

Abstract
CRN2 (synonyms: coronin 1C, coronin 3) functions in the re-organization of the actin network and is implicated in cellular processes like protrusion formation, secretion, migration and invasion. We demonstrate that CRN2 is a binding partner and substrate of protein kinase CK2, which phosphorylates CRN2 at S463 in its C-terminal coiled coil domain. Phosphomimetic S463D CRN2 loses the wild-type CRN2 ability to inhibit actin polymerization, to bundle F-actin, and to bind to the Arp2/3 complex. As a consequence, S463D mutant CRN2 changes the morphology of the F-actin network in the front of lamellipodia. Our data imply that CK2-dependent phosphorylation of CRN2 is involved in the modulation of the local morphology of complex actin structures and thereby inhibits cell migration.
Coronins play an essential role in the structural and functional organization of actin-dependent cellular processes like protrusion formation, secretion, migration, and invasion. Phylogenetic analyses have revealed twelve subfamilies of coronin proteins, consisting of seven vertebrate paralogs and five subfamilies in non-vertebrate metazoa, fungi, and protozoa1. Coronins are structured with a rather short, conserved, basic N-terminal signature motif, followed by one, or, in case of the coronin 7 ‘dimer’ subfamily, two 7-repeat WD40 domains which adopt the fold of a seven-bladed β-propeller. A unique C-terminal extension links the WD40 repeat domains with the C-terminal coiled coil domain2,3.
The predominant form of CRN2 is isoform 1 (CRN2i1), a ubiquitously expressed 474 amino acid protein4,5. CRN2 forms homotrimers via the coiled coil domain and has been identified as an actin filament cross-linking and bundling protein4,6. It exists in two different pools, an actin cytoskeleton associated non-phosphorylated pool enriched at lamellipodia and a diffusely distributed phosphorylated cytosolic pool, however, the phosphorylation site and the kinase are unknown4. In the murine brain, CRN2 seems to play a role in morphogenesis and in certain neuronal cell populations in the adult animal7. Recently, CRN2 has also been implicated in human cancer. While normal resting astrocytes do not express CRN2, the number of CRN2-positive tumor cells is correlated with the malignant phenotype in human diffuse gliomas. Knock-down of CRN2 in human glioblastoma cell lines reduces the rate of cell proliferation, motility, and invasion8. Furthermore, CRN2 is aberrantly regulated in melanoma with an increase of CRN2 expression in metastatic tumor cells9. In hepatocellular carcinoma, CRN2 expression levels correlates with clinical progression10. In a recent analysis of primary effusion lymphoma specimens, the CRN2 gene was found to be amplified in one-fourth of the specimens and CRN2 expression levels were elevated in three-fourths of the specimens11. However, a different effect was observed in another study, where a knock-down of CRN2 in colon carcinoma cell lines appeared to induce opposite effects like enhanced cell migration and the increased number of focal adhesions12.
Several reports support a role for CRN2 in signaling pathways that involve small G-proteins. A short sequence stretch that resembles the Cdc42/Rac interactive binding (CRIB) motif is present in CRN2 and could act as a potential binding site for the activated GTP-binding proteins Rac and Cdc42 involved in the regulation of the actin cytoskeleton13. CRN2 has also been found to be a direct binding partner of GDP-Rab27a. GDP-Rab27a was found to increase the F-actin bundling activity of CRN2 and the protein complex was shown to be involved in the insulin secretory membrane endocytosis14,15.
In this study, we demonstrate that CRN2 function is regulated by CK2-dependent phosphorylation. Protein kinase CK2 (synonyms: casein kinase II, CK II) was first described in a mixture with CK1 using casein as an artificial substrate16. It is an evolutionarily highly conserved, ubiquitously expressed, highly pleiotropic, and constitutively active serine and threonine kinase17,18,19,20,21,22. CK2 is involved in the control of a wide variety of cellular functions including transcription, translation, cell cycle, signal transduction, apoptosis, metabolism, virus infection, cell morphology, malignant transformation, and tumor development23,24,25. CK2 primarily exists as a heterotetrameric protein of either 2α2β, 1α1α'2β or 2α'2β subunit composition26,27,28,29,30. In these CK2 complexes, the two regulatory subunits CK2β form a stable dimer linking together the two catalytic subunits, CK2α or CK2α'29.
We show here that a CK2-dependent phosphorylation of CRN2 at residue S463 leads to a loss of CRN2-mediated inhibition of actin polymerization as well as to a loss of its F-actin bundling activity and Arp2/3 complex interaction. Together, these changes affect the architecture of the F-actin network and result in an inhibition of cell migration. Furthermore, this work reveals that bundling of actin filaments occurs via two separate actin binding sites in CRN2 and that the CRN2 coiled coil domain forms a constitutive trimer which either can interact with F-actin or the Arp2/3 complex.
Results
Identification of Ser at position 463 as a specificity determining site in the mammalian and avian CRN2 subfamily
An alignment of 40 mammalian and avian proteins out of a subclassification of 60 orthologs used in previous phylogenetic analyses of coronin family homologs1 was first transformed into a profile hidden Markov model (pHMM) using the HMMER implementations in Unipro-Ugene (http://ugene.unipro.ru/) and visualized with LogoMat-M (http://www.sanger.ac.uk/Software/analysis/logomat-m/). This identified evolutionarily conserved amino acid patterns with the highest probabilities of information content or functional significance. “Specificity determining positions” (SDPs) peculiar to the CRN2 subfamily were identified by SDPclust31, SDPfox (http://bioinf.fbb.msu.ru/SDPfoxWeb/main.jsp), and “type II divergence”32 among 300 coronins representing all seven subfamily groups present in mammals and birds and were marked with an asterisk in the corresponding position of the pHMM sequence logo for CRN2 (Fig. 1A). The concentration of these SDP sites in the N- and C-terminal domains argues for a predominant role of these regions in the functional differentiation of coronin subfamilies. Other highlighted sites, which are known to be susceptible to post-translational modifications, included Tyr-30133,34 and acetylated lysines 391 and 44635 in CRN2 and various other coronin subfamilies. In contrast, the incorporation of Ser at position 463 was confined to the CRN2 subfamily. The emergence and restricted conservation of S463 may thus reflect the selection for a structural and functional feature that allows for the regulation of CRN2 subcellular interactions through phosphorylation. A putative functional role of S463 is further emphasized by its localization within the coiled coil domain of CRN2 known to be responsible for CRN2 homo-trimerization4. A homology model of the trimeric CRN2 coiled coil domain (aa442-472) based on the crystal structure of the trimeric coiled coil of hemagglutinin36 indicated that S463 is surface exposed (Fig. 1B). The location of S463 in a solvent accessible α-helical surface segment also is visualized in a three-dimensional refined model of the human CRN2 protein predicted by I-Tasser37 and rendered with Chimera v1.538 (Fig. S1). Finally, it is important to note that S463 is part of the sequence motif S-K-L-E coinciding with the S/T-X-X-E/D consensus target motif of the protein kinase CK2.
Figure 1. Phosphorylation of S463 within the CRN2 coiled coil does not induce trimer disassembly.
(A) profile hidden Markov model (pHMM) of the CRN2 subfamily. The probability distribution of amino acids within the CRN2 subfamily is reflected by letter height while the “functional significance” predicted by HMMER is given by the full column height at each site. The Ser-463 phosphorylation site is uniquely conserved within the CRN2 subfamily, whereas sites of other predicted post-translational changes (P-Tyr-301, Ac-Lys-391 and Ac-Lys-446) are common to a limited number of other coronin subfamilies. Asterisks mark amino acids identified by SDPfox which show evidence of a conservation pattern able to distinguish individual coronin subfamilies and are therefore taken to confer “functional specificity”. These SDPs localized mainly to regions of the N- and C-terminal domains, in contrast to the “KGD” motif universally conserved in coronin proteins (aa485-487 in CRN2). (B) homology model of the trimeric CRN2 coiled coil (aa442-447, monomer chains A, B, C) based on the crystal structure of the trimeric coiled coil of hemagglutinin (HA2 chain) using residues 74 to 113 from chain B (36; PDB 1eo8). Interactions of phosphorylated S463 (pS463, see C) with R461 and K464 are illustrated. (C) size-exclusion chromatogram of synthetic pS463-CRN2 peptide (18 mg/ml) acquired on a Superose-12 column under normal (100 mM) and high salt (500 mM NaCl) conditions. The peptide eluted as single species whose molecular mass was determined using the online multi-angle light scattering (MALS) detector; theoretical mass of the trimer is 13,959 g/mol.
Phosphorylation of S463 does not induce disassembly of the CRN2 trimer
To address the issue of trimer stability at the experimental level, gel filtration experiments in conjunction with multi-angle light scattering detection (SEC-MALS) were carried out to determine the oligomerization state of synthetic wild-type and phospho-S463 CRN2 coiled coil peptides (aa442-472; 3.7 kDa). Under physiological buffer conditions, the S463 phosphorylated synthetic CRN2 peptide eluted as trimer (Fig. 1C). Furthermore, small-angle X-ray scattering (SAXS) was conducted in solution and also showed the presence of a trimeric particle, independent of the concentration of the sample. The molecular mass and radius of gyration derived from the SAXS experiments were in good agreement with the expected values from the atomic homology model (Tab. S1). The shape restored from the X-ray scattering data in solution was fitted with the atomic homology model of the trimeric coiled coil as rigid body and revealed an excellent agreement (Fig. S2). The goodness of fit between the experimental and theoretical scattering curves was calculated by CRYSOL39 as χ = 5.5. Additional gel filtration analyses were carried out using purified recombinant wild-type, S463D phosphomimetic and S463A phospho-resistant C-terminal polypeptides (aa300-474; 19.9 kDa), full-length wild-type and S463D phosphomimetic proteins purified from insect cells (53.2 kDa), and lysates of mammalian cells over-expressing GFP-fusion proteins of wild-type, S463D and S463A CRN2. These experiments clearly demonstrate a strong CRN2 trimer that is not disassembled by S463 phosphorylation. With ∼40 nM the concentrations of full-length CRN2 were far lower than the lowest concentration (4.5 mg/ml ≙ 1.2 mM) used for of the synthetic peptides and the protein could only be detected by immunoblotting.
CRN2 directly interacts with protein kinase CK2
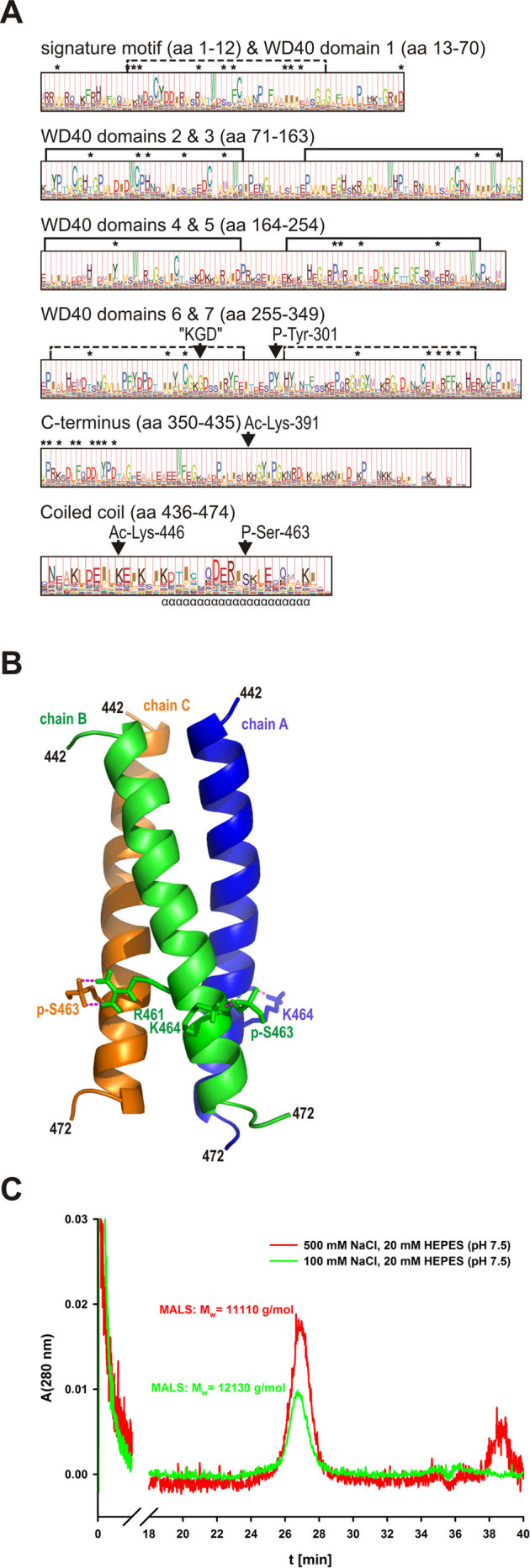
Since our in silico analysis indicated that CRN2 S463 is part of a consensus CK2 target motif, an interaction between the CK2α catalytic subunit and CRN2 was tested by pull-down assays. Experiments employing purified GST-tagged CK2α coupled to glutathione beads as bait and purified soluble CRN2 wild-type or phosphomimetic S463D mutant C-terminal polypeptides as prey showed an interaction between both CRN2 variants and CK2α (Fig. 2A). The experiment was repeated with endogenous as well as GFP-tagged full-length CRN2 proteins from HEK293 cell lysates and showed identical results (Fig. 2B). In both experiments CRN2 also interacted with a dead kinase K68A mutant of CK2α40.
Figure 2. CRN2 directly interacts with CK2α.
(A) pull-down assay employing purified recombinant full-length GST-tagged CK2α coupled to glutathione beads and purified soluble CRN2 wild-type and phosphomimetic S463D C-terminal polypeptides (aa300-474). Both polypeptides bind to CK2α and CK2αdead, the latter lacks kinase activity. (B) pull-down assay employing purified recombinant full-length GST-tagged CK2α coupled to glutathione beads and lysates from HEK293 cells over-expressing endogenous CRN2 (57 kDa) as well as GFP-CRN2 (82 kDa) fusion proteins. Endogenous, wild-type GFP-CRN2, and GFP-CRN2 S463D bind to CK2α. All CRN2 polypeptides were detected with antibody K6-444, and GST-CK2α immunoblotting was done with a rabbit polyclonal GST-antibody86. Asterisk, two additional CK2α bands are the result of degradation. Controls contained beads coated with GST alone. For illustration purposes individual lines from the original western blots were digitally re-arranged.
S463 within the CRN2 coiled coil domain is phosphorylated by CK2α
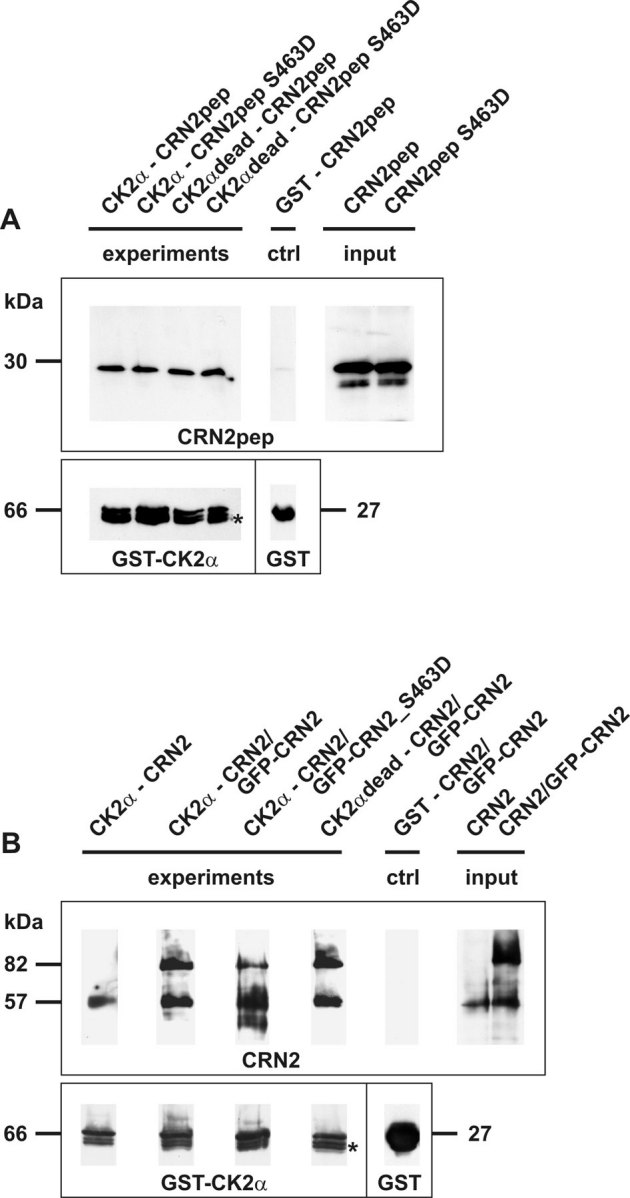
We performed in vitro kinase assays to address the question whether the interaction between CK2α and CRN2 leads to phosphorylation of CRN2. When the wild-type CRN2 C-terminal polypeptide was incubated with CK2α, a CRN2 phosphorylation signal was detected which increased with the incubation time (Fig. 3A). In contrast, the incubation of the S463D mutant resulted in very low levels of phosphate incorporation from [γ-32P]ATP. In addition, kinase assays were performed with recombinant full-length CRN2 proteins purified from insect cells. Wild-type full-length CRN2 was also phosphorylated in a time dependent manner (Fig. 3B). In case of the S463D mutant, phosphorylation signals were hardly detectable at 30 min, and did not reach the intensity of wild-type CRN2 after an extended incubation time of 120 min. These experiments demonstrate that S463 is phosphorylated by CK2α.
Figure 3. CRN2 is a target of CK2α and is phosphorylated at S463.
(A) in vitro kinase assays employing recombinant full-length GST-CK2α (66 kDa, arrow) and His-tagged C-terminal polypeptides of CRN2 (30 kDa, arrowheads). Time course (30, 60, 90 minutes) of phosphate incorporation from [γ-32P]ATP into wild-type as well as S463D mutant CRN2 polypeptides. Controls where the CRN2 polypeptide, [γ-32P]ATP, or CK2α were omitted and use of a kinase-dead CK2α are indicated. Upper panel, autoradiograph of phosphorylation of CRN2 (32P). Lower panel, the corresponding Coomassie brilliant blue stained gel. For all lines containing CK2α identical volumes from the same preparation of purified enzyme were used. Molar concentrations, CK2α 30 nM, CRN2 1.8 µM. Asterisk, contamination by the E. coli 70 kDa chaperone DnaK. Wild-type CRN2 is phosphorylated in a time-dependent manner. Note the minor phosphorylation levels of the mutant polypeptide after 90 min. (B) in vitro kinase assay employing recombinant full-length GST-CK2α (66 kDa, arrow) and GST-His-tagged full-length CRN2 purified from insect cells (90 kDa, arrowheads). Time course (1, 5, 10, 30 minutes) of phosphate incorporation from [γ-32P]ATP into wild-type CRN2. Controls where CRN2, [γ-32P]ATP, or CK2α were omitted and use of a kinase-dead CK2α are indicated. Upper panel, autoradiograph of phosphorylation of CRN2 (32P). Lower panel, the corresponding Coomassie brilliant blue stained gel. For all lines containing CK2α identical volumes from the same preparation of purified enzyme were used. CRN2 is phosphorylated in a time-dependent manner. Note, that only after longer incubation time (120 min) the phosphorylation level of S463D mutant CRN2 reaches the one of wild-type CRN2.
CK2 contributes to the pool of phosphorylated CRN2 in vivo
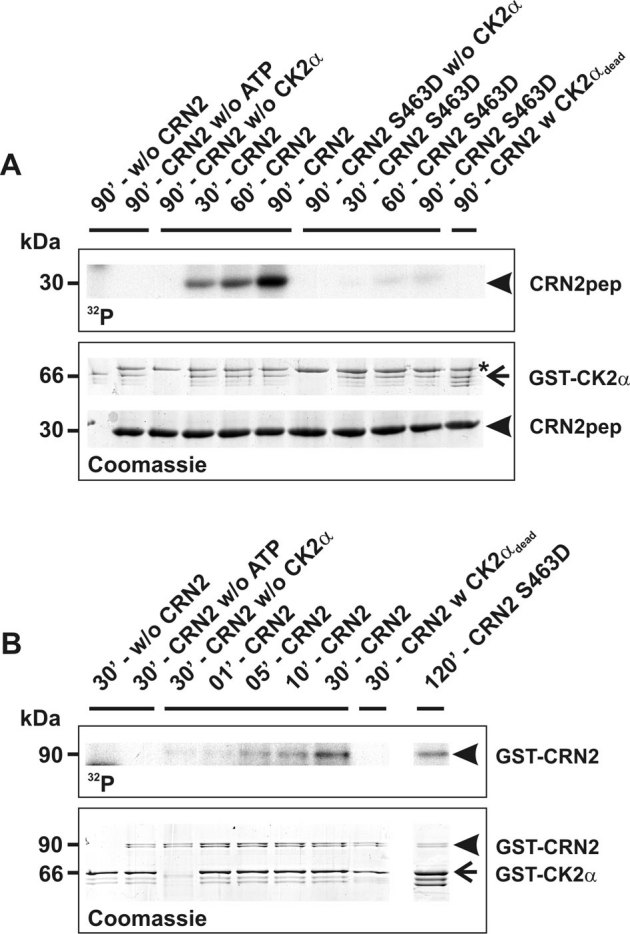
We performed de-phosphorylation experiments to verify the presence of phosphorylated CRN2 within the cell. Lysates from samples of murine tissues were incubated with alkaline phosphatase and analyzed by two-dimensional gel electrophoresis in conjunction with CRN2 immunoblotting. A single spot of CRN2 was detected in untreated samples of skeletal muscle tissue. This spot shifted to a more alkaline pI after treatment with alkaline phosphatase. The distance between both spots was approximately 0.5 pH units and corresponded to a calculated loss of three phosphate residues. Similar results, however with the presence of multiple spots, were obtained for murine brain tissue (Fig. 4A).
Figure 4. CK2 phosphorylates CRN2 in vivo.
(A) presence of a phosphorylated pool of endogenous CRN2. Lysates of murine skeletal muscle (upper three panels) and brain tissue (lower two panels) were separated by 2D-gel electrophoresis. CRN2 was visualized by western blotting using mAb K6-444. The first panel presents the untreated sample (ctrl), the second panel the in vitro de-phosphorylated sample (alkaline phosphatase, AP), and the third panel shows two CRN2 spots resulting from a mixture of the untreated and de-phosphorylated samples (muscle sample only). In contrast to brain tissue (forth and fifth panel), in skeletal muscle only a single spot of CRN2 is detected which corresponds to CRN2 isoform 35. (B) modulation of the in vivo CK2 activity changes the phosphorylation status of CRN2. 293TN cells over-expressing GFP-CRN2 were grown in the presence of the CK2 inhibitor TBB, the CK2 activator 1-ethyl-4,5-dicarbamoylimidazole or solvent control before addition of 32P-orthophosphate. Subsequently, cells were lysed, GFP-CRN2 was immunoprecipitated, samples were separated by SDS-PAGE, proteins were stained by Coomassie brilliant blue (first panel), and gels were dried and used for autoradiography (second panel). Densitometric analysis determined that the CK2 inhibitor decreased the level of CRN2 phosphorylation by 40%, while application of the activator led to an increase of 80% (third panel). Immunoblots for pS13-cdc37, a marker of the in vivo CK2 activity, total cdc37, and CK2α are given as controls (forth to sixth panel). One representative experiment is shown. First and second panel, for illustration purposes the order of lines from the original data was digitally re-arranged. Asterisk, unspecific autoradiography signal which does not correspond to the Coomassie stained CRN2 protein band.
In a next step, we confirmed that phosphorylation of CRN2 also occurs in a CK2-dependent manner in vivo. 293TN cells over-expressing GFP-CRN2 were grown in the presence of the CK2 inhibitor TBB, the CK2 activator 1-ethyl-4,5-dicarbamoylimidazole41,42,43 or solvent control before addition of 32P-orthophosphate. We determined a 40% reduction of CRN2 phosphorylation in case of the CK2 inhibitor and an 80% increase in case of the CK2 activator strongly supporting a physiologically relevant CK2 dependent phosphorylation of CRN2 in vivo (Fig. 4B). Since it has been reported that S13 phosphorylation of the Hsp90 co-chaperone cdc37 is a marker of in vivo CK2 activity44,45, blots were probed for pS13-cdc37, total cdc37, and CK2α for control. We observed a complete suppression of cdc37 phosphorylation in presence of TBB while the levels of cdc37 and CK2 proteins remained unchanged (Fig. 4B, bottom panel). The pattern of an effective TBB induced suppression of cdc37 phosphorylation and an effective 1-ethyl-4,5-dicarbamoylimidazole induced stimulation of CRN2 phosphorylation suggests less CK2-dependent phosphorylation of CRN2 under basal cellular conditions.
CRN2 and CK2 co-localize at the front of lamellipodia
Immunofluorescence analyses were carried out in lamellipodia-rich Pop10 cells to determine the subcellular distribution of CK2 relative to CRN2 and F-actin. Previous studies have shown that the subcellular localization of CRN2 does not change upon CRN2 over-expression6 and that the amount of F-actin is not influenced by the level of CRN2 expression8. In Pop10 cells over-expressing GFP-CRN2 fusion proteins, CRN2 and CK2 were enriched and co-localized in the perinuclear region (Fig. 5, arrowheads) and at the front of lamellipodial structures (Fig. 5, arrows). Lamellipodia showed a co-localization of CRN2, CK2, and F-actin. F-actin stress fibers, which were co-stained by CRN2, did not show any overt enrichment of CK2 (Fig. 5, double-arrowheads). No differences in these patterns were detected, when the cells over-expressed phosphomimetic S463D or phospho-resistant S463A CRN2 instead of the wild-type protein.
Figure 5. CRN2, CK2, and F-actin co-localize in the front of lamellipodia.
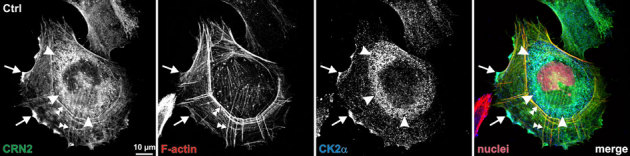
Pop10 cells over-expressing GFP-tagged CRN2 were fixed and CK2α was immunolabeled with primary antibody 1AD9 followed by Alexa-633 labeled secondary antibody; F-actin and nuclei were visualized by TRITC-phalloidin and DAPI, respectively. CRN2 and CK2 co-localize in the peri-nuclear region (arrowheads); CRN2, CK2, and F-actin co-localize in the front of lamellipodia (arrows). Double-arrowheads, F-actin fibers decorated by CRN2.
Wild-type but not S463D phosphomimetic CRN2 inhibits actin polymerization
An influence of CRN2 on actin polymerization was determined in actin polymerization experiments employing G-actin and the Arp2/3 complex together with its activator, the VCA domain of N-WASP. Wild-type CRN2 and the S463A mutant, but not the phosphomimetic S463D CRN2 C-terminal polypeptide, effectively inhibited actin polymerization. CRN2 reduced the velocity of actin filament growth (Fig. 6A; slopes decreased) and the final amount of F-actin (Fig. 6A; plateaus decreased). This inhibitory effect of CRN2 was apparent in the presence or absence of Arp2/3 complex and VCA, although the CRN2 mediated inhibition of actin polymerization always could be antagonized to a limited extent by addition of the Arp2/3 complex (Fig. 6B).
Figure 6. Phosphorylation of serine residue 463 controls the inhibitory effect of CRN2 on actin polymerization.
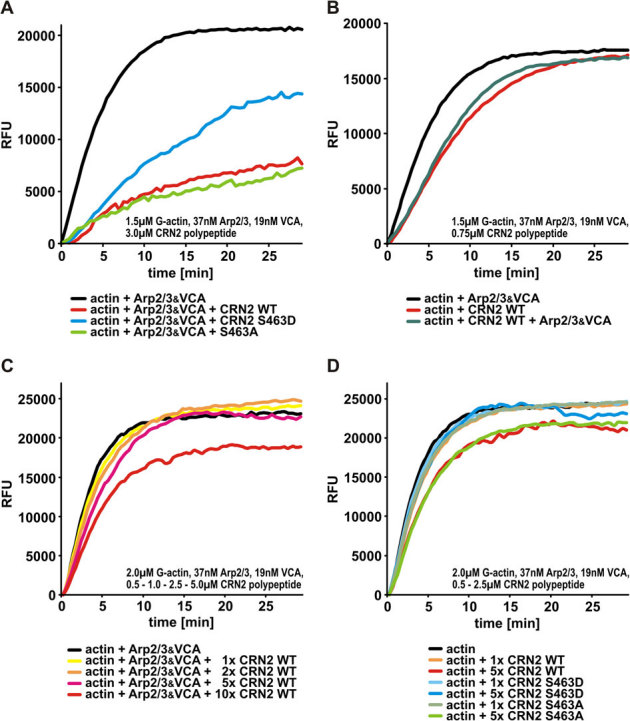
(A) CRN2 wild-type and S643A mutant but not phosphomimetic S463D C-terminal polypeptides effectively inhibit actin polymerization in the presence of Arp2/3 complex. CRN2 reduces the actin polymerization velocity as well as the final amount of F-actin. In this assay a molar ratio of CRN2:actin of 2∶1 was used in order to enhance the effects and visualize differences between the CRN2 polypeptides. For details of the experimental setup see Materials and Methods section. RFU, relative fluorescent units. (B) CRN2 C-terminal polypeptides and the Arp2/3 complex exhibit opposite effects on actin polymerization. The addition of Arp2/3 complex (together with VCA) partially antagonizes the inhibitory effect of CRN2 and increases the actin polymerization velocity. In this assay a molar ratio of CRN2:actin of 1∶2 was used. (C) CRN2 shows a dose-dependent inhibitory effect on actin polymerization. Increasing concentrations of wild-type CRN2 C-terminal polypeptide only decrease the actin polymerization velocity at first (best visible at the time point of 5 min). At the highest CRN2 concentration used the final amount of F-actin also is reduced. In this assay the molar ratio of CRN2:actin was 1∶4 to 2.5∶1. (D) CRN2 wild-type and S463A mutant but not phosphomimetic S463D C-terminal polypeptides affect actin polymerization (in absence of Arp2/3) in a dose-dependent manner. S463D mutant CRN2 neither reduces the actin polymerization velocity nor the final amount of F-actin. In this assay the molar ratio of CRN2:actin was 1∶4 to 1.25∶1.
To verify the specificity of these assays, the CRN2 polypeptides were added to pre-polymerized actin. Here, a small and identical quenching effect of the fluorescence signal of F-actin was observed with every polypeptide tested. It is noteworthy, that the inhibitory effect of CRN2 on actin polymerization was dose-dependent. Low starting concentrations of CRN2 C-terminal polypeptides caused only decreasing velocities of actin polymerization, whereas the highest CRN2 concentration (but lower than the one used in Fig. 6A) additionally reduced the final amount of F-actin (Fig. 6C). A dose-dependent effect was only detected for the wild-type and S463A mutant CRN2 polypeptides, while the phosphomimetic S463D CRN2 C-terminal polypeptide did not show such an effect (Fig. 6D). However, high S463D CRN2 to actin ratios (see figure legend) as used in the experiments shown in Fig. 6A caused a partial inhibition of actin polymerization.
To address the possibility that the inhibitory effect of CRN2 on actin polymerization might result from sequestration of G-actin, we performed fluorescence-based G-actin binding assays. Here, a change in the fluorescence signal of G-actin upon binding of a test protein was only detected for the WH2-domain of CAP2 as control, but not for any of the CRN2 polypeptides. A potential capping effect of CRN2 that might reduce actin polymerization can be excluded due to the high molar ratio of CRN2 vs. G-actin in these assays (see Materials and Methods).
Phosphorylation of CRN2 at S463 affects its interaction with F-actin and Arp2/3 complex
Two-step F-actin co-sedimentation assays were employed to study the interactions of the CRN2 C-terminal polypeptides with F-actin. Wild-type and S463A mutant CRN2 induced the formation of F-actin bundles (Fig. 7A, 10,000xg first pellet), with less binding and co-sedimentation with actin filaments (100,000xg second pellet). S463D phosphomimetic CRN2 demonstrated opposite effects with enrichment in the 100,000xg actin filament pellet and markedly reduced F-actin bundling activity (10,000xg pellet). Addition of Arp2/3 did not change these patterns. However, the CRN2 C-terminal polypeptides competed with the Arp2/3 complex for F-actin binding. Arp2/3 immunoblots of samples derived from 100,000xg F-actin co-sedimentation assays indicated a partial release of the Arp2/3 complex into the supernatant upon the addition of either wild-type or mutant CRN2 polypeptides (Fig. 7B). The interaction of the CRN2 polypeptides as well as of full-length wild-type, S463D and S463A mutant CRN2 proteins with the Arp2/3 complex was further studied by pull-down and co-immunoprecipitation experiments. Both, wild-type and S463A mutant CRN2 bound directly to the Arp2/3 complex, while phosphomimetic S463D CRN2 showed essentially no binding (Fig. 7C,D,E).
Figure 7. Phosphorylation of S463 controls F-actin bundling activity and Arp2/3 interaction of CRN2.
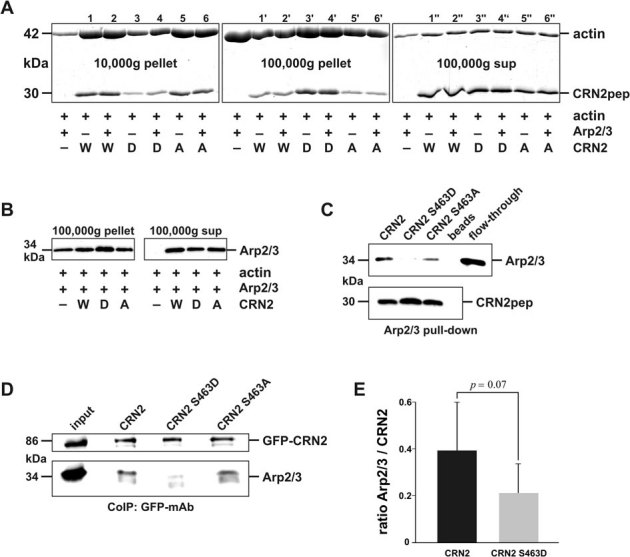
(A) two-step F-actin spin-down assay employing rabbit skeletal muscle G-actin, bovine Arp2/3 complex and purified recombinant CRN2 wild-type (W), phosphomimetic S463D (D), and S463A (A) mutant C-terminal fragments. Coomassie brilliant blue stained SDS-PAGE gels are shown. The left panel (10,000xg first pellet) demonstrates F-actin bundling activity of wild-type and S463A mutant CRN2 in comparison to reduced bundling activity of S463D mutant CRN2. Vice versa the middle panel (100,000xg second pellet) shows increased co-sedimentation of S463D mutant CRN2. Arp2/3 does not influence the results. Right panel, 100,000xg supernatant given as control. As further control, intensities of actin bands were analyzed by densitometry and the measurements demonstrated equal sums for the six triplets n+n'+n''. (B) Arp2/3 immunoblot of a 100,000xg F-actin spin-down experiment. Presence of all CRN2 polypeptides releases Arp2/3 from F-actin into the supernatant. This experiment only allows a qualitative assessment due to difficulties to completely dissolve the pellets in SDS sample buffer and transfer the proteins onto the blot membrane88. (C) pull-down assay employing purified recombinant His-tagged CRN2 wild-type as well as S463A and phosphomimetic S463D mutant C-terminal polypeptides coupled to Ni-beads and soluble purified Arp2/3 complex. In comparison to wild-type and S463A mutant CRN2, the S463D mutant shows reduced direct binding to Arp2/3. Arp2/3, p34 immunoblot; CRN2pep, mAb K6-444 immunoblot. Beads, Ni-sepharose beads lacking CRN2; flow-through, Arp2/3 flow-through from these blank beads. (D) co-immunoprecipitations using GFP mAb K3-167-26 coupled to Protein G coated beads and lysates from 293TN cells expressing GFP-tagged full-length wild-type CRN2 as well as S463D and S463A mutants. Immunoblotting was performed with CRN2 mAb K6-444 and p34 pAb (Upstate #07-227). S463D mutant GFP-CRN2 shows reduced interaction with the Arp2/3 complex. Prior to preparation of the lysates cells were treated with latrunculin B to prevent unspecific co-precipitation of proteins tied together via F-actin bridges. (E) bar chart, densitometry analysis of the Arp2/3 signal intensity from three independent experiments, where GFP-CRN2 wild-type and S463D mutant were parallelly immunoprecipitated; one experiment is shown in D. Arp2/3 values are normalized to the respective GFP-CRN2 values.
Phosphomimetic S463D CRN2 changes the architecture of the F-actin network in the front of lamellipodia and inhibits cell migration
To demonstrate a functional role of CRN2 phosphorylation at S463, U373 human glioblastoma cells with a stable shRNA-mediated knock-down of endogenous CRN2 were transfected with GFP-tagged, shRNA resistant wild-type, phosphomimetic S463D or phospho-resistant S463A mutant CRN2 expression constructs. Replacement of the endogenous CRN2 by the S463A mutant CRN2 led to cells with a smooth and regular co-distribution of CRN2, F-actin, and Arp2/3 complex at the lamellipodia (Fig. 8A). In contrast, cable-like enrichments of CRN2 and F-actin and a disrupted distribution of the Arp2/3 complex were detected in the front of lamellipodial extensions in case of the S463D mutant (Fig. 8B). Furthermore, the latter cells showed a thinner region of CRN2 and Arp2/3 complex (p34-Arc antibody) co-localization, which was restricted to the very tip of lamellipodia (Fig. 8A,B, right panels, distance labels). Cells that expressed GFP-tagged wild-type CRN2 displayed a combination of both phenotypes; a statistical analysis is given in Fig. S3. The sole reduction of the CRN2 expression level in U373 cells did not affect the morphology of lamellipodia (see Fig. 3 in reference8).
Figure 8. Expression of phosphomimetic S463D mutant CRN2 changes the F-actin network in the front of lamellipodia.
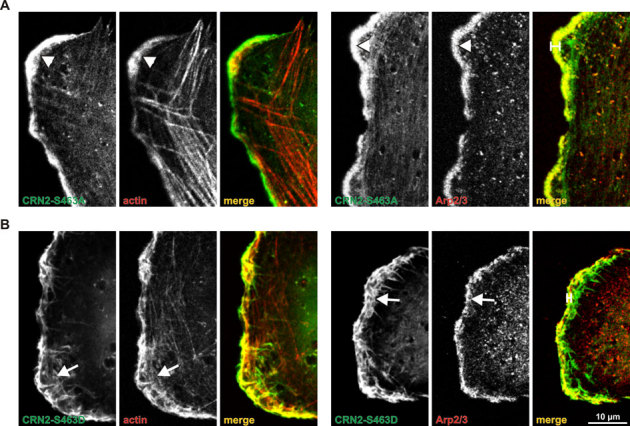
Wild-type (not shown) as well as S463A (A) and S463D (B) mutant GFP-CRN2 was expressed in U373 glioblastoma cells where the endogenous CRN2 was knocked down (95% efficiency, shown in8). The CRN2 over-expression constructs are resistant to the CRN2 specific shRNA used for the knock-down (see Materials and Methods). A,B, left three panels, double stainings of CRN2 (GFP-fluorescence) and actin (TRITC-phalloidin fluorescence). A,B, right three panels, double stainings of CRN2 (GFP-fluorescence) and Arp2/3 (indirect immunofluorescence). Lamellipodia of cells expressing S463D mutant CRN2 demonstrate actin filaments within their fronts which were re-organized into irregular spiky structures in conjunction with irregularly distributed Arp2/3 complex (arrows) and a thinner region of CRN2 and Arp2/3 complex co-localization (distance labels). In contrast S463A induced regular patterns of F-actin and Arp2/3 in the front of lamellipodia (arrowheads). The patterns observed for wild-type CRN2 expressing cells varied between the ones detected for S463A and S463D CRN2 expressing cells and are not shown; see also Fig. S3.
In order to evaluate if these lamellipodial alterations lead to changes in cell migration we monitored the formation of cellular protrusions of HEK293 cells stably expressing the GFP-tagged CRN2 variants. Compared to the wild-type and S463A mutant CRN2 situation, the expression of S463D phosphomimetic CRN2 led to a reduction in the number of cellular protrusions by a factor of two (Fig. 9A). In addition, confluent monolayers of HEK293 cells were used for in vitro wound healing assays. A reduced velocity in wound closure was detected in case of S463D phosphomimetic CRN2 (23 µm/h), compared to wild-type (29 µm/h) and S463A CRN2 (28 µm/h) expressing cells (Fig. 9B).
Figure 9. Phosphorylation of serine residue 463 controls protrusion formation and cell migration.
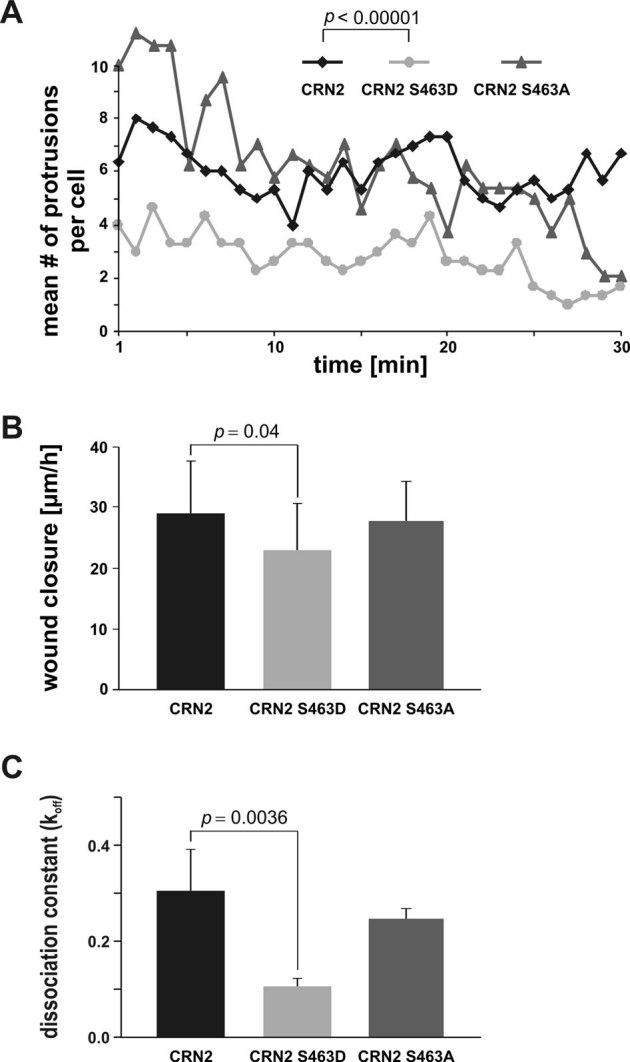
(A) live cell imaging of formation and retraction of cell protrusions. Single 293TN cells expressing wild-type, S463D or S463A mutant CRN2 were monitored. Expression of the S463D mutant led to a distinct reduction (∼50%) of the number of cellular protrusions. CRN2 wild-type vs. CRN2 S463D mutant: mean no. of cellular protrusions 6.0 vs. 2.8, standard deviation 2.1 vs. 2.2, 90 measurements each, Student's t-test p = 4×10−19. (B) in vitro wound healing assays employing 293TN cells expressing full-length wild-type, S463D or S463A mutant CRN2. Cells expressing the S463D mutant show a small but statistically significant defect in wound closure velocity. CRN2 wild-type vs. CRN2 S463D mutant: mean wound closure velocity 29 µm/h vs. 23 µm/h, standard deviation 8.6 µm/h vs. 7.5 µm/h, 60 measurements for each, Student's t-test p = 0.04. (C) GFP-fused CRN2 wild-type, S463D, or S463A was transiently expressed in primary human macrophages. FRAP experiments were performed to determine protein turn-over rates and dissociation constants in F-actin-rich podosome cores (for details see Fig. S7). Note the reduced turn-over rate of the S463D variant (lower koff value of 0.11 s−1; standard deviation 0.017 s−1; Mann-Whitney test wild-type vs. S463D mutant p = 0.0036) compared to wild-type (0.31 s−1; standard deviation 0.09 s−1) and S463A mutant CRN2 (0.25 s−1; standard deviation 0,021 s−1). Each bar represents mean value and standard deviation from 15 measurements from podosomes of at least 3 different cells.
S463D CRN2 displays a delayed integration into the podosome core structure
Alterations in the molecular composition of podosomes, which are prominent adhesion and invasion structures that play an important role in the migration of macrophages and other cell types, further illustrate the cellular relevance of CRN2 S463 phosphorylation. Expression of GFP-CRN2 constructs in primary human macrophages demonstrated an enrichment of wild-type, S463D and S463A mutant CRN2 at podosomes, and all three CRN2 species co-localized with the F-actin core structure (Fig. S4A-C, ctrl). Various podosomal parameters were investigated in the transfected macrophages. Determination of the number, morphology, size, subcellular distribution, and F-actin content of podosomes revealed no differences with respect to the three different CRN2 constructs. Also, a nearly complete knock-down of the endogenous CRN2 demonstrated that podosomes are assembled independently of CRN2 (Fig. S5). However, when the podosomes were disrupted by treatment with the Src family kinase inhibitor PP2 and allowed to re-form after washout of the inhibitor, the phospho-resistant S463A mutant CRN2 was in most cases excluded from the re-assembled podosomes (Fig. S4C, re-formation; Fig. S6). Further, fluorescence recovery after photobleaching (FRAP) experiments were carried out and showed that CRN2 is a fully mobile component (plateau after recovery approximately reaches pre-bleach intensity) of the podosomal structure (Fig. S7, left graphs). An analysis of the fluorescence recovery via bi-exponential equation resulted in fitted curves which indicated a significantly reduced koff value of 0.11 s−1 for S463D mutant CRN2 compared to 0.31 s−1 and 0.25 s−1 for wild-type and S463A mutant CRN2, respectively (Fig. 9C). The dissociation constants were used for the calculation of the half-life times which accordingly showed an elevated half life time (t1/2) of 6.56 s for the S463D mutant in contrast to half-life times of 2.27 s for wild-type and 2.81 s for S463A mutant CRN2 (Fig. S7, right graphs).
Discussion
We identified CRN2 as a novel direct binding partner and substrate of CK2α. This interaction results in phosphorylation of S463 within the coiled coil domain of CRN2. Although it has been shown that CK2 phosphorylation sites in many proteins overlap with sites of caspase cleavage46, this is not the case for S463 of CRN2, since this residue is not part of the consensus motif of caspase 3. Instead, the S463 phosphorylation inhibits the actin filament crosslinking activity and the Arp2/3 binding capacity of CRN2.
Phosphorylation is a common mechanism to regulate coronin protein activity. CRN1 (synonyms: coronin 1B, coronin 2) and CRN4 (synonyms: coronin 1A, coronin 1) are substrates of protein kinase C (PKC). In the case of CRN1 PKC phosphorylates serine 2, a residue that is not present in CRN2, and thereby inhibits the interaction between Arp2/3 and CRN1. As a consequence, cell migration velocity is reduced and the PMA-induced membrane ruffling is suppressed47. For CRN4 the specific PKC phosphorylation site is unknown. However, phosphorylation of CRN4 leads to its dissociation from phagosomes and a role of CRN4 in the maturation of phagosomes has been postulated48.
Thus far, only three studies have reported a specific interaction of CK2 with actin or actin-associated proteins in mammalian cells. First, rabbit skeletal muscle G-actin directly binds to the CK2α subunit and inhibits the activity of CK2 in a dose-dependent manner in vitro49. More importantly, CK2 phosphorylates the VCA domain of WASP at serine residues 483 and 484, which in turn enhances the interaction of VCA domain with the Arp2/3 complex and thereby increases the velocity of Arp2/3 mediated actin polymerization50. Furthermore, CK2 synergizes with CKIP-1 to inhibit the actin capping protein CapZ at the barbed ends of actin filaments in actin de-polymerization assays and actin polymerization assays starting from spectrin-F-actin seeds, but neither protein has an influence on the dissociation of CapZ from F-actin in uncapping assays51. In summary, CK2 is able to promote actin polymerization and reduce the formation of crosslinked actin filaments (Fig. 10).
Figure 10. Scheme summarizing effects of CK2 and CRN2 on actin dynamics.
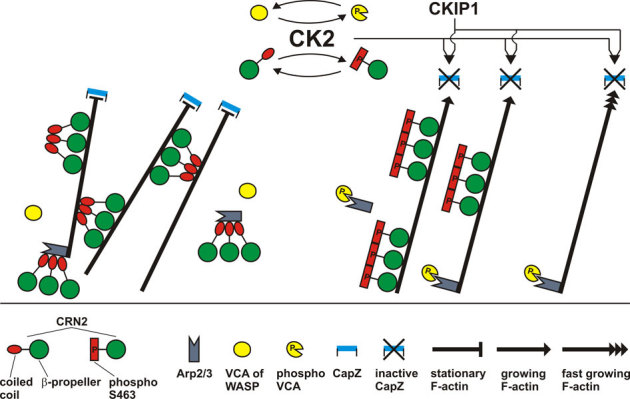
CK2 phosphorylates CRN2 at S463; CRN2 constitutively forms trimers independent of its phosphorylation state; CRN2 and phospho-CRN2 both bind to F-actin; CRN2 strongly inhibits actin polymerization, whereas phospho-CRN2 is nearly inactive; CRN2 strongly and phospho-CRN2 to a much lesser extent bundles F-actin; CRN2, but not phospho-CRN2 interacts with the Arp2/3 complex; both CRN2 and phospho-CRN2 compete with Arp2/3 for F-actin binding. Full-length CRN2 contains a large actin filament binding region formed by the β-propeller and a second distinct actin filament binding site within the coiled coil domain. Arp2/3 mediated branching of actin filaments is not included in this scheme. Data on the interactions of CK2 with VCA domain of WASP, and with CKIP-1 and CapZ were obtained from other studies50,51 and embedded into this scheme. Taken together, a picture emerges where CK2 acts in an integrative manner and promotes actin polymerization and suppresses actin filament bundling.
From our results we conclude that wild-type, S463D and S463A mutant CRN2 polypeptides do not exert any capping effect, but most likely bind to the side facing the actin filaments as described for CRN452. However, only the direct binding of wild-type and S463A mutant CRN2 polypeptides to F-actin leads to a reduced polymerization velocity. All CRN2 polypeptides were able to partially displace the Arp2/3 complex at actin filaments and, moreover, wild-type and S463A mutant, but not the phosphomimetic S463D CRN2 C-terminal polypeptides, were found to interact with free Arp2/3 complex. Thus, the wild-type and S463A mutant CRN2 polypeptides may additionally inhibit actin polymerization in an indirect manner (Fig. 10).
A competition of the Arp2/3 complex binding to F-actin has also been reported for CRN1, which results in a reduced F-actin density in the front of lamellipodia and a disturbed formation and persistence of cell protrusions53. The exact binding site of the Arp2/3 complex on CRN2 is unknown, but our pull-down and CoIP experiments employing CRN2 polypeptides and full-length protein, respectively, suggest that Arp2/3 binds to a motif in the coiled coil domain of CRN2 harboring S463. For mammalian CRN4 and yeast CRN11 (synonym: Crn1p) the Arp2/3 binding site has also been mapped to the coiled coil region3,54,55. CRN2 probably inactivates the Arp2/3 complex in a similar way as it has been described for CRN4, CRN1 and yeast CRN11, where coronin holds the Arp2/3 complex in an inactive open conformation away from the actin filaments55,56,57,58.
Previous studies indicated that full-length CRN2 possesses binding sites for F-actin in the conserved WD40-repeat domain forming the seven-bladed β-propeller6,59 and in the conserved part of the C-terminal linker region3,4,59,60,61. More specifically, a conserved arginine residue, R30 in CRN1 and R28 in CRN2, which is surface exposed and located within the seventh β-propeller blade3, has turned out to be essential for F-actin binding of both coronin proteins12,62. A belt-shaped actin binding region on the surface of the β-propeller has been identified in yeast CRN1163, which is conserved in all coronin proteins. Moreover, for mammalian CRN4 a specific F-actin binding motif was shown in the C-terminal linker region (aa400-416)61, which is conserved in CRN2 (aa398–418, 59% similarity, and 32% identity). Subsequent studies have demonstrated that the residues of CRN4 that bind to actin span over the entire molecule and specifically locate to the β-propeller and the C-terminal linker region52,54. The corroborative data from all these reports indicate that the scattered positions of actin binding sites in coronin proteins map from the N-terminal part to the C-terminal linker and form one large F-actin binding region which can make contacts to several actin molecules. In this scenario one coronin molecule apparently makes contacts to three actin molecules, namely between two actin molecules of one and an additional actin molecule of the second “substrand” of the two-start helix of an actin filament52,64.
Apart from this widespread actin binding region another actin binding site has been detected in the coiled coil domain of CRN4 and yeast CRN1160,65. Our data show that CRN2 also contains an additional actin binding site in the coiled coil domain. The C-terminal CRN2 polypeptides used in our in vitro analyses were proven to fold properly6 and comprise a part of this large belt-shaped actin binding region as well as the coiled coil domain. Phosphorylation of S463 within the coiled coil inactivated this second actin binding site so that the actin filament crosslinking activity of CRN2 was reduced but the F-actin binding activity retained. Thus, the second actin binding site within the coiled coil domain of CRN2 is a CK2 dependent regulatory element for actin filament crosslinking (Fig. 10).
Our data strongly suggest that the coiled coil domain of CRN2 has three major functions, i) formation of a constitutively trimeric CRN2 quaternary structure, ii) binding to actin filaments, and iii) interaction with the Arp2/3 complex. However, only trimerization and F-actin binding, or alternatively trimerization and Arp2/3 interaction of (non-phosphorylated) CRN2, can occur at the same time. A simultaneous binding to F-actin and Arp2/3 apparently is not possible as CRN2 was found to displace the Arp2/3 complex from actin filaments. This agrees with results of the FRAP experiments in this study, which show a 3.5-fold increased half-life time of the phosphomimetic CRN2 S463D mutant in podosome F-actin cores. As these cores are rich in Arp2/3 complex the S463D mutant CRN2 lacking the ability to interact with the Arp2/3 complex exhibited a prolonged interaction with F-actin.
Based on our data we assume that in vivo the cellular fraction of non-phosphorylated CRN2, previously described as cytoskeleton associated pool4, inhibits actin polymerization, bundles actin filaments, binds to and inactivates Arp2/3 complexes, and accordingly leads to the formation of ‘stabilized’ F-actin structures (Fig. 10). Phosphorylation of CRN2 by CK2 inhibits these CRN2 functions. This is supported by our observations where the presence of S463D phosphomimetic full-length CRN2 correlated with an accumulation of F-actin structures and an irregular re-distribution of the Arp2/3 complex in the front of lamellipodia, a reduced velocity of cell migration, and a decreased number of cell protrusions.
Methods
Molecular modeling
The sequence of CRN2 aa315-474 was subjected to multimeric coiled coil prediction by the program MultiCoil66, which predicted, in addition to experimental evidence4, a trimeric coiled coil for the peptide sequence CRN2 aa438-467 with a probability of 62%. While the three-dimensional crystal structure of the coiled coil domain of CRN4 has been determined67, the amino acid sequence alignment of the CRN4 and CRN2 coiled coil domains showed that CRN4 lacks a hexapeptide in this region. We have therefore re-constructed the trimeric coiled coil of hemagglutinin (HA2 chain) using residues 74 to 113 from chain B in PDB entry 1eo836, and used this as a template for comparative modeling. Twenty independent models for CRN2 aa435-474 were generated with MODELLER68 and the one with the lowest energy was selected. The overall geometry was scrutinized using PROCHECK69.
Molecular cloning and protein expression
A human CRN2 cDNA fragment coding for a C-terminal polypeptide cloned into the pQE30 (Qiagen) vector (aa300-4746,) and a full-length CRN2 cDNA cloned into pEGFP-C1 (Invitrogen) vector (aa1-4744,) were used as templates. Using the QuickChange Site-Directed Mutagenesis kit (Stratagene) in conjunction with primer pairs i) CRN2mutS463Dfor 5′-GCAATCAAGATGAGCGTATTTCCAAGTTAGAACAGCAGATGGC-3′ and CRN2mutS463Drev 5′-GCCATCTGCTGTTCTAACTTGTCAATACGCTCATCTTGATTGC-3′, ii) CRN2mutS463Afor 5′-CAATCAAGATGAGCGTATTGCCAAGTTAGAACAGCAGATGGC-3′ and CRN2mutS463Arev 5′-GCCATCTGCTGTTCTAACTTGGCAATACGCTCATCTTGATTGC-3′, and iii) CRN2shRNA77resistFor 5′-GAATCCCCGTACGTGCACTATCTCAATACATTTAGCAGCAAG-3′ and CRN2shRNA77resistRev 5′-CTTGCTGCTAAATGTATTGAGATAGTGCACGTACGGGGATTC-3′ the following expression constructs were generated and verified by sequencing: pQE30-CRN2-S463D, pQE30-CRN2-S463A, pEGFP-CRN2-S463D, pEGFP-CRN2-S463A, pEGFP-CRN2res-WT, pEGFP-CRN2res-S463D, and pEGFP-CRN2res-S463A. The ‘CRN2res’ expression constructs were used for transfections of cells in which a stable expression of the CRN2 specific shRNA oligo cgtccactacctcaacacatt led to a 95% reduction of the endogenous CRN2 levels8. Expression and purification of CRN2 WT, S463D, S463A C-terminal polypeptides from M15 E. coli cells was carried out according to4.
Full-length human GST-His6-tagged CRN2 wild-type and S463D mutant proteins were purified from Sf9 insect cell cultures according to the manufacturer's protocol (bac-to-bac expression system, Invitrogen). In brief, the DH10Bac E. coli strain was transformed with a pDEST10 donor vector containing a GST-His6-CRN2 cassette and subsequently used for the purification of recombinant bacmid DNA. The latter was used to transfect Sf9 cells and resulting baculoviruses were amplified several times in order to finally infect Sf9 cells cultivated in spinner flasks (Techne) using Sf-900 III SFM medium (Invitrogen).
pGEX-2T based vectors for expression of GST-tagged chicken CK2α and CK2α kinase dead (K68A mutant) were kindly provided by Drs. Odile Filhol-Cochet (iRTSV - CEA, Grenoble, France) and Yves Goldberg (University Joseph Fourier, Grenoble, France), and purified as described earlier70,71. Identity of all purified proteins was confirmed by mass spectrometry.
Mammalian cell culture and life cell imaging
HEK293 human embryonic kidney (ATCC: CRL-1573), 293TN human pseudoviral particle producer (BioCat/SBI: LV900A-1), U373 human glioblastoma (ECACC: 89081403), and Pop10 human hepatocarcinoma72 cells were grown in Dulbecco's modified Eagle's Medium (DMEM, 4.5 g/l glucose, Sigma) supplemented with 10% fetal calf serum (Biochrom), 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 2 mM L-glutamine (Sigma), 100 units/ml penicillin G, and 100 µg/ml streptomycin (Invitrogen). Cells were grown in 5% CO2 at 37°C. For transfections of HEK293, 293TN, and Pop10 cells at 50–80% confluence the lipofectamine 2000 reagent (Invitrogen) and for electroporation of U373 cells the Nucleofector II device (Amaxa/Lonza) were used. Generation of U373 cells stably expressing the CRN2 specific shRNA oligo cgtccactacctcaacacatt is described in8. In vitro wound healing and single cell protrusion assays were performed according to6. Primary human macrophages were generated as described in73. Macrophages were transiently transfected using a MicroPorator MP-100 electroporation device (PeqLab) by applying two pulses with a pulse voltage of 1000 V and pulse width of 40 ms and subsequently seeded on cover-slips or glass-bottom cell culture dishes.
Gel filtration analyses
Purified CRN2 C-terminal polypeptides expressed in E. coli, purified GST-tagged or GST-cleaved full-length CRN2 expressed in insect cells, and GFP-tagged CRN2 expressed in mammalian cells were used for gel filtration assays. Purified polypeptides and proteins or the cell lysates were pre-cleared at 100,000xg for 60 min, injected into Superdex G-75 or G-200 columns, and analyzed using the SMART system (Amersham Biosciences). 50-µl fractions collected at the end of each run were analyzed by immunoblotting.
The phosphorylated CRN2 coiled coil aa435-474 with pS463 was obtained as synthetic peptide from Metabion, Germany, and subjected to gel filtration analyses with multi-angle light scattering detection (SEC-MALS) after separation by a Superose-12 column. The samples had concentrations of 4.5, 9 and 18 mg/ml, and the buffer conditions were 20 mM HEPES (pH 7.5) and 100 mM NaCl (normal salt) or 500 mM NaCl (high salt). The SEC-MALS combination consisted of a BioRad DuoFlow HPLC coupled to a Wyatt miniDawn TREOS light scattering detector and a Shimadzu RID-10A refractive index detector. MALS analysis was carried out using the Wyatt ASTRA software.
Small-angle X-ray scattering
Small-angle X-ray scattering data of the sythetic CRN2 coiled coil peptide (435–474) was acquired on a SAXSess instrument (Anton Paar, Austria) with a sealed tube microsource (Cu-Kα). Data collection was performed by multiple 10 s exposures at 20°C in re-usable thermostated quartz capillaries, which were placed in the integrated vacuum chamber of the camera. Datasets were recorded on a CCD detector and software CCDquant, covering a momentum transfer range of 0.003 < q < 2.8 Å−1, where q = (4πsinθ)/λ, 2θ is the scattering angle, and λ is the radiation wavelength. The measured data were corrected for dark current, background scattering from 100 mM NaCl, 20 mM Hepes (pH 7.5) was subtracted, and the data were de-smeared using the instrument-specific profile (SAXSquant). A water sample was used as calibration standard. Data were recorded at ρ*, ρ*/2 and ρ*/4, with ρ* = 18 mg ml−1.
The radius of gyration Rg and the intensity of forward scattering I(0) were determined by Guinier analysis using PRIMUS74. Both parameters were also determined together with the distance distribution function p(r) by the program GNOM75. The experimental molecular mass was calculated from the intensity of forward scattering I(0) using the formula: M = I(0)×NA×ρ*−1×Δρ−2; Δρ = 2×1010 cm g−1; NA = 6.022×1023 mol−1, where Δρ is the excess scattering length per unit mass of the protein and NA is Avogadro's number.
Ab initio models of the scattering particles were obtained by shape restoration with DAMMIN76. For each dataset, twenty independent models were generated and averaged using DAMAVER and DAMFILT76. The final shape obtained for dataset ρ* = 18 mg ml−1 was used to fit the atomic model of the triple phosphorylated coiled coil peptide from the MD simulation (see above). Model fitting was performed with the program SAFIR from the PCSB collection77; theoretical scattering curves and their fit to experimental data were obtained with CRYSOL39.
GST pull-down analyses
Cell extracts were prepared by lysing HEK293 cells with buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, 0.5% NP40) and E. coli cells with buffer B (200 mM NaCl, 5% glycerine, 100 μg/ml lysozyme, 0.5% NP40, 10 mM DTT, 0.5 mM PMSF, 2 mM Benzamidin, 10 μg/ml aprotinin and leupeptin) on ice followed by sonication. Clarified soluble extracts were obtained by centrifugation at 100,000xg for 5 min at 4°C. In parallel, GST-CK2α fusion proteins purified from bacteria were mixed with 100 μl of equilibrated glutathione sepharose beads (GE Healthcare) and incubated for 2 h at 4°C. After 4 washes with buffer C (4.3 mM NaH2PO4, 1.47 mM KH2PO4, 1.37 mM NaCl, 2.7 mM KCl), aliquots of the beads were incubated together with the supernatants from either HEK293 cells expressing GFP-tagged CRN2 proteins or bacteria containing His6-tagged CRN2 C-terminal polypeptides. Further incubation for 2 h at 4°C was carried out to pull-down respective proteins. Finally, the beads were washed 3 times with wash buffer D (4.3 mM NaH2PO4, 1.47 mM KH2PO4, 1.37 mM NaCl, 2.7 mM KCl, 5% glycerin), and proteins were eluted with SDS sample buffer and analyzed by immunoblotting. Control experiments were performed with GST-coated beads or soluble GFP protein alone.
In vitro kinase assays
In vitro CK2α kinase assays were performed in phosphorylation buffer (50 mM MOPS pH 7.0, 150 mM NaCl, 10 mM MgCl2, ATP and [γ-32P]ATP (10 µCi/µl, Amersham)) according to78. His6-tagged CRN2 C-terminal polypeptides (∼4 µg) as well as full-length GST-tagged CRN2 (∼0.2 µg) proteins were added to 50 µl of 2x phosphorylation buffer without [γ-32P]ATP. The reaction was initiated by addition of CK2α (∼0.2 µg) with the final addition of 5 µl ATP mix (6 µl 2 mM ATP, 2.4 µl 10 µCi/µl [γ-32P]ATP, 51.6 µl H2O). After adjusting the reaction volume to 100 µl, the reaction mixtures were incubated for 1, 5, 10, 30, 60, 90, and 120 min at 30°C. The reactions were terminated by addition of 20 µl 60 mM EDTA, 30 µl 5x SDS sample buffer, and boiling for 10 min at 95°C. All samples were analyzed by SDS-PAGE followed by Coomassie brilliant blue staining and autoradiography.
In vitro de-phosphorylation assays and 2D gel electrophoresis
Alkaline phosphatase is able to hydrolyze phosphate esters of primary and secondary alcohols, amines, and phenols, including serine, threonine and tyrosine residues in proteins. For in vitro de-phosphorylation 20 mg of murine skeletal muscle tissue was pulverized on dry ice, dissolved in de-phosphorylation buffer (100 mM Tris/HCl pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT, 1∶200 protease inhibitor (Sigma)), and centrifuged for 10 min at 16,000xg. 5 µl (5 U) alkaline phosphatase (Roche) were added to the supernatant and incubated for 30 min at 30°C. Positive controls additionally contained p-nitrophenylphosphate (pNPP), negative controls lacked the phosphatase. Samples were subjected to two-dimensional gel electrophoresis in conjunction with immunoblotting according to79. CRN2 protein spots were visualized with enhanced chemiluminescence followed by exposure to x-ray films (Kodak).
In vivo kinase assays
Day 0: 293TN cells were seeded into 10 cm culture dishes. Day 1: Three dishes with a cell confluence of 30% were selected for lipofectamine 2000 transfections with the EGFP-CRN2-WT construct. Day 3: The growth medium was replaced by 10 ml phosphate-free DMEM (Gibco 11971) supplemented with 10% fetal calf serum (Biochrom), 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 2 mM L-glutamine (Sigma), 100 units/ml penicillin G, and 100 µg/ml streptomycin (Invitrogen). After 2 hours of incubation the medium was exchanged for 4 ml of the above phosphate-free medium without fetal calf serum (serum may attenuate or inhibit the efficiency of TBB). Either 40 µl of DMSO (control), CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB; final concentration 100 µM; 10 mM stock in DMSO)80,81,82, or CK2 activator 1-ethyl-4,5-dicarbamoylimidazole41,42,43 (final concentration 20 nM; 20 mM stock in H2O) and DMSO were added to the three culture dishes. After 2 hours of pre-incubation with the drugs 1.25 mCi (230 µl) 32P-orthophosphate (PerkinElmer, NEX053) were added to each dish. Day 4: Cells were harvested, washed three times with phosphate- and serum-free medium, lysed in 1 ml lysis buffer (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1% Triton X-100), centrifuged, and GFP-CRN2 was immunoprecipitated from the 16,000xg supernatant using 50 µl Anti-GFP-tag MicroBeads (Miltenyi Biotec). Beads were collected in M-columns (void volume 80 µl) and washed twice with 600 µl lysis buffer, three times with wash buffer 1 (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 0.1% Triton X-100), three times with wash buffer 2 (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS), and finally once with buffer 3 (20 mM Tris/HCl pH 7.5). The lysis buffer contained phosphatase (Sigma) and protease (Roche) inhibitor cocktails in double concentration. Immunoprecipitated GFP-CRN2 was eluted with 70 µl elution buffer (50 mM Tris/HCl pH 6.8, 50 mM DTT, 1% SDS, 1 mM EDTA, 10% glycerol, 0.005% bromphenol blue), subjected to SDS-PAGE, Coomassie brilliant blue staining, and autoradiography. A duplicate set of three plates was treated in parallel in order to harvest cells for immunoblotting. The duplicates were not incubated with32P-orthophosphate but with medium containing phosphate at the appropriate steps of the experiment.
Actin polymerization assays
Actin polymerization assays were performed using the Actin polymerization Biochem kit (BK003, Cytoskeleton) in which the rate of pyrene-labelled G-actin conversion into F-actin was monitored. Pyrene fluorescence signals were monitored in black flat bottom 96 well plates (Nunc) using an Infinite M1000 device (Tecan) equipped with Tecan i-control (version 1.6.19.2) with the following settings: all samples and device equilibrated to 25°C, fluorescence top reading, excitation 350 nm with 20 nm bandwidth, emission 407 nm with 20 nm bandwidth, gain 85, 20 flashes at 400 Hz, 20 µs integration time, 200 ms settle time, 20198 µm Z-position, 30 s measurement interval, polymerization start by dispension of 13 µl 10x polymerization buffer (500 mM KCl, 20 mM MgCl2, 10 mM ATP), final volume per well 125 µl. Pyrene-labelled rabbit skeletal muscle G-actin, bovine brain Arp2/3 complex (RP01, Cytoskeleton), recombinant human GST-tagged VCA-domain of WASP (VCG03, Cytoskeleton), and His6-tagged CRN2 C-terminal polypeptides were prepared in G-buffer (5 mM Tris-HCl pH 8.0, 0.2 mM CaCl2). Final concentrations were: G-actin 1.5 µM (Fig. 6A,B,E) or 2 µM (Fig. 6C,D), Arp2/3 complex 37 nM in conjunction with VCA-domain 19 nM, CRN2 polypeptides 3 µM (Fig. 6A,E), 0.75 µM (Fig. 6B), 0.5 to 5 µM (Fig. 6C), 0.5 and 2.5 µM (Fig. 6D). Several different experiments were conducted for all combinations of proteins. To exclude a potential quenching effect of the CRN2 polypeptides, identical volumes of G-buffer or CRN2 polypeptides were added to separate wells after polymerization of actin. In order to verify the amounts of the CRN2 polypeptides added to the reactions, samples from all wells were taken at the end of each experiment, separated by SDS-PAGE, and proteins were stained by Coomassie brilliant blue.
G-actin binding assays
Three-dimensional fluorescence spectrometry was employed to investigate a potential interaction of CRN2 and G-actin. Changes of the fluorescence signals of pyrene-labelled G-actin upon addition of identical amounts of the CRN2 C-terminal polypeptides were monitored using the Infinite M1000 device (Tecan) with the following settings: all samples and device equilibrated to 25°C, fluorescence top reading, excitation start 310 nm, excitation end 364 nm, excitation step size 2 nm, excitation bandwidth 5 nm, emission start 372 nm, emission end 444 nm, emission step size 2 nm, emission bandwidth 5 nm, gain 100, 20 flashes at 400 Hz, 20 µs integration time, 20198 µm Z-position, volume per well 125 µl. Pyrene-labelled rabbit skeletal muscle G-actin, His6-tagged CRN2 C-terminal polypeptides, BSA (present in all assays), a WH2 domain from CAP2 (positive control for G-actin interaction generously provided by Dr. Vivek Peche) were prepared in G-buffer. Final concentrations were: G-actin 2 µM, CRN2 polypeptides, BSA, and CAP2WH 2 µM. Negative controls lacked the CRN2 C-terminal polypeptides or both, CRN2 polypeptides and the BSA. All assays were also carried out in absence of pyrene-labelled G-actin to determine the intrinsic fluorescence of the CRN2 C-terminal polypeptides.
Actin spin-down assays
75 µl samples derived from the actin polymerization assays were used for sequential low (10,000xg) and high speed (100,000xg) centrifugation steps in order to investigate the actin binding and bundling activity, respectively, of the CRN2 C-terminal polypeptides according to83. Equal amounts of the two pellets and the final supernatant were resolved by SDS-PAGE and proteins were visualized by Coomassie brilliant blue staining. Controls contained only the CRN2 polypeptides. Another 25 µl samples derived from the polymerization assays were separated into 100,000xg supernatant and pellet to determine the competitive influence of the CNR2 C-terminal polypeptides on the F-actin – Arp2/3 interaction according to84. Pellets and supernatants were resolved by SDS-PAGE and analyzed by immunoblotting.
Arp2/3 pull-down assays
Equal amounts of His6-tagged CRN2 C-terminal polypeptides were incubated with 50 μl of Anti-His-tag MicroBeads (Miltenyi Biotec) in 250 µl lysis buffer (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1% Triton-X-100) for 1 h on ice. Incubation was further prolonged for 1 h with addition of 2 µg bovine Arp2/3 complex (RP01, Cytoskeleton). The mixture was passed through µ-columns (void volume 30 µl) prepared according to the manufacturer's instructions. After rinsing the columns with 4x 200 μl of wash buffer 1 (50 mM Tris/HCl pH 8.0, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS,) and 1x 100 μl of wash buffer 2 (20 mM Tris/HCl pH 7.5), proteins were eluted with 120 μl of pre-heated elution buffer (50 mM Tris/HCl pH 6.8, 50 mM DTT, 1% SDS, 1 mM EDTA, 0.005% bromphenol blue, 10% glycerol) and analyzed by SDS-PAGE in conjunction with immunoblotting.
Co-immunoprecipitation experiments
HEK293 cells transfected with pEGFP-CRN2-WT, pEGFP-CRN2-S463D, or pEGFP-CRN2-S463A were treated with latrunculin B according to6, harvested and lysed on ice with immunoprecipitation (IP) buffer (20 mM HEPES pH 7.0, 100 mM KCl, 0.5% NP-40, 1 mM EDTA, 1 mM PMSF, 10 µg/ml 1,10-pheanthroline, 10 µg/ml aprotinin, 10 µg/ml leupeptin, 10 mM NaF, 2 mM sodium orthovanadate) followed by sonication. The lysates were cleared at 13,000xg for 5 min and incubated for 2 h with 500 μl of concentrated GFP mAb K3-167-26. Protein G coated sepharose beads (Zymed) were washed extensively with IP buffer, pre-blocked with 5% BSA, added to the cell lysates and further incubated overnight. Collected immune complexes were washed several times with IP buffer before the beads were boiled in SDS sample buffer. Proteins were separated by SDS PAGE and analyzed by immunoblotting.
Immunofluorescence analyses, immunoblotting, and antibodies
Direct and indirect immunofluorescence analyses as well as immunoblotting were performed as described in85. Immunofluorescence images were captured on a Leica TCS SP5/AOBS/tandem scanning system equipped with the Leica LAS-AF software (version 2.2.1 build 4842) and further processed using CorelDraw Graphics Suite X4. Visualization of immunoblots was done by enhanced chemiluminescence in conjunction with the imaging system Fluorchem SP (Alpha Innotech) or exposure to x-ray films (Kodak). Antibodies used in this study specifically recognized CRN2 (mAb K6-444-44,), Arp2/3 complex (p34-Arc subunit, rabbit polyclonal Ab, Upstate), GST (rabbit polyclonal antibody86,), CK2α (mAb 1AD9, KinaseDetect), and cdc37 phosphorylated at serine 13 (45; note, that the rabbit polyclonal Ab ab61797 (Abcam) also recognizes non-phosphorylated cdc37); F-actin was labelled with 200 ng/ml TRITC-phalloidin (Sigma).
Fluorescence recovery after photobleaching (FRAP) experiments
Transfected primary human macrophages grown in glass-bottom culture dishes were placed in a heating insert P covered with a small incubator S-2 for warm air incubation and CO2-control (Pecon) mounted on a Leica TCS SP2 confocal microscope. Living cells were kept at 37°C in a humidified atmosphere with 5% CO2 and observed using an oil immersion HCX PL APO 63x/1.4-0.6 lambda blue objective lens. Subcellular localization of GFP-tagged proteins was monitored using the 488 nm laser line of an argon ion laser. Photobleaching experiments were performed as follows: before the bleaching event 10–15 frames were acquired, then, by illumination of a selected region with 100% laser light intensity using 10 bleaching iterations, GFP fluorescence has been diminished. The recovery of fluorescence was recorded over time as indicated. FRAP movies were analyzed by using Leica LCS Lite software (version 2.61) for the measurement of fluorescence signal intensities in defined regions of interest (photobleached podosomes, background, control). Obtained raw data were corrected for background fluorescence and acquisition photobleaching, normalized, and further processed using Excel 2004 for Mac (Microsoft) and Systat SigmaPlot (version 10) in order to calculate recovery and regression curves as described in87.
Podosome re-formation experiments
Transfected macrophages expressing mRFP-Lifeact and GFP-CRN2 constructs were seeded on coverslips and cultivated for 6 h in complete culture medium containing RPMI1640, 20% human serum, and penicillin/streptomycin. Cells were then washed twice with plain RPMI1640 medium and cultivated in basal medium overnight. The next day, control cells were fixed with 3.7% formaldehyde directly before application of the Src family kinase inhibitor PP2 (4-Amino-5-(4-chlorophenyl)-7-(t-Butyl)pyrozolo[3-4-d]pyrimidine; Calbiochem). To monitor the re-formation of podosomes, the culture medium was replaced by 25 µM PP2 in plain RPMI1640. After 30 min almost all macrophage podosomes were disassembled, although cells were still adherent. Then, the inhibitor was washed out with plain RPMI1640 and the cells were again incubated in full culture medium in order to stimulate re-formation of podosomes. Macrophages were fixed after 45 min for the analysis of GFP-CRN2 re-incorporation into podosomes using a Zeiss Axiovert 200 M fluorescence microscope equipped with an EC Plan-Neofluar 40x/0.75 objective. In several fields of view the total number of cells expressing GFP-CRN2 proteins were counted and correlated with the number of cells showing a clear co-localization of GFP-CRN2 constructs with mRFP-Lifeact at podosomes. Graphs were generated using Excel 2004 for Mac (Microsoft) and Prism 5.0c for Mac (GraphPad).
Author Contributions
CPX, RHR, MB, MS, MH, ROM, MPF, CW, AO, AH, SL, and CSC designed, performed, and analyzed experimental work. YM and RAG provided new methods and essential chemicals. LE, AAN and CSC designed experiments and evaluated the overall data. CSC wrote the main manuscript text and prepared the figures. All authors reviewed the manuscript.
Supplementary Material
Supplementary Information
Acknowledgments
CK2α expression plasmids were generously provided by Drs. Odile Filhol-Cochet and Yves Goldberg (University Joseph Fourier, Grenoble, France), recombinant WH2 domain from CAP2 by Dr. Vivek Peche (University of Cologne, Cologne, Germany). We are grateful to Kevin Jack (University of Queensland, St. Lucia, Australia) for assistance with data acquisition on the in-house SAXS instrument. We thank Dr. Lorenzo A. Pinna (University of Padua, Padua, Italy) for helpful discussions. This work was supported by grants of the German Research Foundation awarded to C.S.C and A.A.N. (DFG: NO 113/22-1 and CL 381/2-1). Grant BFU2007-67876 support for R.O.M. and M.P.F. came from the Spanish Ministry of Science and Innovation (MICINN).
References
- Morgan R. O. & Fernandez M. P. in The Coronin Family of Proteins Vol. 48 Subcellular Biochemistry (eds C. S. Clemen, L. Eichinger, & V. Rybakin) Landes Bioscience & Springer, 2008. http://www.landesbioscience.com/curie/chapter/3820 [DOI] [PubMed]
- Rybakin V. & Clemen C. S. Coronin proteins as multifunctional regulators of the cytoskeleton and membrane trafficking. Bioessays 27, 625–632 (2005). [DOI] [PubMed] [Google Scholar]
- McArdle B. & Hofmann A. in The Coronin Family of Proteins Vol. 48 Subcellular Biochemistry (eds C. S. Clemen, L. Eichinger, & V. Rybakin) Landes Bioscience & Springer, 2008. http://www.landesbioscience.com/curie/chapter/3821 [DOI] [PubMed]
- Spoerl Z., Stumpf M., Noegel A. A. & Hasse A. Oligomerization, F-actin interaction, and membrane association of the ubiquitous mammalian coronin 3 are mediated by its carboxyl terminus. J Biol Chem 277, 48858–48867 (2002). [DOI] [PubMed] [Google Scholar]
- Xavier C. P. et al. Structural and functional diversity of novel coronin 1C (CRN2) isoforms in muscle. J Mol Biol 393, 287–299 (2009). [DOI] [PubMed] [Google Scholar]
- Rosentreter A. et al. Coronin 3 involvement in F-actin-dependent processes at the cell cortex. Exp Cell Res 313, 878–895 (2007). [DOI] [PubMed] [Google Scholar]
- Hasse A. et al. Coronin 3 and its role in murine brain morphogenesis. Eur J Neurosci 21, 1155–1168 (2005). [DOI] [PubMed] [Google Scholar]
- Thal D. et al. Expression of coronin-3 (coronin-1C) in diffuse gliomas is related to malignancy. J Pathol 214, 415–424 (2008). [DOI] [PubMed] [Google Scholar]
- Roadcap D. W., Clemen C. S. & Bear J. E. in The Coronin Family of Proteins Vol. 48 Subcellular Biochemistry (eds C. S. Clemen, L. Eichinger, & V. Rybakin) Landes Bioscience & Springer, 2008. http://www.landesbioscience.com/curie/chapter/3798 [DOI] [PubMed]
- Wu L. et al. Coronin-1C is a novel biomarker for hepatocellular carcinoma invasive progression identified by proteomics analysis and clinical validation. J Exp Clin Cancer Res 29, 17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan S. L. et al. Primary effusion lymphoma: genomic profiling revealed amplification of SELPLG and CORO1C encoding for proteins important for cell migration. J Pathol 222, 166–179 (2010). [DOI] [PubMed] [Google Scholar]
- Samarin S. N., Koch S., Ivanov A. I., Parkos C. A. & Nusrat A. Coronin 1C negatively regulates cell-matrix adhesion and motility of intestinal epithelial cells. Biochem Biophys Res Commun 391, 394–400 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier C.-P., Eichinger L., Fernandez M. P., Morgan R. O. & Clemen C. S. in The Coronin Family of Proteins Vol. 48 Subcellular Biochemistry (eds C. S. Clemen, L. Eichinger, & V. Rybakin) Landes Bioscience & Springer, 2008. http://www.landesbioscience.com/curie/chapter/3808
- Kimura T., Taniguchi S. & Niki I. Actin assembly controlled by GDP-Rab27a is essential for endocytosis of the insulin secretory membrane. Arch Biochem Biophys 496, 33–37 (2010). [DOI] [PubMed] [Google Scholar]
- Kimura T. et al. The GDP-dependent Rab27a effector coronin 3 controls endocytosis of secretory membrane in insulin-secreting cell lines. J Cell Sci 121, 3092–3098 (2008). [DOI] [PubMed] [Google Scholar]
- Burnett G. & Kennedy E. P. The enzymatic phosphorylation of proteins. J Biol Chem 211, 969–980 (1954). [PubMed] [Google Scholar]
- Allende J. E. & Allende C. C. Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J 9, 313–323 (1995). [DOI] [PubMed] [Google Scholar]
- Canton D. A. & Litchfield D. W. The shape of things to come: an emerging role for protein kinase CK2 in the regulation of cell morphology and the cytoskeleton. Cell Signal 18, 267-275 (2006). [DOI] [PubMed] [Google Scholar]
- Guerra B. & Issinger O. G. Protein kinase CK2 in human diseases. Curr Med Chem 15, 1870–1886 (2008). [DOI] [PubMed] [Google Scholar]
- Pinna L. A. A historical view of protein kinase CK2. Cell Mol Biol Res 40, 383–390 (1994). [PubMed] [Google Scholar]
- Pinna L. A. The raison d'etre of constitutively active protein kinases: the lesson of CK2. Acc Chem Res 36, 378–384 (2003). [DOI] [PubMed] [Google Scholar]
- Salvi M., Sarno S., Cesaro L., Nakamura H. & Pinna L. A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta 1793, 847–859 (2009). [DOI] [PubMed] [Google Scholar]
- Meggio F. & Pinna L. A. One-thousand-and-one substrates of protein kinase CK2? FASEB J 17, 349–368 (2003). [DOI] [PubMed] [Google Scholar]
- Olsten M. E. & Litchfield D. W. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol 82, 681–693 (2004). [DOI] [PubMed] [Google Scholar]
- Deshiere A., Theis-Febvre N., Martel V., Cochet C. & Filhol O. Protein kinase CK2 and cell polarity. Mol Cell Biochem 316, 107–113 (2008). [DOI] [PubMed] [Google Scholar]
- Guerra B. & Issinger O. G. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis 20, 391–408 (1999). [DOI] [PubMed] [Google Scholar]
- Guerra B. et al. CK2: a protein kinase in need of control. Pharmacol Ther 82, 303–313 (1999). [DOI] [PubMed] [Google Scholar]
- Valero E. et al. Quaternary structure of casein kinase 2. Characterization of multiple oligomeric states and relation with its catalytic activity. J Biol Chem 270, 8345–8352 (1995). [DOI] [PubMed] [Google Scholar]
- Niefind K., Guerra B., Ermakowa I. & Issinger O. G. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J 20, 5320–5331 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester N., Yu I. J. & Marshak D. R. Identification and characterization of protein kinase CKII isoforms in HeLa cells. Isoform-specific differences in rates of assembly from catalytic and regulatory subunits. J Biol Chem 270, 7501–7514 (1995). [DOI] [PubMed] [Google Scholar]
- Mazin P. V. et al. An automated stochastic approach to the identification of the protein specificity determinants and functional subfamilies. Algorithms Mol Biol 5, 29 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X. Statistical methods for testing functional divergence after gene duplication. Mol Biol Evol 16, 1664–1674 (1999). [DOI] [PubMed] [Google Scholar]
- Rikova K. et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 (2007). [DOI] [PubMed] [Google Scholar]
- Rush J. et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23, 94–101 (2005). [DOI] [PubMed] [Google Scholar]
- Choudhary C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 (2009). [DOI] [PubMed] [Google Scholar]
- Fleury D., Daniels R. S., Skehel J. J., Knossow M. & Bizebard T. Structural evidence for recognition of a single epitope by two distinct antibodies. Proteins 40, 572–578 (2000). [PubMed] [Google Scholar]
- Roy A., Kucukural A. & Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F. et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- Svergun D., Barberato C. & Koch M. CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. Journal of Applied Crystallography 28, 768–773 (1995). [Google Scholar]
- Lebrin F., Chambaz E. M. & Bianchini L. A role for protein kinase CK2 in cell proliferation: evidence using a kinase-inactive mutant of CK2 catalytic subunit alpha. Oncogene 20, 2010–2022 (2001). [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay K. & Gjerset R. A. Protein kinase CK2 is a central regulator of topoisomerase I hyperphosphorylation and camptothecin sensitivity in cancer cell lines. Biochemistry 50, 704–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikhardt B. A., Kulikova O. G., Borisova G. Y., Aleksandrova I. Y. & Sapronov N. S. Status of the “protein kinase CK2-HMG14” system in age-related amnesia in rats. Neurosci Behav Physiol 33, 799–804 (2003). [DOI] [PubMed] [Google Scholar]
- Reikhardt B. A., Kulikova O. G. & Sapronov N. S. Protein kinase CK2 and regulation of Ca2+-ATPase activity in brain neuron chromatin in rats during aging. Bull Exp Biol Med 133, 565–567 (2002). [DOI] [PubMed] [Google Scholar]
- Miyata Y. Protein kinase CK2 in health and disease: CK2: the kinase controlling the Hsp90 chaperone machinery. Cell Mol Life Sci 66, 1840–1849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y. & Nishida E. Evaluating CK2 activity with the antibody specific for the CK2-phosphorylated form of a kinase-targeting cochaperone Cdc37. Mol Cell Biochem 316, 127–134 (2008). [DOI] [PubMed] [Google Scholar]
- Duncan J. S. et al. Regulation of cell proliferation and survival: convergence of protein kinases and caspases. Biochim Biophys Acta 1804, 505–510 (2010). [DOI] [PubMed] [Google Scholar]
- Cai L., Holoweckyj N., Schaller M. D. & Bear J. E. Phosphorylation of coronin 1B by protein kinase C regulates interaction with Arp2/3 and cell motility. J Biol Chem 280, 31913–31923 (2005). [DOI] [PubMed] [Google Scholar]
- Itoh S. et al. The role of protein kinase C in the transient association of p57, a coronin family actin-binding protein, with phagosomes. Biol Pharm Bull 25, 837–844 (2002). [DOI] [PubMed] [Google Scholar]
- Karino A., Tanoue S., Fukuda M., Nakamura T. & Ohtsuki K. An inhibitory effect of actin on casein kinase II activity in vitro. FEBS Lett 398, 317–321 (1996). [DOI] [PubMed] [Google Scholar]
- Cory G. O., Cramer R., Blanchoin L. & Ridley A. J. Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell 11, 1229–1239 (2003). [DOI] [PubMed] [Google Scholar]
- Canton D. A. et al. The pleckstrin homology domain-containing protein CKIP-1 is involved in regulation of cell morphology and the actin cytoskeleton and interaction with actin capping protein. Mol Cell Biol 25, 3519–3534 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin V. E. et al. Coronin-1A stabilizes F-actin by bridging adjacent actin protomers and stapling opposite strands of the actin filament. J Mol Biol 376, 607–613 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Makhov A. M., Schafer D. A. & Bear J. E. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell 134, 828–842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton B. A., Wu P. & Wiesmann C. The crystal structure of murine coronin-1: a regulator of actin cytoskeletal dynamics in lymphocytes. Structure 14, 87–96 (2006). [DOI] [PubMed] [Google Scholar]
- Humphries C. L. et al. Direct regulation of Arp2/3 complex activity and function by the actin binding protein coronin. J Cell Biol 159, 993–1004 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Marshall T. W., Uetrecht A. C., Schafer D. A. & Bear J. E. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell 128, 915–929 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foger N., Rangell L., Danilenko D. M. & Chan A. C. Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science 313, 839–842 (2006). [DOI] [PubMed] [Google Scholar]
- Rodal A. A. et al. Conformational changes in the Arp2/3 complex leading to actin nucleation. Nat Struct Mol Biol 12, 26–31 (2005). [DOI] [PubMed] [Google Scholar]
- Oku T. et al. Two regions responsible for the actin binding of p57, a mammalian coronin family actin-binding protein. Biol Pharm Bull 26, 409–416 (2003). [DOI] [PubMed] [Google Scholar]
- Liu C. Z., Chen Y. & Sui S. F. The identification of a new actin-binding region in p57. Cell Res 16, 106–112 (2006). [DOI] [PubMed] [Google Scholar]
- Gatfield J., Albrecht I., Zanolari B., Steinmetz M. O. & Pieters J. Association of the leukocyte plasma membrane with the actin cytoskeleton through coiled coil-mediated trimeric coronin 1 molecules. Mol Biol Cell 16, 2786–2798 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Makhov A. M. & Bear J. E. F-actin binding is essential for coronin 1B function in vivo. J Cell Sci 120, 1779–1790 (2007). [DOI] [PubMed] [Google Scholar]
- Gandhi M., Jangi M. & Goode B. L. Functional surfaces on the actin-binding protein coronin revealed by systematic mutagenesis. J Biol Chem (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes K. C., Popp D., Gebhard W. & Kabsch W. Atomic model of the actin filament. Nature 347, 44–49 (1990). [DOI] [PubMed] [Google Scholar]
- Gandhi M., Achard V., Blanchoin L. & Goode B. L. Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol Cell 34, 364–374 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf E., Kim P. S. & Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci 6, 1179–1189 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer R. A. et al. A conserved trimerization motif controls the topology of short coiled coils. PNAS 102, 13891–13896 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A. & Blundell T. L. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- Laskowski R., MacArthur M., Moss D. & Thornton J. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst 26, 283–291 (1993). [Google Scholar]
- Filhol O. & Cochet C. Protein kinase CK2 in health and disease: Cellular functions of protein kinase CK2: a dynamic affair. Cell Mol Life Sci 66, 1830–1839 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heriche J. K. et al. Regulation of protein phosphatase 2A by direct interaction with casein kinase 2alpha. Science 276, 952–955 (1997). [DOI] [PubMed] [Google Scholar]
- Quasdorff M. et al. A concerted action of HNF4alpha and HNF1alpha links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol 10, 1478–1490 (2008). [DOI] [PubMed] [Google Scholar]
- Kopp P. et al. The kinesin KIF1C and microtubule plus ends regulate podosome dynamics in macrophages. Mol Biol Cell 17, 2811–2823 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J. & Svergun D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography 36, 1277-1282 (2003). [Google Scholar]
- Semenyuk A. & Svergun D. GNOM: a program package for small-angle scattering data processing. Journal of Applied Crystallography 24, 537–540 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. & Svergun D. Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography 36, 860–864 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann A. & Wlodawer A. PCSB--a program collection for structural biology and biophysical chemistry. Bioinformatics 18, 209–210 (2002). [DOI] [PubMed] [Google Scholar]
- Skjerpen C. S., Nilsen T., Wesche J. & Olsnes S. Binding of FGF-1 variants to protein kinase CK2 correlates with mitogenicity. EMBO J 21, 4058–4069 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemen C. S. et al. Hsp27-2D-gel electrophoresis is a diagnostic tool to differentiate primary desminopathies from myofibrillar myopathies. FEBS Lett 579, 3777–3782 (2005). [DOI] [PubMed] [Google Scholar]
- Duncan J. S. et al. An unbiased evaluation of CK2 inhibitors by chemoproteomics: characterization of inhibitor effects on CK2 and identification of novel inhibitor targets. Mol Cell Proteomics 7, 1077–1088 (2008). [DOI] [PubMed] [Google Scholar]
- Pagano M. A. et al. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J Med Chem 47, 6239–6247 (2004). [DOI] [PubMed] [Google Scholar]
- Ruzzene M., Penzo D. & Pinna L. A. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem J 364, 41–47 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung E., Fucini P., Stewart M., Noegel A. A. & Schleicher M. Linking microfilaments to intracellular membranes: the actin-binding and vesicle-associated protein comitin exhibits a mannose-specific lectin activity. EMBO J 15, 1238–1246 (1996). [PMC free article] [PubMed] [Google Scholar]
- Morita T., Mayanagi T., Yoshio T. & Sobue K. Changes in the balance between caldesmon regulated by p21-activated kinases and the Arp2/3 complex govern podosome formation. J Biol Chem 282, 8454–8463 (2007). [DOI] [PubMed] [Google Scholar]
- Clemen C. S. et al. Strumpellin is a novel valosin-containing protein binding partner linking hereditary spastic paraplegia to protein aggregation diseases. Brain (2010). [DOI] [PubMed] [Google Scholar]
- Xiong H. et al. Dictyostelium Sun-1 connects the centrosome to chromatin and ensures genome stability. Traffic 9, 708–724 (2008). [DOI] [PubMed] [Google Scholar]
- Himmel M. et al. Control of high affinity interactions in the talin C terminus: how talin domains coordinate protein dynamics in cell adhesions. J Biol Chem 284, 13832–13842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew C. S. et al. Lasp-1 binds to non-muscle F-actin in vitro and is localized within multiple sites of dynamic actin assembly in vivo. J Cell Sci 115, 4787–4799 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information