

Abstract
With the rapid increase of protein sequences in the post-genomic age, it is challenging to develop accurate and automated methods for reliably and quickly predicting their subcellular localizations. Till now, many efforts have been tried, but most of which used only a single algorithm. In this paper, we proposed an ensemble classifier of KNN (k-nearest neighbor) and SVM (support vector machine) algorithms to predict the subcellular localization of eukaryotic proteins based on a voting system. The overall prediction accuracies by the one-versus-one strategy are 78.17%, 89.94% and 75.55% for three benchmark datasets of eukaryotic proteins. The improved prediction accuracies reveal that GO annotations and hydrophobicity of amino acids help to predict subcellular locations of eukaryotic proteins.
Introduction
Researches on subcellular location of proteins are important for elucidating their functions involved in various cellular processes, as well as in understanding some disease mechanisms and developing novel drugs. Since experimental determinations of the localization are time-consuming, tedious and costly, especially for the rapid accumulation of protein sequences, it is highly desirable to develop effective computational methods for accurately and quickly predicting their subcellular attributes.
In the past few years, many computational methods have been developed for this purpose [1], [2], [3], [4]. These methods can be divided into two main categories [5]. Methods in the first category are based on the observation that amino acid compositions of extracellular and intracellular proteins are significantly different [6]. Along this line, many computational approaches based on amino acid composition, dipeptide composition [7] and gapped amino acid pairs [8] were proposed. Meanwhile, to incorporate more sequence information, many other features were incorporated, such as amphiphility of amino acids [9], functional domain composition [10], psi-blast profile [11], [12] and so on. Methods in the second category are based on a certain sorting signals [13], [14], including signal peptides, chloroplast transit peptides and mitochondrial targeting peptides. For example, Emanuelsson et al. [14] provided detailed instructions for the use of SignalP and ChloroP in prediction of cleavage sites for secretory pathway signal peptides and chloroplast transit peptides. However, the reliability of these methods is highly dependent on protein N-terminal sequence assignments, and the molecular mechanisms related to sorting signals are rather complex and not interpreted clearly.
Not only protein sequence information but also prediction algorithms could affect the accuracy of the subcellular localization prediction. So far, many computational techniques, such as the hidden Markov models (HMM) [15], [16], neural network [17], K-nearest neighbor (KNN) [18] and support vector machine (SVM) [5], [19] were introduced for the prediction of protein subcellular localization. However, most of the current predictors are based on a single theory which could have its own inherent defects, so their predictions are not satisfactory. For example, the number of parameters that need to be evaluated in an HMM is large [20]. The neural network can suffer from multiple local minima [21]. Besides, quite a few ensemble classifiers [7], [22], [23] for prediction of protein subcellular localizations have been proposed. However, many of the ensemble classifiers were actually engineered only by a single algorithm, such as the fuzzy KNN [7], KNN [22], and Bayesian [23]. Other ensemble classifiers, such as CE-PLoc [24] and the KNN-SVM ensemble classifier proposed by Zhang [25], were engineered by different algorithms, mostly including SVM and KNN. Along this line, an ensemble classifier making use of the classical SVM and KNN algorithms was developed in this article to predict subcellular localization of eukaryotic proteins.
We apply our method to three widely used eukaryotic protein datasets. By the jackknife cross-validation test [26], [27], [28], [29], the ensemble classifier shows high accuracies and may play an important complementary role to existing methods.
Materials and Methods
1. Datasets
In order to evaluate the performance of the proposed method and compare it with current methods, we introduced three widely used datasets into this study. The first dataset was constructed by Chou [30]. This dataset (denoted as iLoc8897) consists of 8,897 locative protein sequences (7,766 different proteins), which divided into 22 subcellular locations. Among the 7,766 different eukaryotic proteins, 6,687 belong to one subcellular location, 1,029 to two locations, 48 to three locations, and 2 to four locations. None of the proteins has ≥25% sequence identity to any other in the same subset. The second benchmark dataset was constructed by Park and Kanehisa [8]. This dataset (denoted as Euk7579) contains 7579 proteins, which are divided into 12 subcellular locations. Proteins in this dataset have the pairwised sequence similarity below 80%. The third dataset was constructed by Shen and Chou [31]. This dataset (denoted as Hum3681) consists of 3,681 locative protein sequences (3,106 different human proteins), which are divided into 14 human subcellular locations. Among the 3,106 different proteins, 2,580 belong to one subcellular location, 480 to two locations, 43 to three locations, and 3 to four locations. None of the proteins has ≥25% sequence identity to any other in the same subcellular location. The detailed information of the three datasets are listed in Table 1 .
Table 1. Three benchmark datasets used to train and test our predictor.
| iLoc8897 | Euk7579 | Hum3681 | |||
| Subcellular location | Number of proteins | Subcellular location | Number of proteins | Subcellular location | Number of proteins |
| Acrosome | 14 | Chloroplast | 671 | Centriole | 77 |
| Cell membrane | 697 | Cytoplasm | 1241 | Cytoplasm | 817 |
| Cell wall | 49 | Cytoskeleton | 40 | Cytoskeleton | 79 |
| Centrosome | 96 | Endoplasmic reticulum | 114 | Endosome | 24 |
| Chloroplast | 385 | Extracell | 861 | Endoplasmic reticulum | 229 |
| Cyanelle | 79 | Golgi apparatus | 47 | Extracell | 385 |
| Cytoplasm | 2186 | Lysosomal | 93 | Golgi apparatus | 161 |
| Cytoskeleton | 139 | Mitochondrion | 727 | Lysosome | 77 |
| Endoplasmic reticulum | 457 | Nucleus | 1932 | Microsome | 24 |
| Endosome | 41 | Peroxisomal | 125 | Mitochondrion | 364 |
| Extracell | 1048 | Plasma membrane | 1674 | Nucleus | 1021 |
| Golgi apparatus | 254 | Vacuolar | 54 | Peroxisome | 47 |
| Hydrogenosome | 10 | - | - | Plasma membrane | 354 |
| Lysosome | 57 | - | - | Synapse | 22 |
| Melanosome | 47 | - | - | - | - |
| Microsome | 13 | - | - | - | - |
| Mitochondrion | 610 | - | - | - | - |
| Nucleus | 2320 | - | - | - | - |
| Peroxisome | 110 | - | - | - | - |
| Spindle pole body | 68 | - | - | - | - |
| Synapse | 47 | - | - | - | - |
| Vacuole | 170 | - | - | - | - |
| Total | 8897 | Total | 7579 | Total | 3681 |
2. Gene Ontology
Gene Ontology (GO) is a major bioinformatics initiative. It meets the need for consistent descriptions of gene products in different databases. Gene Ontology database is established on the three criteria: molecular function, cellular component and biological process. It has been developed to manage the overwhelming mass of current biological data from a computational perspective and become a standard tool to annotate gene products for various databases [32], [33]. Accordingly, GO annotation has been being used for diverse sequence-based prediction tasks, such as analyzing the pathogenic gene function with human squamous cell cervical carcinoma [34], mapping molecular responses to xenoestrogens [35], predicting the enzymatic attribute of proteins [36], predicting the transcription factor DNA binding preference [37], and predicting the eukaryotic protein subcellular localization [38]. In particular, the growth of Gene Ontology databases has increased the effectiveness of GO-based features [39]. As a result, Gene Ontology could be used to improve the predictive performance of protein subcellular localization [22], [40].
We downloaded all GO data at ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/UNIPROT/(released on March 15, 2010), and searched the GO terms for all the protein entries in the three datasets. We eliminate those proteins, which have no corresponding GO terms and the number (60, 127 and 4 for the iLoc8897, Euk7579 and Hum3681 datasets) are relatively small compared to the total datasets. We consider this would not have a great influence on its final accuracy. After this step, we got a list of GO terms for each protein entry of the three datasets. For example, the human protein entry “Q9H400” in the Hum3681 dataset corresponds to four GO numbers, i.e., GO: 0005886, GO: 0006955, GO: 0016020 and GO: 0016021, while the protein entry “P81084” in the Euk7579 dataset corresponds to six GO numbers, i.e., GO: 0000166, GO: 0005524, GO: 0006950, GO: 0009507, GO: 0009536 and GO: 0009570. So as to handle these GO numbers efficiently, a compression procedure was proposed to renumber them. For example, all involved GO numbers for the eukaryotic proteins in the Euk7579 dataset are GO: 0000001, GO: 0000002, GO: 0000003, GO: 0000006, GO: 00000009, GO: 0000011, GO: 0000012, …, GO: 0090184. They are renamed as GO_compress: 0000001, GO_compress: 0000002, GO_compress: 0000003, GO_compress: 0000004, GO_compress: 0000005, GO_compress: 0000006, GO_compress: 0000007, ……, GO_compress: 0006533, respectively. When this treatment finished, we got the GO_compress database that contained 6533 numbers. We numbered those data from 1 to 6533. The total numbers of GO terms that appeared for the iLoc8897, Euk7579 and Hum3681 datasets were 7871, 6533 and 5553.
As we know, if we want to describe all possible GO terms for a certain dataset, the simplest way to vector represent a protein was using a binary feature component for a protein. We used value 1 if the corresponding GO number appears and value 0 if it does not appear. For example, the human protein entry “Q8TDM5” in the Hum3681 dataset corresponds to seven GO numbers in the GO database, i.e., GO: 0001669, GO: 0005515, GO: 0005886, GO: 0007155, GO: 0016020, GO: 0031225 and GO: 0031410, which corresponded to GO_compress: 0000212, GO_compress: 0001037, GO_compress: 0001203, GO_compress: 0001722, GO_compress: 0002543, GO_compress: 0003360, GO_compress: 0003398 in the GO_compress database. So the 212th, 1037th, 1203rd, 1722nd, 2543rd, 3360th, and 3398th components of the feature vector were assigned the value 1 and the rest 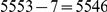 components with the value 0. At last, we transformed the GO terms annotated for each human protein into a 5553-dimension input vector.
components with the value 0. At last, we transformed the GO terms annotated for each human protein into a 5553-dimension input vector.
3. Amphiphilic pseudo amino acid composition
In a protein, the hydrophobicity and hydrophilicity of the native amino acids play an important part in its folding, interior packing, catalytic mechanism, as well as its interaction with other molecules in the environment [41]. Therefore, the two indices may be used to effectively reflect the subcellular locations of proteins. Both the hydrophobicity and hydrophilicity are introduced in the concept of AmPseAAC. As we know, the concept of AmPseAAC proposed by Chou [22] was widely used by many researchers in improving the prediction quality for protein subcellular localization [42], [43]. Following the concept of AmPseAAC, a protein sample could be descripted by a 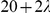 dimensional feature vector, where
dimensional feature vector, where 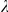 is equal to
is equal to 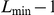 , where
, where 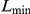 is the length of the shortest protein sequence in the dataset. The
is the length of the shortest protein sequence in the dataset. The 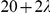 dimensional feature vector for a protein comprises 20 features of the conventional amino acid composition (AAC), and the rest
dimensional feature vector for a protein comprises 20 features of the conventional amino acid composition (AAC), and the rest 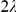 components reflect its sequence-order pattern through the amphiphilic feature. The protein representation is called the “amphiphilic pseudo amino acid composition” or “AmPseAAC” for short. In order to get more local sequence information, we incorporated 400 dipeptide components to the AmPseAAC. Then the new AmPseAAC is constructed and the dimension is increased to
components reflect its sequence-order pattern through the amphiphilic feature. The protein representation is called the “amphiphilic pseudo amino acid composition” or “AmPseAAC” for short. In order to get more local sequence information, we incorporated 400 dipeptide components to the AmPseAAC. Then the new AmPseAAC is constructed and the dimension is increased to 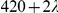 , which are
, which are 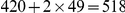 ,
, 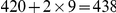 , and
, and 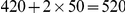 for the iLoc8897, Euk7579 and Hum3681 datasets, respectively. Then we combined the new AmPseAAC and Gene Ontology as the features for protein subcellular localization prediction. As a result, the dimensions of the final input feature vectors are
for the iLoc8897, Euk7579 and Hum3681 datasets, respectively. Then we combined the new AmPseAAC and Gene Ontology as the features for protein subcellular localization prediction. As a result, the dimensions of the final input feature vectors are 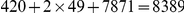 ,
, 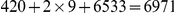 , and
, and 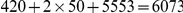 for the iLoc8897, Euk7579 and Hum3681 datasets.
for the iLoc8897, Euk7579 and Hum3681 datasets.
4. Feature extraction
Due to the limited numbers of learning examples, learning with a small number of features often leads to a better generalization of machine learning algorithms (Occam's razor) [44]. Additionally, with the increase of the dimension of the feature vector, the computational loads for some machine-learning tools, e.g., Support Vector Machine [45] and Neural Network [46], are seriously affected. As a result, we used the “fselect.py” in Libsvm software package to reduce the dimensionality. The fselect.py is a simple python script used F-score to select features. After running the python script, one could get an output file called “.fscore”, in which each feature was given a score to describe the importance of it and all features were sorted by their scores. Then we chose the top features with the highest contribution scores ( Figs. 1 , 2 , and 3 ).
Figure 1. This graph shows the contribution scores of top 45 features on the iLoc8897 dataset.

Figure 2. This graph shows the contribution scores of top 45 features on the Euk7579 dataset.

Hydrophobicity: 6, 2, 5 … stand for the 6th, 2nd, 5th … elements in the hydrophobicity vectors respectively.
Figure 3. This graph shows the contribution scores of top 45 features on the Hum3681 dataset.

5. The KNN-SVM ensemble classifier
A wide variety of machine learning methods have been proposed for predicting protein subcellular localization in recent years [47], [48], [49], [50], such as Markov chain models [51], neural networks [46], K-Nearest Neighborhood (KNN) [18], and Support Vector Machines (SVM) [52], [53]. In these methods, KNN and SVM are two popular classifiers in machine learning task. Previous studies presented that each algorithm has its own advantage and the ensemble classifier of different algorithms is the future direction of protein subcellular localization prediction. So, in this paper we proposed an ensemble classifier of KNN and SVM based on one-versus-one strategy and a voting system (
Fig. 4
). LIBSVM still has a few tunable parameters which affect the accuracy of the subcellular localization prediction and need to be determined. In this article, “grid.py” was used in the iLoc8897 dataset to select the parameter  and the regularization parameter
and the regularization parameter 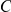 in LIBSVM [24]. Here, the iLoc8897 dataset was selected for optimization of the parameters of the classification models due to the following reasons: (i) compared to the other datasets, this dataset has the largest number of proteins, so it possesses a distinct statistical significance for training; (ii) sequences in this dataset have relatively low pairwise sequence homology; (iii) this dataset covers enough subcellular locations and was widely adopted for evaluating a new proposed method [30], [38].
in LIBSVM [24]. Here, the iLoc8897 dataset was selected for optimization of the parameters of the classification models due to the following reasons: (i) compared to the other datasets, this dataset has the largest number of proteins, so it possesses a distinct statistical significance for training; (ii) sequences in this dataset have relatively low pairwise sequence homology; (iii) this dataset covers enough subcellular locations and was widely adopted for evaluating a new proposed method [30], [38].
Figure 4. This graph shows the flow chart for application of KNN and LIBSVM algorithms.

Prediction of protein subcellular localization is a multi-class classification problem. Here, the class number is equal to 22 for iLoc8897 dataset, 12 for Euk7579 dataset and 14 for Hum3681 dataset, respectively. A simple way to deal with the multi-class classification is to reduce the multi-classification to a series of binary classifications. During this study, we adopted the one-versus-one method, i.e., 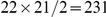 ,
, 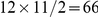 , and
, and 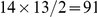 binary classification tasks were constructed for the iLoc8897, Euk7579 and Hum3681 datasets. Compared to the one-versus-one approach, the one-versus-rest strategy has the shortage that the numbers of positive and negative training data points are not symmetric [54]. For each binary classification, the predictor (KNN or SVM) with the higher output accuracy was selected, and the free parameters, i.e.,
binary classification tasks were constructed for the iLoc8897, Euk7579 and Hum3681 datasets. Compared to the one-versus-one approach, the one-versus-rest strategy has the shortage that the numbers of positive and negative training data points are not symmetric [54]. For each binary classification, the predictor (KNN or SVM) with the higher output accuracy was selected, and the free parameters, i.e., 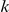 for KNN and
for KNN and 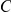 and
and  for LIBSVM, are optimized by the iLoc8897 dataset.
for LIBSVM, are optimized by the iLoc8897 dataset.
Take the Hum3681 dataset as an example. Following the one-versus-one strategy, 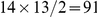 binary classification tasks were constructed for this dataset. For each binary classification task, the KNN and SVM are used to predict the attribute of each protein. As a result, we chose the predictor with the higher output accuracy, where the parameters of KNN and SVM were optimized by the iLoc8897 dataset. Then a score function was generated by the KNN-SVM ensemble classifier formed by fusing the 91 individual binary classifiers through a voting system (see Eqs. 1
–
3). Each protein was assigned to the subcellular location where the score function has the maximum value. Suppose that the predicted classification results for the query human protein
binary classification tasks were constructed for this dataset. For each binary classification task, the KNN and SVM are used to predict the attribute of each protein. As a result, we chose the predictor with the higher output accuracy, where the parameters of KNN and SVM were optimized by the iLoc8897 dataset. Then a score function was generated by the KNN-SVM ensemble classifier formed by fusing the 91 individual binary classifiers through a voting system (see Eqs. 1
–
3). Each protein was assigned to the subcellular location where the score function has the maximum value. Suppose that the predicted classification results for the query human protein 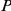 for the 91 binary classifiers are
for the 91 binary classifiers are 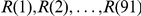 , that is
, that is
| (1) |
where 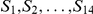 represent the 14 subcellular locations. The voting score for the protein
represent the 14 subcellular locations. The voting score for the protein 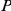 belonging to class
belonging to class  is defined as
is defined as
| (2) |
where the 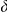 function in Eq. 2 is given by
function in Eq. 2 is given by
| (3) |
Subsequently, the query protein 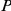 was assigned to the class that gives the highest score for Eq. 2 of the 91 binary classifiers. We can assume that there are five subsets and
was assigned to the class that gives the highest score for Eq. 2 of the 91 binary classifiers. We can assume that there are five subsets and 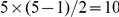 binary classification tasks are constructed. If the predicted classification results for a query protein
binary classification tasks are constructed. If the predicted classification results for a query protein 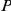 with the ten binary classifiers are
with the ten binary classifiers are 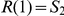 ,
, 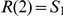 ,
, 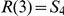 ,
, 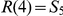 ,
, 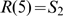 ,
, 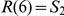 ,
, 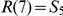 ,
, 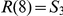 ,
, 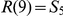 ,
, 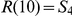 that is, classifiers 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 assign protein
that is, classifiers 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10 assign protein 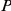 to subsets 2, 1, 4, 5, 2, 2, 5, 3, 5 and 4, respectively. As a result, the voting scores for protein
to subsets 2, 1, 4, 5, 2, 2, 5, 3, 5 and 4, respectively. As a result, the voting scores for protein 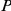 are
are 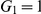 ,
, 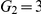 ,
, 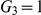 ,
, 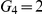 ,
, 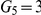 . Then protein
. Then protein 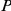 was predicted to classes 2 and 5, which both give the highest score of
was predicted to classes 2 and 5, which both give the highest score of 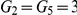 .
.
6. Assessment of prediction performances
The prediction quality is examined by the jackknife test currently. Three methods, i.e., the jackknife test, sub-sampling test, and independent dataset test are often used for examining the accuracy of a statistical prediction method. The jackknife test is deemed the most objective and rigorous one [55], [56].
The accuracy, the overall accuracy, the “absolute true” overall accuracy and Matthew's Correlation Coefficient (MCC) [57] for each subcellular location calculated for assessment of the prediction system are formulated as
| (4) |
| (5) |
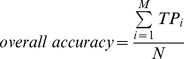 |
(6) |
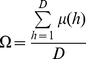 |
(7) |
 |
(8) |
| (9) |
where 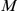 is the class number,
is the class number, 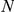 is the total number of locative proteins,
is the total number of locative proteins, 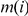 and
and 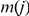 are the numbers of the locative proteins in classes
are the numbers of the locative proteins in classes  and
and 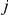 ,
, 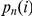 and
and 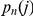 are the numbers of the correctly predicted locative proteins of class
are the numbers of the correctly predicted locative proteins of class  and class
and class 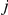 by binary classifier
by binary classifier  .
. 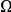 is the so-called “absolute true” overall accuracy.
is the so-called “absolute true” overall accuracy. 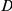 is the number of total proteins investigated.
is the number of total proteins investigated. 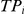 ,
, 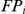 ,
, 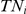 , and
, and 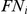 are the numbers of true positives, false positives, true negatives, and false negatives in class
are the numbers of true positives, false positives, true negatives, and false negatives in class  by the KNN-SVM ensemble classifier, respectively.
by the KNN-SVM ensemble classifier, respectively.
Results and Discussion
1. Selection of algorithms and parameters
It is important to point out that the best combination of parameters  and
and 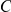 depends on the dimension
depends on the dimension 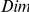 of the protein top feature vector. In the present work, we select the parameters
of the protein top feature vector. In the present work, we select the parameters  and
and 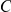 when parameter
when parameter 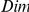 varied from 10 to 50. As seen in
Table 2
, the highest prediction accuracy was 78.01% at
varied from 10 to 50. As seen in
Table 2
, the highest prediction accuracy was 78.01% at 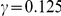 ,
, 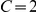 and
and 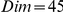 . While the prediction accuracy obtained by KNN changed as parameter
. While the prediction accuracy obtained by KNN changed as parameter 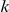 varied from 1 to 9, and the highest prediction accuracy (74.70%) was obtained at
varied from 1 to 9, and the highest prediction accuracy (74.70%) was obtained at  and
and 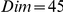 for the iLoc8897 dataset. Then the same parameters, i.e.,
for the iLoc8897 dataset. Then the same parameters, i.e., 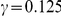 ,
, 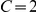 ,
,  and
and 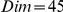 were used for all the three datasets.
were used for all the three datasets.
Table 2. Prediction performance of different top-N features on the iLoc8897 dataset by LIBSVM.
| Top10 | Top15 | Top20 | Top25 | Top30 | Top35 | Top40 | Top45 | Top50 | |

|
0.03125 | 0.5 | 0.5 | 0.125 | 0.125 | 0.125 | 0.125 | 0.125 | 0.125 |
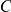
|
512 | 0.03125 | 0.03125 | 2 | 2 | 2 | 2 | 2 | 2 |
| Overall accuracy (%) | 51.14 | 73.08 | 75.12 | 74.18 | 74.40 | 77.46 | 77.65 | 78.01 | 77.98 |
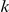
|
- | - | - | - | - | - | - | 5 | - |
| Overall accuracy (%) | - | - | - | - | - | - | - | 74.70 | - |
Because the Hum3681 dataset has 14 subcellular locations, a total of 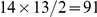 binary classification tasks were constructed. For each one-versus-one classification task, the algorithm (KNN or SVM), which gave a higher prediction accuracy for Eq. 4, was adopt as the final classifier. For example, the 6th, 21st, 26th, 32nd, 34th, 42nd, 43rd, 76th, 82nd, 84th and 90th binary classifiers (11 of 91 classifiers) was based on the KNN method, because the accuracy of KNN method was higher than LIBSVM method by jackknife test, while the rest
binary classification tasks were constructed. For each one-versus-one classification task, the algorithm (KNN or SVM), which gave a higher prediction accuracy for Eq. 4, was adopt as the final classifier. For example, the 6th, 21st, 26th, 32nd, 34th, 42nd, 43rd, 76th, 82nd, 84th and 90th binary classifiers (11 of 91 classifiers) was based on the KNN method, because the accuracy of KNN method was higher than LIBSVM method by jackknife test, while the rest 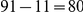 binary classifiers were based on LIBSVM, because the accuracy of LIBSVM method was higher than KNN method by jackknife test.
binary classifiers were based on LIBSVM, because the accuracy of LIBSVM method was higher than KNN method by jackknife test.
In addition, most of the existing methods for predicting protein subcellular localization are limited to a single location. It is instructive to note that the KNN-SVM ensemble classifier can effectively deal with multiple-location proteins as well, that is, the predicted result for a query protein 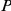 may be attributed to two or more subcellular locations. For example, the real subcellular locations of the protein entry “Q05329” in iLoc8897 dataset are
may be attributed to two or more subcellular locations. For example, the real subcellular locations of the protein entry “Q05329” in iLoc8897 dataset are 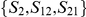 , and the predicted subcellular locations for “Q05329” by the KNN-SVM ensemble classifier are also
, and the predicted subcellular locations for “Q05329” by the KNN-SVM ensemble classifier are also 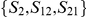 , because
, because 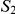 ,
, 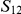 ,
, 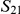 give the highest score (
give the highest score (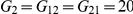 ) according to Eq. 2.
) according to Eq. 2.
2. Comparison with other methods
In order to check the performance of our method, we made comparisons with the following methods: iLoc-Euk [30], Euk-mPLoc 2.0 [38], Hum-mPLoc 2.0 [31], LOCSVMPSI [58], Complexity-based method [59], and the method proposed by Park and Kanehisa [8] which are also based on the Euk7579 dataset. We also compared our method with the KNN binary classifiers, LIBSVM binary calssifiers, and the KNN-SVM ensemble classifier [25]. The comparison is summarized in Tables 3 , 4 , 5 , and 6 .
Table 3. Performance comparisons for eukaryotic protein subcellular location prediction method based on the iLoc8897 dataset.
| Subcellular location | Euk-mPLoc 2.0 (2010) (Chou and Shen 2010) | iLoc-Euk (2011) (Chou et al. 2011) | LIBSVM | KNN | The proposed method | |||
| Jackknife | Jackknife | Jackknife | Jackknife | Jackknife | ||||
| Accuracy (%) | Accuracy (%) | Accuracy (%) | MCC | Accuracy (%) | MCC | Accuracy (%) | MCC | |
| Acrosome | 7.14 | 7.14 | 57.14 | 0.8526 | 71.43 | 0.8449 | 64.29 | 0.8659 |
| Cell membrane | 64.85 | 80.49 | 84.52 | 0.9123 | 96.67 | 0.8558 | 85.09 | 0.9121 |
| Cell wall | 12.24 | 16.33 | 91.84 | 0.8750 | 85.71 | 0.8981 | 91.84 | 0.8750 |
| Centrosome | 22.92 | 69.79 | 86.17 | 0.8650 | 92.55 | 0.6513 | 88.30 | 0.8688 |
| Chloroplast | 82.60 | 87.79 | 99.73 | 0.9943 | 99.73 | 0.9873 | 99.73 | 0.9943 |
| Cyanelle | 59.49 | 64.56 | 100.00 | 1.0000 | 98.73 | 1.0000 | 100.00 | 1.0000 |
| Cytoplasm | 64.87 | 76.72 | 45.24 | 0.9399 | 90.34 | 0.8198 | 45.70 | 0.9361 |
| Cytoskeleton | 31.65 | 27.34 | 50.36 | 0.7629 | 6.47 | 0.8318 | 49.64 | 0.7640 |
| Endoplasmic reticulum | 76.15 | 89.06 | 87.72 | 0.9529 | 84.65 | 0.9457 | 87.72 | 0.9542 |
| Endosome | 4.88 | 7.32 | 21.95 | 0.7272 | 19.51 | 0.8163 | 21.95 | 0.7497 |
| Extracell | 81.87 | 90.46 | 91.82 | 0.9812 | 88.64 | 0.9902 | 91.92 | 0.9824 |
| Golgi apparatus | 22.05 | 63.39 | 76.59 | 0.8997 | 46.83 | 0.9633 | 77.38 | 0.9131 |
| Hydrogenosome | 20.00 | 0.00 | 100.00 | 1.0000 | 70.00 | 1.0000 | 100.00 | 1.0000 |
| Lysosome | 45.61 | 31.58 | 87.72 | 0.8813 | 57.89 | 0.9851 | 87.72 | 0.8813 |
| Melanosome | 0.00 | 2.13 | 76.60 | 0.9474 | 14.89 | 1.0000 | 76.60 | 0.9474 |
| Microsome | 7.69 | 0.00 | 69.23 | 0.8579 | 15.38 | 1.0000 | 69.23 | 0.8579 |
| Mitochondrion | 70.00 | 77.05 | 78.03 | 0.9749 | 80.66 | 0.9688 | 78.20 | 0.9750 |
| Nucleus | 64.70 | 87.93 | 93.69 | 0.8865 | 50.65 | 0.9943 | 93.60 | 0.8873 |
| Peroxisome | 50.91 | 54.55 | 100.00 | 0.9650 | 74.55 | 1.0000 | 100.00 | 0.9650 |
| Spindle pole body | 33.82 | 66.18 | 95.59 | 0.9110 | 4.41 | 1.0000 | 95.59 | 0.9181 |
| Synapse | 0.00 | 38.30 | 80.85 | 0.7918 | 25.53 | 0.8399 | 80.85 | 0.7918 |
| Vacuole | 59.41 | 71.76 | 95.88 | 0.9399 | 80.59 | 0.9819 | 93.53 | 0.9606 |
| Overall accuracy | 64.17 | 79.06 | 78.01 | - | 74.70 | - | 78.17 | - |
|
- | 71.27 | 75.54 | - | 72.84 | - | 75.64 | - |
Table 4. Performance comparisons for eukaryotic protein subcellular location prediction method based on the Euk7579 dataset.
| Subcellular location | Park et al. (2003) (Park and Kanehisa 2003) | LOCSVMPSI (2005) (Xie et al. 2005) | Complexity-based method (2009) (Zheng et al. 2009) | LIBSVM | KNN | The proposed method | ||||
| Jackknife | 5-Fold cross | 5-Fold cross | Jackknife | Jackknife | Jackknife | Jackknife | ||||
| Accuracy (%) | Accuracy (%) | Accuracy (%) | Accuracy (%) | Accuracy (%) | MCC | Accuracy (%) | MCC | Accuracy (%) | MCC | |
| Chloroplast | 57 | 72.3 | 76.5 | 86.4 | 93.21 | 0.9982 | 85.52 | 0.9689 | 93.21 | 0.9982 |
| Cytoplasm | 88 | 72.2 | 76.4 | 81.6 | 87.81 | 0.9035 | 89.13 | 0.7444 | 87.81 | 0.9013 |
| Cytoskeleton | 44 | 58.5 | 60.0 | 77.5 | 12.82 | 1.0000 | 35.90 | 0.9660 | 35.90 | 0.9660 |
| Endoplasmic reticulum | 31 | 46.5 | 61.4 | 78.9 | 59.82 | 0.9708 | 27.68 | 0.9276 | 59.82 | 0.9708 |
| Extracell | 57 | 78.0 | 89.7 | 84.0 | 91.01 | 0.9746 | 85.92 | 0.8879 | 91.01 | 0.9739 |
| Golgi apparatus | 12 | 14.6 | 46.8 | 61.7 | 33.33 | 1.0000 | 22.22 | 0.9127 | 33.33 | 0.9682 |
| Lysosomal | 54 | 61.8 | 62.4 | 73.1 | 67.74 | 0.9691 | 16.13 | 0.9392 | 67.74 | 0.9691 |
| Mitochondrion | 42 | 57.4 | 68.2 | 62.9 | 87.02 | 0.9502 | 70.99 | 0.9017 | 87.15 | 0.9494 |
| Nucleus | 73 | 89.6 | 91.5 | 84.4 | 95.94 | 0.8710 | 81.85 | 0.9441 | 95.94 | 0.8741 |
| Peroxisomal | 4 | 25.2 | 41.6 | 62.4 | 66.94 | 0.9648 | 20.16 | 0.8446 | 66.94 | 0.9648 |
| Plasma membrane | 91 | 92.2 | 94.7 | 86.7 | 93.07 | 0.9647 | 93.98 | 0.9140 | 93.07 | 0.9647 |
| Vacuolar | 25 | 25.0 | 40.7 | 66.7 | 50.94 | 0.9648 | 0.00 | - | 50.94 | 0.9330 |
| Overall accuracy | 75 | 78.2 | 83.5 | 81.6 | 89.80 | - | 81.60 | - | 89.94 | - |
|
- | - | - | - | 89.65 | - | 81.60 | - | 89.73 | - |
Table 5. Performance comparisons for human protein subcellular location prediction method based on the Hum3681 dataset.
| Subcellular location | Hum-mPLoc 2.0 (2009) (Shen and Chou 2009) | LIBSVM | KNN | The proposed method | |||
| Jackknife | Jackknife | Jackknife | Jackknife | ||||
| Accuracy (%) | Accuracy (%) | MCC | Accuracy (%) | MCC | Accuracy (%) | MCC | |
| Centriole | - | 93.51 | 0.9240 | 93.51 | 0.8867 | 94.81 | 0.9249 |
| Cytoplasm | - | 39.66 | 0.9151 | 91.43 | 0.7218 | 41.37 | 0.9007 |
| Cytoskeleton | - | 51.90 | 0.8138 | 8.86 | 0.8816 | 51.90 | 0.8232 |
| Endosome | - | 54.17 | 0.7012 | 33.33 | 0.7552 | 54.17 | 0.7417 |
| Endoplasmic reticulum | - | 78.85 | 0.9046 | 79.30 | 0.8960 | 78.85 | 0.9043 |
| Extracell | - | 86.23 | 0.9705 | 82.60 | 0.9029 | 86.23 | 0.9689 |
| Golgi apparatus | - | 70.19 | 0.8853 | 39.75 | 0.9284 | 70.19 | 0.8887 |
| Lysosome | - | 93.51 | 0.9407 | 57.14 | 0.9777 | 93.51 | 0.9407 |
| Microsome | - | 50.00 | 0.8008 | 0.00 | - | 50.00 | 0.8008 |
| Mitochondrion | - | 84.89 | 0.9569 | 81.04 | 0.9763 | 83.79 | 0.9596 |
| Nucleus | - | 91.67 | 0.8876 | 50.15 | 0.9833 | 91.77 | 0.8932 |
| Peroxisome | - | 97.87 | 0.9380 | 51.06 | 0.9605 | 97.87 | 0.9481 |
| Plasma membrane | - | 84.66 | 0.8887 | 60.80 | 0.9618 | 84.66 | 0.8870 |
| Synapse | - | 86.36 | 0.8487 | 27.27 | 0.8657 | 86.36 | 0.8487 |
| Overall accuracy | 62.7 | 75.22 | - | 67.75 | - | 75.55 | - |
|
- | 72.22 | - | 65.19 | - | 72.25 | - |
Table 6. Performance comparisons for eukaryotic protein subcellular location prediction method based on the Euk6181 dataset.
| Subcellular location | Euk-mPloc | KNN-SVM ensemble classifier (2010) | The proposed method | ||||
| Jackknife | Jackknife | Resubstitution | Jackknife | ||||
| Accuracy(%) | Accuracy(%) | MCC | Accuracy(%) | MCC | Accuracy(%) | MCC | |
| Acrosome | - | 41.2 | 0.641 | 76.5 | 0.874 | 76.47 | 0.9308 |
| Cell wall | - | 67.9 | 0.711 | 88.7 | 0.903 | 92.45 | 0.9028 |
| Centriole | - | 62.5 | 0.690 | 81.3 | 0.786 | 89.06 | 0.8857 |
| Chloroplast | - | 97.4 | 0.879 | 99.0 | 0.918 | 97.80 | 0.9956 |
| Cyanelle | - | 91.8 | 0.957 | 91.8 | 0.957 | 100.00 | 1.0000 |
| Cytoplasm | - | 88.2 | 0.640 | 91.8 | 0.729 | 82.64 | 0.7946 |
| Cytoskeleton | - | 24.3 | 0.491 | 41.9 | 0.645 | 0.00 | 0.0000 |
| Endoplasmic reticulum | - | 79.7 | 0.776 | 86.8 | 0.839 | 77.20 | 0.8906 |
| Endosome | - | 62.9 | 0.770 | 67.4 | 0.812 | 65.17 | 0.7867 |
| Golgi apparatus | - | 74.0 | 0.802 | 79.5 | 0.828 | 81.89 | 0.8355 |
| Hydrogenosome | - | 38.5 | 0.620 | 69.2 | 0.692 | 100.00 | 1.0000 |
| Lysosome | - | 65.0 | 0.662 | 72.5 | 0.772 | 98.75 | 0.9106 |
| Melanosome | - | 53.9 | 0.733 | 84.6 | 0.880 | 76.92 | 1.0000 |
| Microsome | - | 19.4 | 0.380 | 41.9 | 0.647 | 9.68 | 0.5996 |
| Mitochondrion | - | 85.1 | 0.872 | 87.5 | 0.910 | 89.91 | 0.9425 |
| Nucleus | - | 84.6 | 0.824 | 85.7 | 0.862 | 61.97 | 0.9642 |
| Peroxisome | - | 37.1 | 0.589 | 74.2 | 0.860 | 98.97 | 0.9896 |
| Plasma membrane | - | 81.4 | 0.766 | 84.4 | 0.817 | 71.86 | 0.9373 |
| Extracell | - | 83.3 | 0.864 | 85.9 | 0.894 | 92.81 | 0.9537 |
| Spindle pole body | - | 50.0 | 0.669 | 75.0 | 0.850 | 72.22 | 0.8679 |
| Synapse | - | 66.7 | 0.816 | 66.7 | 0.816 | 53.33 | 1.0000 |
| Vacuole | - | 42.2 | 0.610 | 82.4 | 0.865 | 92.16 | 0.9181 |
| Overall accuracy | 67.4 | 70.5 | - | 77.6 | - | 79.14 | - |
|
- | - | - | - | - | 77.62 | - |
For the iLoc8897 dataset, the absolute true overall accuracy of the current approach is 75.64%, which is 4.37% higher than the iLoc-Euk method, though the overall accuracy is only 0.89% lower than it. In addition, our method achieves the best performances among the 22 subcellular locations except for the locations of Cytoplasm and Endoplasmic reticulum. Meanwhile, our method also performs better than Euk-mPLoc 2.0 [38] which is also based on the same dataset. For the Euk7579 dataset, the overall accuracy of the current approach is 89.94%, which is also higher than those achieved using the methods listed in Table 4 (from 6.44% to 14.94%). Meanwhile, our method also performs better than some other classifiers such as LOCSVMPSI [58] and complexity-based method [59]. As shown in Table 5 , our method also achieves better performances than Hum-mPLoc 2.0. For the Hum3681 dataset, the overall accuracy of the current approach is 75.55%, which is 12.85% higher than the Hum-mPLoc 2.0 method. It is worth noting that all the three datasets (Euk-mPLoc 2.0, iLoc-Euk and Hum-mPLoc 2.0), which also extract sequence features from the Gene Ontology information to represent the query protein, get the comparable accuracies to the present method. This demonstrates that the Gene Ontology information provides a better source of information for the prediction of protein subcellular location. As shown in Table 6 , the proposed method, examined by the jackknife test, also performs better than Euk-mPLoc and the KNN-SVM ensemble classifier [25]. For the Euk6181 dataset [60], the overall accuracy of the proposed method is 79.14%, which is 11.74% and 8.64% higher than Euk-mPLoc and the KNN-SVM ensemble classifier respectively [25].
As illustrated by some researchers, protein sequence similarity within the datasets has a significant effect on the prediction performance of protein subcellular location, i.e., accuracies will be overestimated when using high-similarity datasets. To avoid this problem, two low-similarity datasets, i.e., the iLoc8897 dataset and Hum3681 dataset were used to evaluate the performance of our method. The results also show that our method achieves good performances and the prediction accuracies are higher than those achieved using the methods listed in Table 3 and Table 5 .
3. A case study
To evaluate the performance of the proposed method, it was also used to predict the subcellular locations of some proteins used in our laboratory. Take two proteins for example. The first example is fibronectin (FN) [61], [62], which is an “extracell” protein and abundant in the extracellular matrix and participates in many cellular processes, including osteoblastic differentiation/mineralization, tissue repair, embryogenesis, cell migration/adhesion, and blood clotting. The accession number for FN is shown in Table 7 . According to our ensemble classifier, this protein was predicted as “extracell” protein, which is in accordance with the annotation in Swiss-Prot database. The second is cadherin 11 (CDH 11) [61], [62], which is a plasma membrane protein preferentially expressed in osteoblasts. CDH 11 can promote cells to form specialized cell junctions and enhanced crosstalk between adjacent osteocytes. The accession number for CDH 11 is also shown in Table 7 . We also predicted it correctly. More examples are list in Table 7 . As is shown, 10 of all the 11 proteins are predicted in accordance with the Swiss-Prot annotations by the proposed method. While only 8 of 11 eukaryotic proteins and 2 of 4 human proteins are predicted correctly by iLoc-Euk and Hum-mPLoc2.0 respectively.
Table 7. Examples to show the predicted results by three predictors.
| Accession number | Entry name | Swiss-Prot annotation | iLoc-Euk (2011) | Hum-mPLoc 2.0 (2009) | The proposed method |
| Trained by iLoc8897 dataset | |||||
| P55287 | Cad11_human | Plasma membrane | Plasma membrane | Plasma membraneCytoplasmExtracell | Plasma membrane |
| P02751 | Finc_human | Extracell | Extracell | Extracell | Extracell |
| Q8IZC6 | Cora1_human | Extracell | Extracell | Extracell | |
| Q9EPU7 | Z354c_rat | Nucleus | Nucleus | - | Nucleus |
| Q5QNQ9 | Cora1_mouse | Extracell | Extracell | - | Extracell |
| Q5BKR2 | Nhdc2_mouse | Mitochondrion | Plasma membrane | - | Mitochondrion |
| P12645 | Bmp3_human | Extracell | Extracell | Extracell | Extracell |
| P51690 | Arse_human | Golgi apparatus | Cytoplasm | Lysosome | Golgi apparatus |
| Q8C341 | Ospt_mouse | Endoplasmic reticulum | Plasma membrane | - | Cytoplasm |
| P00922 | Cah2_sheep | Cytoplasm | Cytoplasm | - | Cytoplasm |
| Q30D77 | Cooa1_mouse | Extracell | Extracell | - | Extracell |
We also used iLoc-Euk, Hum-mPLoc 2.0 and the proposed method to predict the subcellular locations of some multiple-location proteins. As can be seen from Table 8 , all subcellular locations of the protein Q05329 was correctly identified by the proposed method and iLoc-Euk, but not entirely correctly by Hum-mPLoc 2.0. The second protein P58335 was identified completely correctly by the proposed method, but according to iLoc-Euk and Hum-mPLoc 2.0, it was assigned to only one of its real subcellular locations. The third protein P30622 simultaneously exists at “Cytoplasm” and “Cytoskeleton” in Swiss-Prot. Both iLoc-Euk and Hum-mPLoc 2.0 only identified one location correctly. Although the proposed method incorrectly predicted P30622 as belonging to “endosome”, yet it successfully identified two of its subcellular locations.
Table 8. Examples to show the predicted results by three predictors on multiple-location proteins.
| Accession number | Entry name | Swiss-Prot annotation | iLoc-Euk (2011) | Hum-mPLoc 2.0 (2009) | The proposed method |
| Trained by iLoc8897 dataset | |||||
| Q05329 | DCE2_human | Plasma membraneGolgi apparatusSynapse | Plasma membraneGolgi apparatusSynapse | CytoplasmMitochondrionSynapse | Plasma membraneGolgi apparatusSynapse |
| P58335 | Antr2_human | Endoplasmic reticulumPlasma membraneExtracell | Extracell | Endoplasmic reticulum | Endoplasmic reticulumPlasma membraneExtracell |
| P30622 | Clip1_human | CytoplasmCytoskeleton | Cytoplasm | CytoskeletonEndosome | CytoplasmCytoskeletonEndosome |
| P13395 | Sptca_drome | CytoskeletonGolgi apparatusPlasma membrane | Golgi apparatus | - | CytoskeletonGolgi apparatus |
| P11279 | Lamp1_human | EndosomeLysosomePlasma membrane | Plasma membrane | Lysosome | Plasma membraneLysosomeMelanosome |
| Q15942 | Zyx_human | CytoplasmCytoskeleton | Cytoskeleton | Plasma membrane | CytoplasmCytoskeletonNucleus |
4. Conclusions
In this study, a KNN-SVM ensemble classifier by fusing the GO attributes and hydrophobicity features was investigated to predict subcellular location of eukaryotic proteins. Three widely used benchmark datasets were adopted in our work. To improve the prediction quality, the following strategies were applied: (i) representing protein samples by using Gene Ontology could effectively grasp the core features to indicate the subcellular localization, (ii) adopting the one-versus-one strategy and two most popular classifiers in machine learning task, i.e., LIBSVM and KNN to predict protein subcellular location, (iii) capturing the top features and learning with a small number of features might lead to a better generalization of machine learning algorithms (Occam's razor). In summary, the results of the predictions performed by KNN-SVM ensemble classifier indicate that our method is very promising and may play an important complementary role to existing methods.
Acknowledgments
The authors thank Ning Huang and Yan Yu whose constructive comments are very helpful for strengthening the presentation of this paper.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the National Natural Science Foundation of China (No. 30901512 and No. 31100953) and the Shanghai Leading Academic Discipline Project (No. S30405). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Laurila K, Vihinen M. PROlocalizer: integrated web service for protein subcellular localization prediction. Amino Acids. 2011;40:975–980. doi: 10.1007/s00726-010-0724-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu NY, Wagner JR, Laird MR, Melli G, Rey S, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen YQ, Burger G. TESTLoc: protein subcellular localization prediction from EST data. BMC Bioinformatics. 2010;11:563. doi: 10.1186/1471-2105-11-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou KC, Shen HB. Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One. 2010;5:e11335. doi: 10.1371/journal.pone.0011335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang W, Geng X, Dou Y, Liu T, Zheng X. Predicting protein subcellular localization by pseudo amino acid composition with a segment-weighted and features-combined approach. Protein Pept Lett. 2011;18:480–487. doi: 10.2174/092986611794927947. [DOI] [PubMed] [Google Scholar]
- 6.Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J Mol Biol. 1994;238:54–61. doi: 10.1006/jmbi.1994.1267. [DOI] [PubMed] [Google Scholar]
- 7.Gu Q, Ding YS, Jiang XY, Zhang TL. Prediction of subcellular location apoptosis proteins with ensemble classifier and feature selection. Amino Acids. 2010;38:975–983. doi: 10.1007/s00726-008-0209-4. [DOI] [PubMed] [Google Scholar]
- 8.Park KJ, Kanehisa M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics. 2003;19:1656–1663. doi: 10.1093/bioinformatics/btg222. [DOI] [PubMed] [Google Scholar]
- 9.Rao HB, Zhu F, Yang GB, Li ZR, Chen YZ. Update of PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2011;39(Suppl 2):W385–390. doi: 10.1093/nar/gkr284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia P, Qian Z, Zeng Z, Cai Y, Li Y. Prediction of subcellular protein localization based on functional domain composition. Biochem Biophys Res Commun. 2007;357:366–370. doi: 10.1016/j.bbrc.2007.03.139. [DOI] [PubMed] [Google Scholar]
- 11.Guo J, Pu X, Lin Y, Leung H. Protein subcellular localization based on PSI-BLAST and machine learning. J Bioinform Comput Biol. 2006;4:1181–1195. doi: 10.1142/s0219720006002405. [DOI] [PubMed] [Google Scholar]
- 12.Bhasin M, Raghava GP. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414–419. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 14.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 15.Rashid M, Saha S, Raghava GP. Support Vector Machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinformatics. 2007;8:337. doi: 10.1186/1471-2105-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin TH, Murphy RF, Bar-Joseph Z. Discriminative motif finding for predicting protein subcellular localization. IEEE/ACM Trans Comput Biol Bioinform. 2011;8:441–451. doi: 10.1109/TCBB.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou L, Wang Z, Huang J. Prediction of subcellular localization of eukaryotic proteins using position-specific profiles and neural network with weighted inputs. J Genet Genomics. 2007;34:1080–1087. doi: 10.1016/S1673-8527(07)60123-4. [DOI] [PubMed] [Google Scholar]
- 18.Wang T, Yang J. Predicting subcellular localization of gram-negative bacterial proteins by linear dimensionality reduction method. Protein Pept Lett. 2010;17:32–37. doi: 10.2174/092986610789909494. [DOI] [PubMed] [Google Scholar]
- 19.Liao B, Jiang JB, Zeng QG, Zhu W. Predicting Apoptosis Protein Subcellular Location with PseAAC by Incorporating Tripeptide Composition. Protein Pept Lett. 2011;18:1086–1092. doi: 10.2174/092986611797200931. [DOI] [PubMed] [Google Scholar]
- 20.Mount DW. Using hidden Markov models to align multiple sequences. Cold Spring Harb Protoc. 2009;2009:pdb top41. doi: 10.1101/pdb.top41. [DOI] [PubMed] [Google Scholar]
- 21.Marinov M, Weeks DE. The complexity of linkage analysis with neural networks. Hum Hered. 2001;51:169–176. doi: 10.1159/000053338. [DOI] [PubMed] [Google Scholar]
- 22.Shen HB, Yang J, Chou KC. Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids. 2007;33:57–67. doi: 10.1007/s00726-006-0478-8. [DOI] [PubMed] [Google Scholar]
- 23.Bulashevska A, Eils R. Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinformatics. 2006;7:298. doi: 10.1186/1471-2105-7-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan A, Majid A, Hayat M. CE-PLoc: an ensemble classifier for predicting protein subcellular locations by fusing different modes of pseudo amino acid composition. Comput Biol Chem. 2011;35:218–229. doi: 10.1016/j.compbiolchem.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Li LQ, Kuang H, Zhang Y, Zhou Y, Wang KF, et al. Prediction of eukaryotic protein subcellular multilocalisation with a combined KNN-SVM ensemble classifier. Journal of Computational Biology and Bioinformatics Research. 2011;3:15–24. [Google Scholar]
- 26.Yu X, Zheng X, Liu T, Dou Y, Wang J. Predicting subcellular location of apoptosis proteins with pseudo amino acid composition: approach from amino acid substitution matrix and auto covariance transformation. Amino Acids. 2011 doi: 10.1007/s00726-011-0848-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 27.Wang P, Hu L, Liu G, Jiang N, Chen X, et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS One. 2011;6:e18476. doi: 10.1371/journal.pone.0018476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang T, Chen L, Cai YD, Chou KC. Classification and analysis of regulatory pathways using graph property, biochemical and physicochemical property, and functional property. PLoS One. 2011;6:e25297. doi: 10.1371/journal.pone.0025297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang T, Wan S, Xu Z, Zheng Y, Feng KY, et al. Analysis and prediction of translation rate based on sequence and functional features of the mRNA. PLoS One. 2011;6:e16036. doi: 10.1371/journal.pone.0016036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou KC, Wu ZC, Xiao X. iLoc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS One. 2011;6:e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen HB, Chou KC. A top-down approach to enhance the power of predicting human protein subcellular localization: Hum-mPLoc 2.0. Anal Biochem. 2009;394:269–274. doi: 10.1016/j.ab.2009.07.046. [DOI] [PubMed] [Google Scholar]
- 32.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lei Z, Dai Y. Assessing protein similarity with Gene Ontology and its use in subnuclear localization prediction. BMC Bioinformatics. 2006;7:491. doi: 10.1186/1471-2105-7-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo MJ, Bae SM, Kim YW, Hur SY, Ro DY, et al. New approaches to pathogenic gene function discovery with human squamous cell cervical carcinoma by gene ontology. Gynecol Oncol. 2005;96:621–629. doi: 10.1016/j.ygyno.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 35.Currie RA, Orphanides G, Moggs JG. Mapping molecular responses to xenoestrogens through Gene Ontology and pathway analysis of toxicogenomic data. Reprod Toxicol. 2005;20:433–440. doi: 10.1016/j.reprotox.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Cai YD, Zhou GP, Chou KC. Predicting enzyme family classes by hybridizing gene product composition and pseudo-amino acid composition. J Theor Biol. 2005;234:145–149. doi: 10.1016/j.jtbi.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 37.Qian Z, Cai YD, Li Y. A novel computational method to predict transcription factor DNA binding preference. Biochem Biophys Res Commun. 2006;348:1034–1037. doi: 10.1016/j.bbrc.2006.07.149. [DOI] [PubMed] [Google Scholar]
- 38.Chou KC, Shen HB. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mPLoc 2.0. PLoS One. 2010;5:e9931. doi: 10.1371/journal.pone.0009931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang WL, Tung CW, Huang HL, Ho SY. Predicting protein subnuclear localization using GO-amino-acid composition features. Biosystems. 2009;98:73–79. doi: 10.1016/j.biosystems.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Mei S, Fei W, Zhou S. Gene ontology based transfer learning for protein subcellular localization. BMC Bioinformatics. 2011;12:44. doi: 10.1186/1471-2105-12-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahu SS, Panda G. A novel feature representation method based on Chou's pseudo amino acid composition for protein structural class prediction. Comput Biol Chem. 2010;34:320–327. doi: 10.1016/j.compbiolchem.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Khan A, Majid A, Choi TS. Predicting protein subcellular location: exploiting amino acid based sequence of feature spaces and fusion of diverse classifiers. Amino Acids. 2010;38:347–350. doi: 10.1007/s00726-009-0238-7. [DOI] [PubMed] [Google Scholar]
- 43.Zhou XB, Chen C, Li ZC, Zou XY. Improved prediction of subcellular location for apoptosis proteins by the dual-layer support vector machine. Amino Acids. 2008;35:383–388. doi: 10.1007/s00726-007-0608-y. [DOI] [PubMed] [Google Scholar]
- 44.Smith TF. Occam's razor. Nature. 1980;285:620. doi: 10.1038/285620a0. [DOI] [PubMed] [Google Scholar]
- 45.Qiu JD, Luo SH, Huang JH, Sun XY, Liang RP. Predicting subcellular location of apoptosis proteins based on wavelet transform and support vector machine. Amino Acids. 2010;38:1201–1208. doi: 10.1007/s00726-009-0331-y. [DOI] [PubMed] [Google Scholar]
- 46.Cai YD, Chou KC. Using neural networks for prediction of subcellular location of prokaryotic and eukaryotic proteins. Mol Cell Biol Res Commun. 2000;4:172–173. doi: 10.1006/mcbr.2001.0269. [DOI] [PubMed] [Google Scholar]
- 47.Yu NY, Laird MR, Spencer C, Brinkman FS. PSORTdb–an expanded, auto-updated, user-friendly protein subcellular localization database for Bacteria and Archaea. Nucleic Acids Res. 2011;39:D241–244. doi: 10.1093/nar/gkq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pierleoni A, Martelli PL, Casadio R. MemLoci: predicting subcellular localization of membrane proteins in eukaryotes. Bioinformatics. 2011;27:1224–1230. doi: 10.1093/bioinformatics/btr108. [DOI] [PubMed] [Google Scholar]
- 49.Xu Q, Pan SJ, Xue HH, Yang Q. Multitask learning for protein subcellular location prediction. IEEE/ACM Trans Comput Biol Bioinform. 2011;8:748–759. doi: 10.1109/TCBB.2010.22. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Li C, Wang E, Wang X. An FPT approach for predicting protein localization from yeast genomic data. PLoS One. 2011;6:e14449. doi: 10.1371/journal.pone.0014449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan Z. Prediction of protein subcellular locations using Markov chain models. FEBS Lett. 1999;451:23–26. doi: 10.1016/s0014-5793(99)00506-2. [DOI] [PubMed] [Google Scholar]
- 52.Shi R, Xu C. Prediction of rat protein subcellular localization with pseudo amino acid composition based on multiple sequential features. Protein Pept Lett. 2011;18:625–633. doi: 10.2174/092986611795222768. [DOI] [PubMed] [Google Scholar]
- 53.Panwar B, Raghava GP. Predicting sub-cellular localization of tRNA synthetases from their primary structures. Amino Acids. 2011 doi: 10.1007/s00726-011-0872-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 54.Kim JK, Raghava GPS, Bang SY, Choi SJ. Prediction of subcellular localization of proteins using pairwise sequence alignment and support vector machine. Pattern Recognition Letters. 2006;27:996–1001. [Google Scholar]
- 55.Chou KC, Shen HB. Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 56.Shi SP, Qiu JD, Sun XY, Huang JH, Huang SY, et al. Identify submitochondria and subchloroplast locations with pseudo amino acid composition: approach from the strategy of discrete wavelet transform feature extraction. Biochim Biophys Acta. 2011;1813:424–430. doi: 10.1016/j.bbamcr.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 57.Ansari HR, Raghava GP. Identification of NAD interacting residues in proteins. BMC Bioinformatics. 2010;11:160. doi: 10.1186/1471-2105-11-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie D, Li A, Wang M, Fan Z, Feng H. LOCSVMPSI: a web server for subcellular localization of eukaryotic proteins using SVM and profile of PSI-BLAST. Nucleic Acids Res. 2005;33:W105–110. doi: 10.1093/nar/gki359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng X, Liu T, Wang J. A complexity-based method for predicting protein subcellular location. Amino Acids. 2009;37:427–433. doi: 10.1007/s00726-008-0172-0. [DOI] [PubMed] [Google Scholar]
- 60.Chou KC, Shen HB. Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res. 2007;6:1728–1734. doi: 10.1021/pr060635i. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Xiang Q, Dong S, Li C, Zhou Y. Fabrication and characterization of a recombinant fibronectin/cadherin bio-inspired ceramic surface and its influence on adhesion and ossification in vitro. Acta Biomater. 2010;6:776–785. doi: 10.1016/j.actbio.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Zhou Y, Zhu J, Dong S, Li C, et al. Effect of a novel recombinant protein of fibronectinIII7-10/cadherin 11 EC1-2 on osteoblastic adhesion and differentiation. Biosci Biotechnol Biochem. 2009;73:1999–2006. doi: 10.1271/bbb.90187. [DOI] [PubMed] [Google Scholar]