experimental groups. All experiments were approved by the animal studies committee at Washington University in St. Louis, MO.
Controlled Cortical Impact TBI
The experimental TBI methods were performed as previously described (39). Briefly, a 5-mm craniotomy was performed on the left hemisphere by a motorized trephine. Experimental TBI was induced by impacting a 3.0-mm-diameter metal tip onto the cortex. Impact was centered at 3.0 mm anterior to lambda and 2.7 mm to the left of midline. A 2.0-mm impact below the dura was chosen, as this injury severity not only results in moderate damage to the cortex and underlying hippocampus ipsilateral to the injury, but also causes robust total and phosphorylated tau accumulations in injured axons (37). Sham injured mice went through identical procedures but were not injured. Duration of anesthesia exposure for sham group was approximately 15 minutes ± 1 minute vs. 18 minutes ± 1 minute for the TBI group. Mice were kept at 37°C via a rectal temperature probe throughout the surgery and allowed to recover on a warming pad to prevent hypothermia-induced hyperphosphorylation of tau (40).
Antibodies
All antibodies used are listed in the Table. Monoclonal 3D6 antibody was biotinylated using NHS-LC-biotin from Pierce (Rockford, IL).
Table.
Antibodies
| Protein | Antibody | Epitope | Host/Application Dilution | Source/Cat # |
|---|---|---|---|---|
| PKA | PKA | α and β catalytic subunits | Goat/WB 0.4 μg/ml | Santa Cruz Biotechnology, Santa Cruz, CA (sc-30668) |
| p-PKA | pT197 of α and β catalytic subunits | Rabbit/WB, IHC 0.4 μg/ml | Santa Cruz Biotechnology (sc-32968) | |
| ERK1/2 | p44/42 MAPK | Full-length, C-terminus | Rabbit/WB 1:500 | Cell Signaling, Danvers, MA (4695) |
| p-p44/42 MAPK | pT202 and pY204 | Rabbit/WB, IHC 1:500 | Cell Signaling (4370) | |
| GSK3β | GSK3β | Full-length | Rabbit/WB 1:1000 | Cell Signaling (9315) |
| p-GSK3β | pS9 | Rabbit/WB, IHC 1:500 | Cell Signaling (#9323) | |
| p-GSK3 | pY279 of α and pY216 of β subunits | Rabbit/IHC 1:500 | Invitrogen, Carlsbad, CA (44604G) | |
| JNK | JNK | Full-length | Rabbit/WB 1:1000 | Cell Signaling (9258) |
| p-JNK | pT183 and pY185 | Rabbit/WB, IHC 1:500 | Cell Signaling (4668) | |
| CDK5 | CDK5 | Full-length | Rabbit/WB, IHC 1:500 | Cell Signaling (2506) |
| p35/25 | p35/25 | Full-length, C-terminus | Rabbit/WB 1:1000 | Cell Signaling (2680) |
| c-jun | p-c-jun | pS63 | Rabbit/IHC 1:500 | Cell Signaling (2361) |
| Tau | pAb Tau | panTau | Sheep/IHC 1 μg/ml | Pierce, Rockford, IL (PN1000) |
| pS199 | pS199 | Rabbit/IHC 1:2000 | Invitrogen (44734G) | |
| pT231 | pT231 | Rabbit/IHC 1:1000 | Invitrogen (44746G) | |
| PHF1 | pS396 and pS404 | Mouse/IHC 1:500 | Dr. P.Davies, Albert Einstein College of Medicine, Bronx, NY | |
| APP | APP | Full-length, C-terminus | Rabbit/IHC 0.5 μg/ml | Invitrogen (51–2700) |
| Aβ | 3D6 | aa 1–5, requiring free N-terminus | Mouse/IHC 1 μg/ml | Eli Lilly and Co., Indianapolis, IN |
| Tubulin | α-tubulin | α-tubulin | Mouse/WB 1 μg/ml | Sigma, St. Louis, MO (T5168) |
aa, amino acid; Aβ, β-amyloid; APP, amyloid precursor protein; CDK5, cyclin-dependent kinase-5; ERK1/2, extracellular signal-regulated kinase; GSK-3β, glycogen synthase kinase-3β; IHC, immunohistochemistry; JNK, c-jun N-terminal kinase; p, phosphorylated; PKA, protein kinase A; WB, Western blot.
Western Blotting
Mice were killed by deep isoflurane anesthesia, followed by rapid decapitation at 24 hours following sham or TBI procedure. Hippocampi and surrounding white matter, including the fimbria/fornix ipsilateral to the injury site, were dissected, immediately frozen, and stored at −80°C. Tissues were homogenized in modified RIPA buffer containing protease and phosphatase inhibitor tablets (Roche), as described (41). Homogenates were centrifuged at 13,000 rpm for 20 minutes at 4°C, and protein concentrations were determined using the BCA method (Pierce). Equal amounts of each sample (6 μg) were electrophoresed on 10% BisTris NUPAGE gels using MOPS buffer (Invitrogen). Gels were transferred to 0.2-μm nitrocellulose membranes, which were then blocked with Tris-buffered saline containing 0.1% Tween-20 (TBS-T) and 5% non-fat dry milk for 1 hour at room temperature. Membranes were incubated overnight in TBS-T buffer containing 5% BSA and the appropriate primary antibodies. Corresponding anti-rabbit-HRP (GE Healthcare, 1:10,000) or anti-goat-HRP (Invitrogen, 1:10,000) and ECL Advance Western Blotting kit (GE Healthcare) were used for detection. Blots were washed 4 times for 5 minutes each with TBS-T between blocking and applications of antibodies. Blots were scanned and densitometry was performed via Image J (NIH, Bethesda, MD).
Protein Phosphatase Activity Assays
Serine/threonine phosphatase activity assay kits (V2460) were purchased from Promega Corp. (Fitchburg, WI). Assays were performed on a 96-well plate format, per manufacturer's instructions. Briefly, to remove phosphatase inhibitors and endogenous phosphates from hippocampal RIPA lysates, samples were desalted using the Zeba micro spin desalting columns (Pierce, 89877). Each sample was run in duplicate reactions; each contained 2 μl of lysates, 10 μl of appropriate 5× phosphatase reaction buffer, 5 μl of 1 mM phosphopeptide, and 33 μl of deionized H2O. Protein phosphatase 2A (PP2A) reaction buffer contained 250 mM imidazole, 1 mM EGTA, 0.1% β-mercaptoethanol, and 0.5 mg/ml acetylated BSA (Promega, R3961). In addition to the reagents listed for PP2A reaction buffer, PP2B (calcineurin) reaction buffer also included 50 mM MgCl2, 5 mM NiCl2, 250 μg/ml calmodulin (Calbiochem, 208690, EMD Millipore, Billerica, MA). Plates were incubated at 30°C for 30 minutes for phosphatase reactions to take place. Reactions were stopped by addition of 50 μl of Molybdate Dye/Additive mixture to each well. Plates were subsequently incubated at room temperature for 30 minutes to allow the Molybdate Dye to bind to free phosphates released from the reaction. Plates were read using a plate reader with 630 nm filter. Optical densities of the samples were determined based on the optical densities of free phosphate standards. Specific activities for PP2A and PP2B were expressed as pmol phosphates per minute per μg of total protein.
Immunohistochemistry and Double Immunofluorescence
Immunohistochemistry was done as previously reported (37). Mice were killed at 24 hours post-TBI; their brains were fixed for 24 hours in 4% paraformaldehyde and cryoprotected in 30% sucrose for 2 days before sectioning to 50-μm-thick slices via a sliding microtome. To reduce background staining on injured tissues when staining with monoclonal PHF1 antibody, an additional blocking step for 1 hour with unconjugated anti-mouse IgG monovalent Fab fragments (Jackson ImmunoResearch, West Grove, PA, 315-007-003, 130 μg/ml) was performed following blocking with serum.
For double labelling of phospho-tau and activated JNK, sequential applications of primary antibodies were employed. First, sections were incubated with rabbit anti-pS199, followed by goat anti-rabbit secondary antibody conjugated to Alexa Fluor® 488 (Invitrogen, A11008, 2 μg/ml). Sections were blocked again for 30 minutes with 3% normal rabbit serum to saturate open binding sites on the first secondary antibody with IgG. Sections were then incubated for 1 hour in excess of unconjugated goat anti-rabbit IgG monovalent Fab fragments (Jackson ImmunoResearch, 111-007-003, 130 μg/ml). This was done to cover the rabbit IgG so that the second secondary antibody would not bind to it. Rabbit anti-p-JNK was subsequently applied, followed by goat anti-rabbit conjugated to Alexa Fluor 594 (Invitrogen, A11012, 2 μg/ml). Sections were washed with TBS 3 times for 5 minutes each between steps. Images were obtained using LSM 5 Pascal software (Zeiss Physiology Software) coupled to an LSM Pascal Vario 2RGB confocal system (Zeiss).
Quantitative Analyses of Histological Data
All histological analyses were done by an investigator who was blinded to treatment conditions of all mice. A mouse brain atlas was used to identify the ipsilateral fimbria/fornix, thalamus, amygdala, and hippocampal CA1 (42).
Densitometric analysis of different kinase staining was performed on the ipsilateral fimbria/fornix of 4 sections per mouse, with each section separated by 400 μm. Phospho-c-jun staining was performed on the ipsilateral thalamus using 5 sections per mouse. These sections spanned approximately bregma −0.8 mm to −2.6 mm. Slides were scanned using a Nanozoomer HT system (Hamamatsu Photonics, Bridgewater, NJ) to obtain digitized images. Scanned images were exported with the NDP viewer software (Hamamatsu Photonics) and analyzed using the Image J software, as described previously (43). Briefly, images were converted to 8 bit grayscale. The “polygon” selection tool was then used to delineate either the fimbria/fornix or the thalamus. Images were thresholded to highlight stained objects using the automatic “MaxEntropy” thresholding function in ImageJ. The “Analyze Particles” function was subsequently used to quantify the % area occupied by each kinase in the ipsilateral fimbria/fornix and by p-c-jun in the ipsilateral thalamus.
Stereological quantifications were performed via the StereoInvestigator software (version 8.2, MBF Bioscience, Williston, VT). The optical fractionator method was used to quantify total numbers of amyloid precursor protein (APP)-, 3D6-, total tau-, pS199-, PHF1-, and pT231-positive axonal profiles per cubic mm of the fimbria/fornix. Axonal bulbs and swellings with spheroidal or “beads-on-a-string” morphologies that were ≥5 μm in diameter were counted. Axons with multiple, anatomically continuous “beads-on-a-string” varicosities were only counted once. As we have noted previously (44), this method may result in over-counting if 2 apparently discontinuous varicosities represent 2 parts of a single disconnected axon, or undercounting if injured axons do not stain with APP or are <5 μm in diameter. Thus, the quantitative estimates of axonal injury should be regarded as approximate. This optical fractionator method was also used to quantify total numbers of total tau-positive somata in the ipsilateral amygdala. The spherical probe (also known as “space balls”) was used to estimate total tau-positive process length per cubic mm of the contralateral CA1. All parameters used for these stereological methods were as previously reported (37).
JNK Inhibitor Treatment
D-JNKi1 peptide (BML-EI355) and D-TAT control peptide (BML-EI384) were purchased from Enzo Life Sciences International, Inc. (Farmingdale, NY). D-JNKi1 peptide is a specific inhibitor of JNK, which blocks the interaction between JNK (JNK-1, -2, -3) and its substrates (45–48). D-JNKi1 is cell-permeable and has longer half-life than its L-stereoisomer. D-JNKi1 contains a 20-amino acid sequence of the JNK binding domain of the JNK-interaction protein JIP1 covalently linked to the 10-amino acid HIV-TAT sequence. D-TAT control peptide contains only the 10-amino acid HIV-TAT sequence. Prior to craniotomy and TBI induction, a 1-mm burr hole was drilled on the right hemisphere at −0.5 mm posterior to bregma and 1.0 mm lateral to midline. Mice were randomly assigned to receive either D-JNKi1 or D-TAT (5 μg, dissolved in PBS + 0.1% DMSO) immediately post-injury. A 33-gauge needle attached to a Hamilton syringe and KDS310 nano pump system (KD Scientific, Inc.) was lowered −2.2 mm below the dura through the burr hole to deliver peptide solutions (5 μl in total volume) at 0.3 μl/min rate into the right lateral ventricle. Duration of anesthesia exposure for the combined injury and intracerebroventricular injection procedure was similar for D-TAT and D-JNKi1 treated groups: 50 ± 2 minutes. Mice recovered well after this combined surgical procedure. They lost approximately 10% of their original body weight, which was similar to mice that underwent only the TBI procedure.
Statistical Methods
All data were analyzed using Prism 5.0 (GraphPad, San Diego, CA). For pair-wise comparisons of levels of tau kinases via Western blot and immunohistochemistry and phosphatase activity between TBI and sham mice, two-tailed Student t-tests were used; p values of < 0.05 were considered significant. For comparisons of staining areas covered by activated kinases in the fimbria/fornix, a one-way ANOVA with Newman-Keuls post-test was used. For pair-wise comparisons of quantitative histological data of D-JNKi1 experiments (e.g. D-JNKi vs. D-TAT), one-sided Student t-test were used because unidirectional hypotheses were prespecified.
There was a trend toward reduced tau pathology when we first analyzed results from 5 D-JNKi1-and 4 D-TAT-treated mice. Therefore, 4 additional mice were added to each group and data were re-analyzed. As such, statistical significance for these analyses was set to p < 0.025 due to the optional stopping design of the experiment. Values presented are mean ± SEM.
RESULTS
Activities of Tau Kinases and Phosphatases Were Not Different in Hippocampal Homogenates of TBI vs. Sham 3xTg-AD Mice at 24 Hours
Aberrant activation of tau kinase(s) (21–25, 49) or inhibition of protein phosphatases (26–28, 50) are the major proposed mechanisms underlying tau hyperphosphorylation in many tauopathies. We therefore tested whether these mechanisms could account for the observed trauma-induced tau phosphorylation in our experimental TBI model. We studied overall tissue levels of the PKA, ERK1/2, GSK-3β, and JNK (31–35, 51–54). Phosphorylation of the catalytic subunit of PKA is essential for its activation by cAMP (55, 56); ERK1/2 and JNK are directly activated via phosphorylation (57–59). Thus, blots were probed with phospho-specific antibodies to assess the levels of active PKA, ERK1/2, and JNK (Fig. 1A). GSK-3β activity, on the other hand, is controlled via inhibitory phosphorylation of GSK-3β at Ser-9 by Akt/protein kinase B pathways (60). Thus, blots were probed with an antibody against phosphorylated Ser-9 of GSK-3β (Fig. 1A). Another well-characterized tau kinase is the cyclin-dependent kinase 5 (CDK5). Physiological activity of CDK5 is regulated by its association to the regulatory subunit p35, whereas association of CDK5 to p25 results in abnormal kinase activation and contributes to neurodegeneration (21, 22, 49). Therefore, we also measured CDK5, p35, and p25 levels via Western blot to probe for CDK5 activity following TBI (Fig. 1C). Western blot analyses showed no difference in the total and activated levels of all examined kinases from the homogenates of TBI compared to sham mice (p > 0.05, Fig. 1B, D).
Figure 1.

Traumatic brain injury (TBI) did not affect levels of tau kinases and activities of tau phosphatases in hippocampal lysates at 24 hours. (A) Representative Western blots of ipsilateral hippocampal lysates of a sham and an injured (i.e. TBI) mouse detected with antibodies against phosphorylated protein kinase A (p-PKA), phosphorylated extracellular signal-regulated kinase 1/2 (p-ERK1/2), p-glycogen synthase kinase-3β (GSK-3β) at Ser9, and phosphorylated c-jun N-terminal kinase (p-JNK). Blots were stripped and reprobed with antibodies against total PKA, ERK1/2, GSK3β, and JNK. The same blots were finally stripped and reprobed with α-tubulin for loading control. (B) Densitometric quantification of ratios of phosphorylated protein over total protein for each examined kinase from hippocampal lysates of sham and TBI mice expressed as % of sham. None were significantly different from 100%. (C) Representative Western blots of hippocampal lysates of a sham and a TBI mouse probed with antibodies against total cyclin-dependent kinase-5 (CDK5) and its regulatory subunits, p35/p25. CDK5 blots were reprobed with α-tubulin for loading control. (D) Densitometric quantification of bands immunoreactive for CD5, p35, and p25 from hippocampal lysates of sham and TBI mice, expressed as % sham. No significant difference was observed between TBI (n = 4) and sham (n = 4) samples, Student t-test. (E) Activity of protein phosphatase 2A (PP2A) and 2B (PP2B, calcineurin) from hippocampal lysates of sham and TBI mice, expressed as % sham. No significant difference was observed between TBI (n = 6) and sham (n = 6) samples, Student t-tests. Bars are mean ± SEM.
Protein phosphatase 2A (PP2A) and protein phosphatase 2B (PP2B) are major tau phosphatases (26, 61, 62); thus, we measured the activities of these phosphatases from the same hippocampal homogenates of TBI and sham mice using a phosphatase activity assay kit. TBI did not significantly affect activities of PP2A and PP2B when compared to sham mice (p > 0.05, Fig. 1E). In summary, changes in tau kinases and phosphatases could not be detected at the whole tissue homogenate level 24 hours following injury in 3xTg-AD mice.
Altered Localizations of Tau Kinases and Tau post-TBI
Traumatic axonal injury (TAI) is a prominent feature of TBI in many contexts, including pericontusional axonal injury in our mouse model (37, 63–66). TAI is thought to disrupt axonal transport thereby altering the localizations of many proteins (67). As such, it is probable that TAI causes mislocalizations of tau and tau kinases, resulting in the observed TBI-induced tauopathy in our model. We tested this hypothesis by subjecting separate 3xTg-AD mice to TBI or sham injuries and examining their brains immunohistochemically. The brains were stained for activated forms of PKA, ERK1/2, and JNK, and for total CDK5 using the same antibodies used for Western blotting. In a pilot experiment, we did not observe any immunoreactivity in our tissues using antibody directed against phospho-S9 of GSK-3β (Supplemental Fig. 1). Therefore, we used an antibody against phosphorylated tyrosine residues of GSK-3 in this experiment. Tyrosine phosphorylation of GSK-3 is necessary for its functional activity and is enhanced following various insults (68–70).
TBI resulted in immunohistochemically detectible activation of most of the kinases examined, primarily in injured axons of the ipsilateral fimbria/fornix (Fig. 2). JNK (Fig. 2I–J) appeared markedly activated compared to the rest of the examined kinases (Fig. 2A–H). JNK activation was also observed in the ipsilateral cortex and thalamus of injured mice (Fig. 3A–D), and increased immunoreactivity for activated PKA and GSK-3 was observed in the ipsilateral CA1 (Fig. 3E–H). Densitometric analyses showed 7.6 ± 0.8% area covered with phosphorylated (p-)JNK-positive staining and 2.5 ± 0.5% area covered with p-GSK-3 staining in the fimbria/fornix of TBI mice vs. <0.01% p-JNK-positive area and 0.38 ± 0.1% phosphorylated (p-)GSK-3-positive area in sham mice. Areas covered by p-JNK (p < 0.0001) and p-GSK-3 (p < 0.05) were significantly greater in TBI vs. sham mice (Fig. 2K). In comparisons with other examined kinases, p-JNK staining in the fimbria/fornix was the most prominent (p < 0.0001, Newman-Keuls post-test following one-way ANOVA). Furthermore, double immunofluorescence and confocal microscopy revealed that p-JNK colocalized with tau phosphorylated at Ser-199 (pS199) in the fimbria/fornix of injured but not sham mice (Fig. 2L–M).
Figure 2.

c-jun N-terminal kinase (JNK) was markedly activated in the ipsilateral fimbria/fornix of injured 3×Tg-AD mice and colocalized with phospho-tau. (A–J) Representative brain sections from a sham and a traumatic brain injury (TBI) mouse immunostained for phosphorylated protein kinase A (p-PKA) (A, B), phosphorylated extracellular signal-regulated kinase 1/2 (p-ERK1/2) (C, D), Cyclin-dependent kinase-5 (CDK5) (E, F), phosphorylated glycogen synthase kinase-3β (p-GSK-3β) at Y279 and Y216 (G, H), and p-JNK (I, J). Scale bar in A: 50 μm. (K) Percentage of fimbria area occupied by p-PKA, p-ERK1/2, CDK5, p-GSK3, and p-JNK staining. Areas occupied by p-GSK3 and p-JNK were significantly different from sham. * p < 0.05, *** p < 0.001, Student t-test. p-JNK-positive area was significantly more than the rest of the examined kinases. # p < 0.0001, one-way ANOVA with Newman-Keuls post-test, n = 4 mice per group. Bars: mean ± SEM. (L, M) Colocalization of tau phosphorylated at S199 (pS199) and phosphorylated JNK (p-JNK) in a TBI (M) but not sham (L) mouse.
Figure 3.

Activated c-jun N-terminal kinase (JNK), protein kinase A (PKA), and glycogen synthase kinase-3β (GSK-3β) localized in distinct brain regions of injured 3xTg-AD mice at 24 hours. (A–D) Phospho-JNK staining (p-JNK) for activated JNK was observed in the ipsilateral cortex (B) and thalamus (D) of injured but not sham 3xTg-AD mice (A, C). Scale bars in A and C: 500 μm. Insets in A–D: higher magnification of phospho-JNK staining. Scale bar: 50 μm. (E, F) Phospho-PKA (p-PKA) for activated PKA in the ipsilateral (ipsi) CA1 of injured 3xTg-AD mice. Scale bar in E: 50 μm. (G, H) Phospho-GSK3 (p-GSK3, activated) for GSK-3 phosphorylated at Y279 and Y216.
Taken together, these data suggest that axonal co-accumulation and mislocalization of tau and tau kinases, particularly JNK, following TBI could be responsible for post-traumatic axonal tau pathology in 3×Tg-AD mice.
JNK Inhibition by D-JNKi1 Peptide Reduces TBI-Induced Tauopathy
To test the hypothesis that JNK is involved in increasing axonal tau phosphorylation and accumulation following TBI in 3×Tg-AD mice, we treated mice with a specific peptide inhibitor of JNK, D-JNKi1, or control peptide, D-TAT, via intracerebroventricular injection immediately following TBI. D-JNKi1 was chosen over the ATP-competitive inhibitor of JNK, SP600125, because of its high specificity to JNK and its long half-life (45, 46). Mice were killed at 24 hours post-injury and their brains were examined by immunohistochemistry. Because c-jun is a known major target of JNK (71), we stained for c-jun phosphorylated at Ser-63 (p-c-jun) to determine the extent to which JNK activity was inhibited by D-JNKi1 treatment. TBI resulted in c-jun activation in many pericontusional regions, most consistently the ipsilateral thalamus (Fig. 4A–D). We therefore quantified p-c-jun nuclear staining in this region and found that D-JNKi1 treatment reduced p-c-jun immunoreactivity approximately 40% when compared with D-TAT treated mice (p < 0.0001, Fig. 5A–C).
Figure 4.
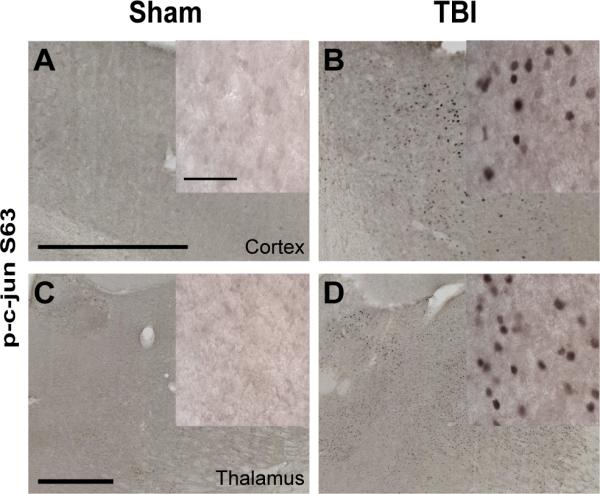
Traumatic brain injury (TBI) caused c-jun N-terminal kinase (JNK) activation in the ipsilateral cortex and thalamus of 3xTg-AD mice. (A–D) Phospho-c-jun staining for activated cjun was detected in the ipsilateral cortex and thalamus of injured mice (B, D), but not sham mice (A, C). Scale bars: 500 μm. Insets: Higher magnification of nuclear c-jun staining. Scale bar: 50 μm.
Figure 5.

D-JNKi1 treatment blocked c-jun activation in the thalamus but did not affect axonal injury as assessed by amyloid precursor protein (APP) immunohistochemistry or β-amyloid (Aβ) accumulation in the fimbria of injured 3xTg-AD mice at 24 hours. (A, B) Nuclear staining of phosphorylated c-jun at S63 (p-c-jun S63) in the thalamus of injured 3xTg-AD mice that received intracerebroventricular injection of D-JNKi1 or D-TAT control peptide. Scale bar in A: 500 μm. Insets in A and B: higher magnification of p-c-jun S63 staining. Scale bar: 50 μm. (C) Percentage of area occupied by p-c-jun S63 staining in D-JNKi1- and D-TAT-treated mice. JNK inhibition by D-JNKi1 significantly reduced the extent of c-jun phosphorylation in the thalamus of injured 3xTg-AD mice: 40% reduction, *** p < 0.001. (D, E) APP staining in the ipsilateral fimbria of injured 3xTg-AD mice treated with D-JNKi1 or D-TAT. Scale bar in D: 50 μm. (F) Stereological quantification showed similar numbers of APP-stained varicosities in mice treated with D-JNKi1 compared to D-TAT. (G, H) Aβ staining using N-terminal antibody 3D6 in the ipsilateral fimbria of injured 3xTg-AD mice treated with D-JNKi1 or D-TAT. I: Stereological quantification showed a non-significant 20% reduction in the numbers of Aβ-stained axonal varicosities between groups. n.s.: not significant. Student t-tests, n = 8–9 mice per treatment group. Bars: mean ± SEM.
APP is a robust marker of axonal injury (72–75); thus, we stained these brains for APP to assess the effects of JNK inhibition on the extent of axonal injury. We also stained for APP proteolytic product Aβ using the 3D6 antibody, which does not recognize APP (76). D-JNKi1 treatment did not significantly affect the degree of axonal injury as determined by the numbers of APP-positive axonal varicosities in the fimbria/fornix (p = 0.31, Fig. 5D–F). D-JNKi1 treatment appeared to reduce the numbers of 3D6-positive varicosities in the fimbria, but the reduction did not reach statistical significance when compared to D-TAT treated mice (p = 0.07, Fig. 5G–I). This finding is not surprising because D-JNKi1 has been shown to reduce Aβ production in vitro (77). We conclude that D-JNKi1 did not affect the severity of axonal injury in this setting.
Although the D-JNKi1 treatment did not fully block c-jun phosphorylation, we nevertheless asked if partial JNK inhibition was sufficient to affect post-traumatic tau pathology in this model. We assessed total tau pathology by staining with a polyclonal antibody that recognizes tau independent of its phosphorylation state (Fig. 6A, B, D, E, G, H). Stereological quantification showed a moderate but significant reduction (35%) of total tau-positive puncta in the ipsilateral fimbria/fornix (p = 0.025, Fig. 6C). As controls, we also quantified total tau-positive somata in the ipsilateral amygdala and tau-positive neurites in the contralateral CA1. These two regions exhibited increased total tau immunoreactivity (37) but lacked p-JNK staining following TBI (Supplemental Fig.2). As expected, stereological quantification showed similar numbers of tau-positive somata and neurites in the amygdala and CA1 of D-JNKi1 and D-TAT treated mice (p > 0.05, Fig. 6F, I).
Figure 6.

c-jun N-terminal kinase (JNK) inhibition by D-JNKi1 peptide reduced axonal tau accumulation but did not affect somatodendritic tau accumulation. (A–I) Total tau staining using polyclonal antibody (pAb) in the ipsilateral fimbria (A, B), ipsilateral amygdala (D, E), and contralateral (contra) CA1 (G, H) of mice that received D-JNKi1 or D-TAT by intracerebroventricular injection. Scale bar in A: 50 μm. Stereological quantification showed DJNKi1 treatment reduced axonal tau accumulation in the fimbria (C), but had no effect on numbers of tau-positive somata in the ipsilateral amygdala (F) or tau-positive process length in the contralateral (contra) CA1 region (I). * p < 0.025, n.s: not significant. Student t-tests, n = 8–9 mice per group. Bars: mean ± SEM.
We next studied effects of JNK inhibition on tau phosphorylation using phospho-specific antibodies against tau phosphorylated at Ser-199 (pS199), Ser-396 and/or Ser-404 (PHF1) (78, 79), and Thr-231 (pT231) (Fig. 7A, B, D, E, G, H). There were significant reductions of numbers of pS199-positive (55%, p < 0.0001, Fig. 7C) and PHF1-positive (34%, p = 0.014, Fig. 7F) puncta in the ipsilateral fimbria/fornix of D-JNKi1 compared to D-TAT treated mice. Numbers of pT231-positive puncta were not statistically different between treatment groups (p = 0.12, Fig. 7I). This is consistent with in vitro findings that JNK preferentially phosphorylates tau at several sites including Ser-396, but not at Thr-231 (80). In summary, we found that moderate reduction of JNK activity could ameliorate the axonal accumulations of total, pS199, and PHF1-tau in injured axons of 3×Tg-AD mice.
Figure 7.

D-JNKi1 treatment reduces tau pathology in injured axons of 3xTg-AD mice. (A–I) Phospho-tau staining using polyclonal antibodies against tau phosphorylated at S199 (pS199, A, B) at S396 and S404 (PHF1, D, E), and T231 (pT231, G, H) in the ipsilateral fimbria/fornix of injured 3xTg-AD mice treated with D-JNKi1 or D-TAT peptide. Scale bar in A: 50 μm. Stereological quantification of pS199-, PHF1-, and pT213-tau puncta in the ipsilateral fimbria of injured mice treated with D-JNKi1 or D-TAT. D-JNKi1 treatment significantly reduced numbers of pS199- (C) and PHF1-tau puncta (F) but had no effect of pT231-tau puncta in the ipsilateral fimbria of injured mice (I). * p < 0.025, ** p < 0.001, ns: not significant, one-sided t-tests, n = 8–9 mice per treatment group. Bars: mean ± SEM.
DISCUSSION
In this study we show that moderately severe TBI resulted in different regional patterns of activation of a number of tau kinases. The primary site of kinase activation and accumulation was within injured axons, particularly the ipsilateral fimbria/fornix. JNK was markedly activated in this region compared to the other examined kinases. Notably, JNK appeared to play a critical role in TBI-induced tau hyperphosphorylation, as activated JNK colocalized with phospho-tau and inhibition of JNK activity reduced tau phosphorylation in injured axons.
Traumatic axonal injury (TAI) is thought to cause axonal transport deficits, resulting in accumulations of various organelles and proteins, including neurofilaments and APP (67). Our data suggest that axonal transport deficits induced by TAI may be responsible for the accumulation and activation of the examined tau kinases and tau. The observations that sciatic nerve ligation resulted in accumulation of total and phosphorylated ERK1/2 and JNK (81–83) lend support to this hypothesis. Nonetheless, this hypothesis can be further tested by treatment of TBI mice with drugs that rescue or reduce transport deficits, such as the microtubule stabilizer epothilone D. Epothilone D has been shown to reduce fast axonal transport defects in CNS axons and lessen axonal degeneration in tau transgenic mice (84).
The distinct spatial distributions of activated kinases, particularly JNK, GSK-3 and PKA, indicate the heterogeneous responses of different brain structures and cellular compartments to TBI. Such selective responses may be best documented using immunohistochemical techniques, which may account for the mismatch between our immunohistochemical and Western blotting data. Nevertheless, it is possible that our semiquantitative densitometric approach used to assess the levels of total and activated protein kinases in hippocampal homogenates may not be sensitive enough to detect modest but functionally important changes. It is also likely that these kinases exhibit transient pattern of activation, which our analysis at 24 hours post-TBI did not capture. Indeed, a study using fluid percussion TBI in rats has reported that activated ERK1/2 and JNK in hippocampal lysates were evident within minutes but no longer detectable within hours post-injury (85). As such, a more thorough analysis in which mice are killed at different time points post-injury will be necessary to resolve the temporal profiles of kinase activations.
Importantly, JNK activation has been documented in contusional TBI in humans (86). This supports the validity of our TBI model. JNK was also reported to be activated in a number of studies using the fluid percussion TBI model in rats (85–89). Together, these data suggest that JNK activation is a general response to brain trauma, which is consistent with the role of JNK in signalling stress signals (90). Furthermore, our findings and those from Raghupathi et al suggest that JNK signalling is complex and may have distinct functions in somata vs. axons (89). In support of this notion many studies provide evidence for the unequivocal roles of JNK and c-jun activation in programmed cell death in neurons (91). Although JNK function in axons has received less attention, recent investigations implicate JNK in signalling axonal injury (92) and in mediating axonal degeneration (93). Because hyperphosphorylated tau is associated with axon degeneration, our findings of JNK's role in tau phosphorylation is in line with previous reports.
Nonetheless, our study has a number of limitations. First, we have not tested the therapeutic window during which D-JNKi1 can affect post-traumatic tau pathology. Borsello et al showed that D-JNKi1 treatment can have beneficial effects if given up to 6 hours following ischemic injury (46). Meanwhile, Miller et al found that JNK inhibition within 3 hours following axotomy of dorsal roots ganglion axons can effectively block JNK-mediated axon degeneration (93). The latter time window of JNK inhibition is perhaps more applicable to our model because axonal injury is a major pathology observed following TBI. Second, we have not systematically tested other doses and methods of delivery of this peptide inhibitor. Third, we have yet to determine which JNK isoform is responsible for induction tau phosphorylation post-injury. JNK1−/−, JNK2−/− and JNK3−/− knockout mice subjected to similar injury paradigm will be useful for this purpose. Fourth, although our study supports JNK activation as a probable mechanism underlying TBI-induced tau pathology, we cannot rule out other mechanisms that may result in tau hyperphosphorylation, such as changes in tau conformation (94) and other post-translational modifications of tau (95, 96). Future studies will be required to assess these alternative mechanisms. Additionally, roles of GSK-3 and PKA in tau phosphorylation will require further investigation, as activated forms of these kinases were found to localize in both axons and ipsilateral CA1 regions of injured mice. Interestingly, inhibition of GSK-3 was recently shown to protect dorsal root ganglion axons from degeneration following axotomy (97). Thus, it is possible that a combined therapy involving JNK, GSK-3, and possibly PKA inhibition may be required to effect functional benefits of blocking tau hyperphosphorylation and axon degeneration. Other kinases and phosphatases not assessed here could also be involved. Lastly, it will also be important to determine if the effects of contusional TBI are similar to or different from the effects of multiple concussive injuries on pathological hyperphosphorylation and accumulation of tau.
In summary, we identified JNK as a likely kinase that phosphorylates tau in vivo in the setting of moderately severe TBI. Future studies will be required to determine whether acute axonal tau accumulation leads to NFT formation, and whether reducing acute tau pathology proves beneficial in contusional TBI.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. David Holtzman for the use of his stereology system, Dr. Frank LaFerla for 3xTg-AD mice, Dr. Peter Davies for the PHF1 antibody, and Eli Lilly and Co for the 3D6 antibody. We thank R.E. Bennett for helpful discussions, and T.J. Esparza for helping with blinding and randomization.
This work was supported by NIH R01 NS065069 (David L. Brody), NIH K08 NS049237 (Brody), a Burroughs Wellcome Career Award in the Biomedical Sciences (David L. Brody), the Thrasher Research Fund (Brody), the Alafi Neuroimaging facility and the NIH Neuroscience Blueprint Interdisciplinary Center Core Grant P30 NS057105 to Washington University, and the Office of Undergraduate Research Award at Washington University (Laura Sanchez).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nature reviews Neuroscience. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 2.Ikonomovic MD, Uryu K, Abrahamson EE, et al. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 3.Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–92. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol epub. 2011 Sep 12; doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corsellis JA. Boxing and the brain. BMJ. 1989;298:105–9. doi: 10.1136/bmj.298.6666.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geddes JF, Vowles GH, Nicoll JA, et al. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta neuropathologica. 1999;98:171–8. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 7.Geddes JF, Vowles GH, Robinson SF, et al. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol. 1996;22:12–6. [PubMed] [Google Scholar]
- 8.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Experimental Neurol. 2009;68:709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–8. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt ML, Zhukareva V, Newell KL, et al. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathologica. 2001;101:518–24. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 11.Tokuda T, Ikeda S, Yanagisawa N, et al. Re-examination of ex-boxers' brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathologica. 1991;82:280–5. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 12.Anderton BH, Callahan L, Coleman P, et al. Dendritic changes in Alzheimer's disease and factors that may underlie these changes. Prog Neurobiol. 1998;55:595–609. doi: 10.1016/s0301-0082(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 13.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathologica. 1994;87:554–67. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 14.Drechsel DN, Hyman AA, Cobb MH, et al. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–54. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso AC, Zaidi T, Grundke-Iqbal I, et al. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–6. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bramblett GT, Goedert M, Jakes R, et al. Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–99. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 17.Merrick SE, Trojanowski JQ, Lee VM. Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons. J Neurosci. 1997;17:5726–37. doi: 10.1523/JNEUROSCI.17-15-05726.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–50. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Ballatore C, Brunden KR, Piscitelli F, et al. Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of tau aggregation. J Med Chem. 2010;53:3739–47. doi: 10.1021/jm100138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunden KR, Trojanowski JQ, Lee VM. Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov. 2009;8:783–93. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noble W, Olm V, Takata K, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–65. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 22.Patrick GN, Zukerberg L, Nikolic M, et al. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–22. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 23.Tseng HC, Zhou Y, Shen Y, et al. A survey of Cdk5 activator p35 and p25 levels in Alzheimer's disease brains. FEBS Lett. 2002;523:58–62. doi: 10.1016/s0014-5793(02)02934-4. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez F, Borrell J, Guaza C, et al. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J Neurochem. 2002;83:1529–33. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- 25.Lucas JJ, Hernandez F, Gomez-Ramos P, et al. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. The EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong CX, Shaikh S, Wang JZ, et al. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–8. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 27.Gong CX, Singh TJ, Grundke-Iqbal I, et al. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–7. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 28.Kins S, Crameri A, Evans DR, et al. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem. 2001;276:38193–200. doi: 10.1074/jbc.M102621200. [DOI] [PubMed] [Google Scholar]
- 29.Litersky JM, Johnson GV. Phosphorylation by cAMP-dependent protein kinase inhibits the degradation of tau by calpain. J Biol Chem. 1992;267:1563–8. [PubMed] [Google Scholar]
- 30.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drewes G, Lichtenberg-Kraag B, Doring F, et al. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992;11:2131–8. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goedert M, Cohen ES, Jakes R, et al. p42 MAP kinase phosphorylation sites in microtubule-associated protein tau are dephosphorylated by protein phosphatase 2A1. Implications for Alzheimer's disease [corrected] FEBS Lett. 1992;312:95–9. doi: 10.1016/0014-5793(92)81418-l. [DOI] [PubMed] [Google Scholar]
- 33.Buee-Scherrer V, Goedert M. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases in intact cells. FEBS Lett. 2002;515:151–4. doi: 10.1016/s0014-5793(02)02460-2. [DOI] [PubMed] [Google Scholar]
- 34.Goedert M, Hasegawa M, Jakes R, et al. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida H, Hastie CJ, McLauchlan H, et al. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–8. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- 36.Vogel J, Anand VS, Ludwig B, et al. The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology. 2009;57:539–50. doi: 10.1016/j.neuropharm.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 37.Tran HT, LaFerla FM, Holtzman DM, et al. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci. 2011;31:9513–25. doi: 10.1523/JNEUROSCI.0858-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 39.Brody DL, Mac Donald C, Kessens CC, et al. Electromagnetic controlled cortical impact device for precise, graded experimental traumatic brain injury. J Neurotrauma. 2007;24:657–73. doi: 10.1089/neu.2006.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Planel E, Miyasaka T, Launey T, et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer's disease. J Neurosci. 2004;24:2401–11. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cirrito JR, May PC, O'Dell MA, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–53. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franklin KB, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; London: 1997. [Google Scholar]
- 43.Kim J, Castellano JM, Jiang H, et al. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–44. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mac Donald CL, Dikranian K, Song SK, et al. Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol. 2007;205:116–31. doi: 10.1016/j.expneurol.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonny C, Oberson A, Negri S, et al. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 46.Borsello T, Clarke PG, Hirt L, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–6. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 47.Borsello T, Croquelois K, Hornung JP, et al. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–85. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 48.Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–97. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- 49.Cruz JC, Tseng HC, Goldman JA, et al. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 50.Arendt T, Holzer M, Fruth R, et al. Phosphorylation of tau, Abeta-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiol Aging. 1998;19:3–13. doi: 10.1016/s0197-4580(98)00003-7. [DOI] [PubMed] [Google Scholar]
- 51.Litersky JM, Johnson GV, Jakes R, et al. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem J. 1996;316:655–60. doi: 10.1042/bj3160655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robertson J, Loviny TL, Goedert M, et al. Phosphorylation of tau by cyclic-AMP-dependent protein kinase. Dementia. 1993;4:256–63. doi: 10.1159/000107331. [DOI] [PubMed] [Google Scholar]
- 53.Sperber BR, Leight S, Goedert M, et al. Glycogen synthase kinase-3 beta phosphorylates tau protein at multiple sites in intact cells. Neurosci Lett. 1995;197:149–53. doi: 10.1016/0304-3940(95)11902-9. [DOI] [PubMed] [Google Scholar]
- 54.Virdee K, Yoshida H, Peak-Chew S, et al. Phosphorylation of human microtubule-associated protein tau by protein kinases of the AGC subfamily. FEBS Lett. 2007;581:2657–62. doi: 10.1016/j.febslet.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Adams JA, McGlone ML, Gibson R, et al. Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry. 1995;34:2447–54. doi: 10.1021/bi00008a007. [DOI] [PubMed] [Google Scholar]
- 56.Smith CM, Radzio-Andzelm E, Madhusudan, et al. The catalytic subunit of cAMP-dependent protein kinase: prototype for an extended network of communication. Prog Biophys Mol Biol. 1999;71:313–41. doi: 10.1016/s0079-6107(98)00059-5. [DOI] [PubMed] [Google Scholar]
- 57.Anderson NG, Maller JL, Tonks NK, et al. Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature. 1990;343:651–3. doi: 10.1038/343651a0. [DOI] [PubMed] [Google Scholar]
- 58.Lisnock J, Griffin P, Calaycay J, et al. Activation of JNK3 alpha 1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry. 2000;39:3141–8. doi: 10.1021/bi992410+. [DOI] [PubMed] [Google Scholar]
- 59.Wada T, Nakagawa K, Watanabe T, et al. Impaired synergistic activation of stress-activated protein kinase SAPK/JNK in mouse embryonic stem cells lacking SEK1/MKK4: different contribution of SEK2/MKK7 isoforms to the synergistic activation. The Journal of biological chemistry. 2001;276:30892–7. doi: 10.1074/jbc.M011780200. [DOI] [PubMed] [Google Scholar]
- 60.Cross DA, Alessi DR, Cohen P, et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 61.Wang JZ, Gong CX, Zaidi T, et al. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem. 1995;270:4854–60. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- 62.Gong CX, Lidsky T, Wegiel J, et al. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem. 2000;275:5535–44. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 63.Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathology and applied neurobiology. 2000;26:105–16. doi: 10.1046/j.1365-2990.2000.026002105.x. [DOI] [PubMed] [Google Scholar]
- 64.Gennarelli TA, Thibault LE, Adams JH, et al. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–74. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- 65.Gentleman SM, Roberts GW, Gennarelli TA, et al. Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol. 1995;89:537–43. doi: 10.1007/BF00571509. [DOI] [PubMed] [Google Scholar]
- 66.Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehab. 2003;18:307–16. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 67.Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma. 1995;12:555–64. doi: 10.1089/neu.1995.12.555. [DOI] [PubMed] [Google Scholar]
- 68.Bhat RV, Shanley J, Correll MP, et al. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc Nat Acad Sci USA. 2000;97:11074–9. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hughes K, Nikolakaki E, Plyte SE, et al. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–8. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leroy K, Boutajangout A, Authelet M, et al. The active form of glycogen synthase kinase-3beta is associated with granulovacuolar degeneration in neurons in Alzheimer's disease. Acta Neuropathol. 2002;103:91–9. doi: 10.1007/s004010100435. [DOI] [PubMed] [Google Scholar]
- 71.Hibi M, Lin A, Smeal T, et al. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–48. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 72.Graham DI, Gentleman SM, Lynch A, et al. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995;21:27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- 73.Sherriff FE, Bridges LR, Gentleman SM, et al. Markers of axonal injury in post mortem human brain. Acta Neuropathol. 1994;88:433–9. doi: 10.1007/BF00389495. [DOI] [PubMed] [Google Scholar]
- 74.Stone JR, Singleton RH, Povlishock JT. Antibodies to the C-terminus of the beta-amyloid precursor protein (APP): a site specific marker for the detection of traumatic axonal injury. Brain Res. 2000;871:288–302. doi: 10.1016/s0006-8993(00)02485-9. [DOI] [PubMed] [Google Scholar]
- 75.Stone JR, Singleton RH, Povlishock JT. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol. 2001;172:320–31. doi: 10.1006/exnr.2001.7818. [DOI] [PubMed] [Google Scholar]
- 76.Johnson-Wood K, Lee M, Motter R, et al. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad U S A. 1997;94:1550–5. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Colombo A, Repici M, Pesaresi M, et al. The TAT-JNK inhibitor peptide interferes with beta amyloid protein stability. Cell Death Differ. 2007;14:1845–8. doi: 10.1038/sj.cdd.4402202. [DOI] [PubMed] [Google Scholar]
- 78.Greenberg SG, Davies P, Schein JD, et al. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–9. [PubMed] [Google Scholar]
- 79.Otvos L, Jr., Feiner L, Lang E, et al. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–73. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 80.Reynolds CH, Utton MA, Gibb GM, et al. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997;68:1736–44. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- 81.Averill S, Delcroix JD, Michael GJ, et al. Nerve growth factor modulates the activation status and fast axonal transport of ERK 1/2 in adult nociceptive neurones. Mol Cell Neurosci. 2001;18:183–96. doi: 10.1006/mcne.2001.1015. [DOI] [PubMed] [Google Scholar]
- 82.Middlemas A, Delcroix JD, Sayers NM, et al. Enhanced activation of axonally transported stress-activated protein kinases in peripheral nerve in diabetic neuropathy is prevented by neurotrophin-3. Brain. 2003;126:1671–82. doi: 10.1093/brain/awg150. [DOI] [PubMed] [Google Scholar]
- 83.Reynolds AJ, Hendry IA, Bartlett SE. Anterograde and retrograde transport of active extracellular signal-related kinase 1 (ERK1) in the ligated rat sciatic nerve. Neuroscience. 2001;105:761–71. doi: 10.1016/s0306-4522(01)00235-4. [DOI] [PubMed] [Google Scholar]
- 84.Brunden KR, Zhang B, Carroll J, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–6. doi: 10.1523/JNEUROSCI.3059-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Otani N, Nawashiro H, Fukui S, et al. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J Cereb Blood Flow Metab. 2002;22:327–34. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- 86.Ortolano F, Colombo A, Zanier ER, et al. c-Jun N-terminal kinase pathway activation in human and experimental cerebral contusion. J Neuropathol Exp Neurol. 2009;68:964–71. doi: 10.1097/NEN.0b013e3181b20670. [DOI] [PubMed] [Google Scholar]
- 87.Mori T, Wang X, Jung JC, et al. Mitogen-activated protein kinase inhibition in traumatic brain injury: in vitro and in vivo effects. J Cereb Blood Flow Met. 2002;22:444–52. doi: 10.1097/00004647-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 88.Otani N, Nawashiro H, Fukui S, et al. Temporal and spatial profile of phosphorylated mitogen-activated protein kinase pathways after lateral fluid percussion injury in the cortex of the rat brain. J Neurotrauma. 2002;19:1587–96. doi: 10.1089/089771502762300247. [DOI] [PubMed] [Google Scholar]
- 89.Raghupathi R, Muir JK, Fulp CT, et al. Acute activation of mitogen-activated protein kinases following traumatic brain injury in the rat: implications for posttraumatic cell death. Exp Neurol. 2003;183:438–48. doi: 10.1016/s0014-4886(03)00166-3. [DOI] [PubMed] [Google Scholar]
- 90.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–69. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 91.Silva RM, Kuan CY, Rakic P, et al. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson's disease. Mov Disord. 2005;20:653–64. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 92.Cavalli V, Kujala P, Klumperman J, et al. Sunday driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–87. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller BR, Press C, Daniels RW, et al. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nature neuroscience. 2009;12:387–9. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kanaan NM, Morfini GA, Lapointe NE, et al. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci. 2011;31:9858–68. doi: 10.1523/JNEUROSCI.0560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. doi: 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–66. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gerdts J, Sasaki Y, Vohra B, et al. Image-based screening identifies novel roles for i{kappa}b kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2011;286:28011–8. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.