

Abstract
X-ray imaging of biological samples is progressing rapidly. In this paper we review the progress to date in high resolution imaging of cellular architecture. In particular we survey the progress in soft X-ray tomography and argue that the field is coming of age and that important biological insights are starting to emerge. We then review the new ideas based on coherent diffraction. These methods are at a much earlier stage of development but, as they eliminate the need for X-ray optics, have the capacity to provide substantially better spatial resolution than zone plate based methods.
Introduction
Much of our knowledge about cell architecture and its remodeling during normal and disease processes has been derived from imaging. As a result, there is an ongoing drive to develop new imaging technologies that will advance our understanding of cell function. For centuries, light microscopy techniques have yielded remarkable information about cell and molecular dynamics. The development of genetically encoded fluorescent protein markers and modern confocal microscopes in the 20th century revolutionized cellular imaging by making it possible to track molecular events in situ to the Abbe diffraction limit, ~200-300 nm for visible light. Several methods for imaging beyond the diffraction limit, known as super-resolution fluorescence microscopy, have been developed during the past decade. These techniques, which enable nanometer scale localization of isolated fluorescent molecules, have been described in detail recently [1,2].
Fluorescence microscopy has significantly advanced our understanding of cellular processes, but one only detects the fluorescent molecules present in the specimen while the vast majority of cellular structures remain invisible. Since cellular constituents are weakly absorbing at visible light wavelengths, transmission light microscopy methods, such as bright field phase contrast imaging, can only provide limited details. Imaging of subcellular structures has been done best with transmission electron microscopy. This technique has elucidated some of the most intricate details of cellular organelles and cytoskeletal proteins, often at molecular resolution. Because of the low penetrating power of electrons, however, it is not possible to examine an intact eukaryotic cell. Cells are typically sectioned into 60-500 nm slices. This process requires initial fixation, dehydration and plastic embedment as well as application of heavy metal stains to generate contrast. Since most eukaryotic cells are at least 10μm diameter, extensive work is required to get three-dimensional structural information of a whole cell, so most studies are limited to small regions of cells. Consequently little is known about the structural organization of eukaryotic cells in the native, unstained state. The importance of imaging cells in the native state has been demonstrated very well by images collected using cryo electron tomography [3], but the technique is limited to specimens less than 500nm thick. Attempts to accomplish the goal of whole cell imaging using cryo-sectioning methods have been discouraging, but ongoing efforts using focused ion beam milling approaches seem promising [4].
Recent advances in X-ray imaging technologies, and the establishment of dedicated facilities, have made X-ray microscopy an important new tool for cellular imaging. X-rays can penetrate thick cells and tissues, eliminating the need to section the specimen [5,6]. The unique properties of photons at soft X-ray wavelengths make it possible to image cells based on the organic composition of unstained subcellular constituents [7,8]. Consequently, soft X-ray tomography (SXT) can generate quantitative three-dimensional images of cells in the near-native state at better than 50nm isotropic resolution [9-12]. While recent advances in the nanofabrication of X-ray optical components have improved achievable resolution to that approaching 10nm [13], such lenses have yet to find routine application to cellular imaging. The recent development of high efficiency lensless imaging methods promises to further extend the capability of biological X-ray imaging. Coherent diffraction imaging techniques offer the possibility of high-resolution X-ray imaging of cells using computation and high speed computers rather than an X-ray optic to phase the image. In this article we discuss the current capabilities of X-ray imaging techniques for imaging cellular structures and show that cellular X-ray imaging has come of age.
Soft X-ray microscopy
The first X-ray microscope, which used grazing-incidence reflective optics to focus the X-rays onto the specimen, was developed in the 1940s [14]. Even though this was an exciting development in the field of X-ray science and materials research, it didn’t generate any noteworthy biological images. It wasn’t until the 1990s that the technology of X-ray microscopy had advanced sufficiently to successfully image sub-cellular details in cells and tissue specimens. At this time, new X-ray optical components, in particular zone plate lenses, became available [15] and began to be used in soft X-ray microscopes employing third generation synchrotrons as the X-ray source [16-18]. The combination of precision nanostructured X-ray lenses, high-efficiency direct detection CCD cameras, and well-designed transmission X-ray microscopes enabled the production of soft X-ray projection images with stunning clarity and contrast from a range of cells and tissue specimens [8].
Soft X-ray microscopes are typically operated using photons with energies in the ‘water window’, i.e. the region of the spectrum that lies between the K shell absorption edges of carbon (284 eV, λ=4.4 nm) and oxygen (543 eV, λ=2.3 nm). Such X-rays are absorbed an order of magnitude more strongly by carbon- and nitrogen-containing organic material than by water [7,8,16]. Consequently, structures in the cell are visualized directly as a function of both their density and biochemical composition while cellular water remains relatively transparent [7,8,12]. Structures such as lipid droplets, for example, absorb significantly more than organelles with high water content (e.g. vacuoles). Lens-based biological X-ray imaging can also be performed using X-rays with energies outside of the water window, where images are generated using phase contrast techniques. However, to date the most compelling images of cells have been obtained using bright field, absorption contrast, transmission X-ray microscopes (TXM) operating at water window wavelengths.
The optical arrangement of a water window TXM is similar in concept to that of a conventional bright-field light microscope [7,8,12]. A condenser zone plate optic focuses X-rays on the specimen, and an objective zone plate optic focuses the transmitted light onto the detector. Greater details about this type of microscope can be found in other publications [19] and at the facility web pages: (http://ncxt.lbl.gov, http://www.helmholtz-berlin.de/forschung/grossgeraete/mikroskopie/index_en.html, http://www.cells.es/Beamlines/XM/)
Soft X-ray tomography
Soft X-ray TXMs, like light and electron microscopes, generate two-dimensional representations of a three-dimensional specimen [20]. For thick biological specimens, this results in structural features being confusingly superimposed in the image [3,7,8,12,21]. However, by collecting projection images of the specimen at incremental angles around a rotation or ‘tilt’ axis, it is possible to mathematically compute a three-dimensional tomographic reconstruction of the specimen [21].
In practice, carrying out three-dimensional tomography on biological specimens required one further technological development. All biological materials are damaged when they are exposed to intense light, whether this is the ultra violet illumination in a fluorescence microscope or the photons in an X-ray microscope [22]. Imaging a typical cell by soft X-ray tomography requires collection of projection images at 1-2° increments through 180° rotation. During this procedure, the specimen receives a dose of radiation large enough to cause a significant degree of structural damage. Fortunately, however, cryo-immobilization of the specimen circumvents this problem. When cells are imaged at liquid nitrogen temperature, more than a thousand soft X-ray projection images can be collected without apparent signs of radiation damage [7,8]. To image at such extreme low temperatures, it was necessary to develop specialized cryogenic tomography stages. One approach, implemented on the X-ray microscope located at BESSY II in Berlin Germany, is the use of a modified electron microscope cryo-tilt stage. Although this was convenient in that it was commercially available, this stage must operate in a vacuum and suffers from the missing wedge problems that compromise the quality of the tomographic reconstruction. The Berkeley microscope employs a novel cryo-rotation stage that is capable of a full 180° degree imaging, generating isotropic resolution tomograms of whole cells [8]. In this stage, the specimen is at atmospheric pressure rather than in a vacuum, and continually cooled by a gentle stream of liquid Nitrogen cooled Helium gas to maintain temperature during imaging.
A major strength of water-window soft X-ray tomography is that it can be applied to virtually any imaging investigation in cell biology: from imaging simple bacteria, to yeast and algae, to advanced eukaryotic cells and tissues. The unique ability to obtain high-resolution images of the subcellular organization of intact, eukaryotic cells in the native state, without any stains or labels, is something that cannot be done with light or electron microscopy. The first reported soft X-ray tomographic reconstruction demonstrating these features was of the algae, Chlamydomonas reinhardtii, [12,23] followed by studies of yeast [9,10,24]. Dramatic improvements in X-ray images were soon noted upon completion of a second generation X-ray microscope, dedicated to biological imaging, constructed by scientists at the National Center for X-ray Tomography (NCXT) at Lawrence Berkeley National Laboratory. Remarkable 50nm resolution images of the internal organization of the fission yeast, S. pombe, can be seen in Figure 1 [10,24]. Another recent study shows the structural reorganization associated with transition through the cell cycle in the budding yeast, Saccharomyces cerevisiae, revealing high contrast details of cellular architecture (Figure 2, Uchida et al, submitted).
Figure 1.
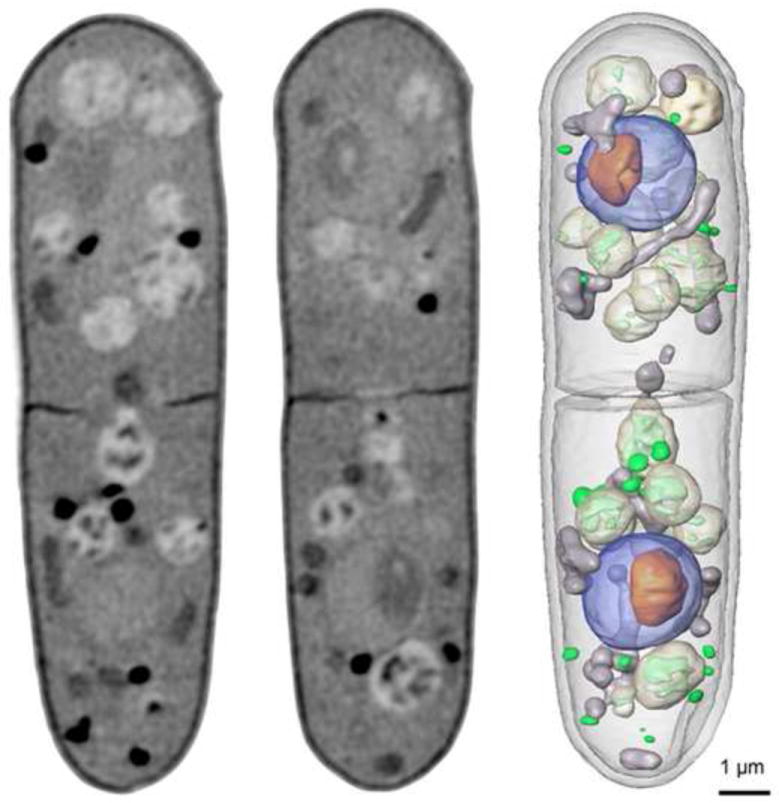
Soft X-ray tomography of a rapidly frozen fission yeast, Schizo-saccharomyces pombe. The data for the complete three-dimensional reconstruction were composed of 90 images, collected through a total of 180° of rotation. Two computer-generated sections through the tomographic reconstruction are shown along with an image showing select organelles that have been segmented and color coded according to type. Key: nucleus, blue; nucleolus, orange; mitochondria, grey; vacuoles, white; lipid-rich vesicles, green. (X-ray tomographic reconstructions are an update of the work outlined in [27, 28]). Scale bar=1 μm.
Figure 2.
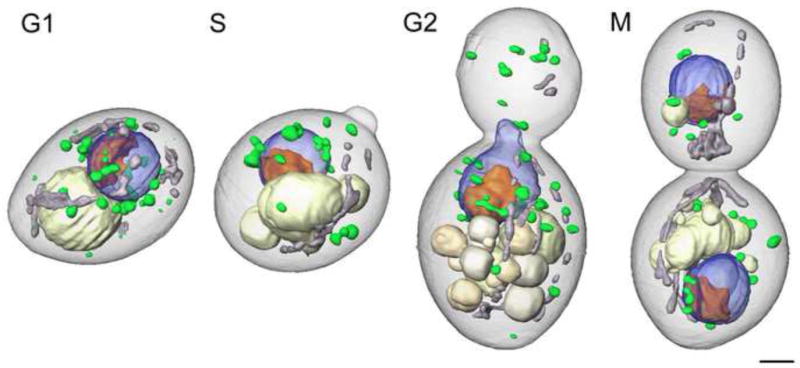
Soft X-ray tomography of rapidly frozen Saccharomyces cerevisiae cells imaged at each phase of cell cycle - G1, S, M, and G2. Organelles are color-coded as follows: blue, nucleus; orange, nucleolus; gray, mitochondria; ivory, vacuoles; green, lipid bodies. Scale bar, 2 μm. (manuscript submitted)
The first demonstration that soft X-ray tomography can play a major role in biomedical research and drug discovery was the examination of the cellular organization of three phenotypes characteristic of the yeast, Candida albicans, (Figure 3) and the altered phenotype resulting from the application of potential anti-fungal components [11]. The hyphal phenotype shown in Figure 3 was 47 μm long, which is greater than the soft X-ray microscope field of view. However, in cases such as this, collecting sequential fields of view allows the entire cell to be imaged. Projection data from each of the fields of view are reconstructed, and then stitched together to form a composite image. Imaging a cell this long using electron tomography is a near impossible challenge (it would require the cell be cryo-sliced into approximately 200 sections, a Herculean task with a high probability of failure at a number key steps). Whereas, with soft X-ray tomography the data necessary to reconstruct a large hyphal cell can be collected in less than 20 minutes [11]. X-ray tomography can also be used to examine larger, more advanced eukaryotic cells, such as white blood cells (Figure 4) and malaria- infected red blood cells (Hanssen et al., submitted). The images from the tomographic reconstruction of the lymphocyte seen in Figure 4 are the first views of the 3-D organization of an advanced eukaryotic cell, and its nuclear architecture, in the near-native state.
Figure 3.

Soft X-ray tomography of the yeast, Candida albicans, showing the three phenotypes displayed by this yeast: (A) Yeast-like cells, (B) pseudo-hyphae, and c) hyphae. The hyphae shown in (C) is 47 μm long. To image this cell – which is significantly larger than the 15 μm microscope field of view - projection series were collected from sequentially overlapping fields of view along the length of the cell. These projection series were individually reconstructed and then computationally stitched together to form a single, seamless tomographic reconstruction. Volumes were segmented and color-coded as follows: blue, nucleus; orange, nucleolus; gray, mitochondria; ivory, vacuoles; green, lipid bodies Scale bar, 2.0 μm. (taken from 11)
Figure 4.
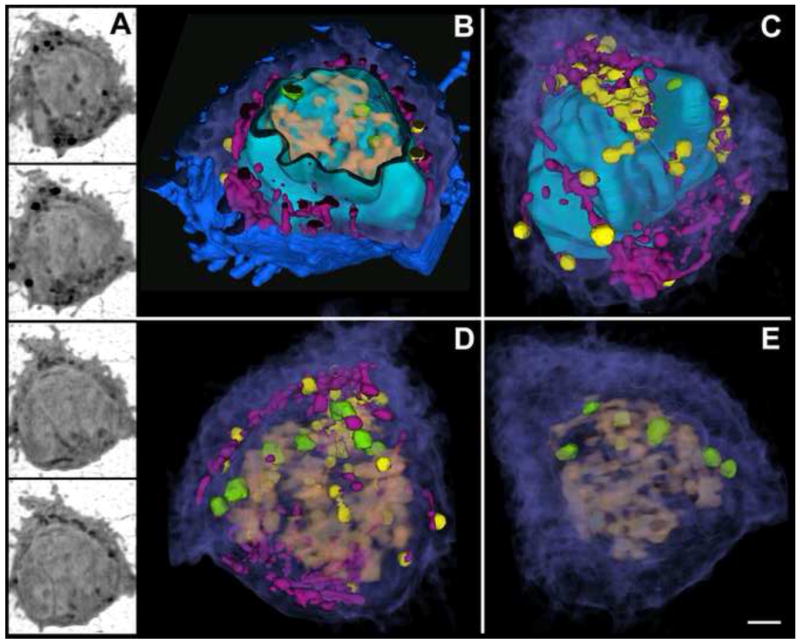
Soft X-ray tomography of a lymphocyte (T-cell). (A) Four orthoslices from the tomographic reconstruction of a cryo-immobilized T-cell. (B-E) Segmented volumes color-coded for identification of internal structures. (B) Cut-away view showing several filopodial extensions on the cell surface (blue) and the internal cytoplasm (purple) containing organelles such as mitochondria (magenta) and highly absorbing vesicles (yellow). A cut-away view of the nuclear envelope (cyan) reveals the chromatin (salmon) and nuclear bodies (green) within. (C) The cell surface has been removed, showing the 3-D organization of cytoplasm and organelles with respect to the intact nuclear envelope, which is highly folded. (D) View of the cell interior in which the nuclear envelope has been made transparent revealing the 3-D organization of the chromatin with respect to cytoplasmic organelles. (E) View of the cell showing the 3-D organization of chromatin and nuclear bodies with respect to cytosol. Scale bar, 1.0 μm.
Coherent diffractive imaging
The methods discussed in the first part of the present paper rely on optical elements, typically zone plates, to produce an image of the sample under investigation. The ongoing improvement of the spatial resolution of these methods is very likely to ultimately hit some fundamental barriers due to the difficulty of fabricating ever finer structures. Coherent diffractive imaging (CDI), sometimes also referred to as coherent diffraction microscopy, is a method that offers the possibility of removing the need for a lens and so obviating the limitations imposed by the state-of-the-art in fabrication technology.
The method has a long history, originating from an early proposal by the crystallographer David Sayre [25] in which it was suggested that the methods of crystallography could be adapted to the imaging of samples that do not display periodicity. The manner in which the image is recovered has more in common with electron imaging methods than it does with X-ray crystallography and, indeed, the phase problem is much easier to solve than it is for the case of crystals. The basic underlying principle for CDI is the realization that a finite sample is uniquely defined by its diffraction pattern [26]. An iterative method based on ideas with origins in electron and visible optics [27] is used to find an object that is consistent with both the measured diffraction pattern and any a-priori knowledge possessed about the object.
The basic arrangement of a CDI experiment is shown schematically in Figure 5(a). In essence, a coherent beam is used to illuminate the sample and its far-field diffraction pattern is measured. As the beam will typically interact only weakly with the sample, the undiffracted component of the beam is dominant and will damage the detector unless a beam-stop is introduced, precluding the measurement of the beam at very low diffraction angles. Ideally, if the angular subtended by the beam-stop is smaller than the characteristic scale of the variation of the diffraction pattern (ie. the characteristic size of the coherent speckle) then there need not be a significant impact on the reconstruction. The first experimental confirmation did not take place until long after Sayre’s proposal [28], and this work used an electron microscope image of the sample to replace the data obscured by the beam-stop. In the intervening decade or so, there has been considerable further progress driven by the rapidly growing access to coherent X-ray photons.
Figure 5.
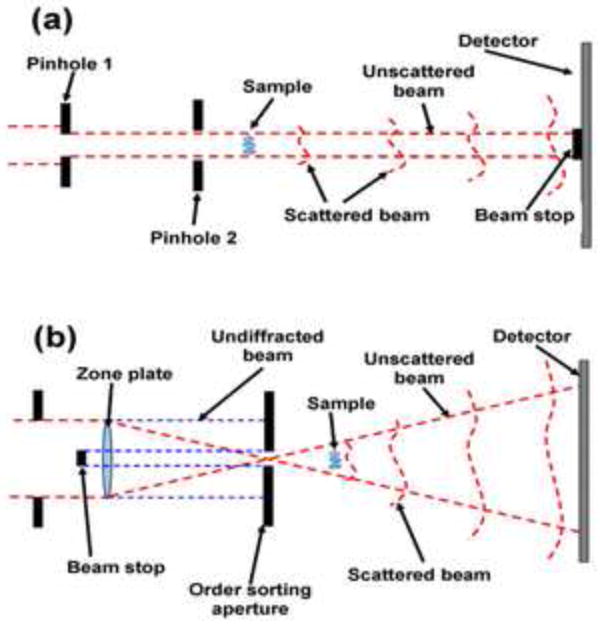
(a) Schematic of a coherent diffractive imaging experiment. The sample is illuminated with a nominally coherent beam of X-rays and the resulting diffraction pattern is captured on a suitable detector placed downstream of the sample. Pinhole 1 is used to select a coherent patch of the beam and pinhole 2 is used to eliminate the scattering from the edges of the first pinhole so as to allow a well-defined coherent beam to be incident on the sample. The object is finite in extent and so must be isolated from other features in the sample. A beam stop is introduced to prevent the direct beam from the synchrotron damaging the detector. (b) Schematic of the configuration used from Fresnel coherent diffractive imaging. A zone plate is here used to create a diverging beam to be incident on the sample. An order sorting aperture (OSA) is used to remove other diffracting orders from the zone plate and also, in conjunction with a beam-top upstream of the OSA, to prevent the undiffracted beam (0th zone plate order, the edges of which are shown as blue dotted lines) from reaching the detector.
A major requirement for flexible microscopy is the ability to image samples without the need for the sample to be spatially isolated – the sample must have a well-defined and finite spatial extent. Williams et al [29] introduced the idea of using a curved incident beam (see Figure 5(b)), where a zone plate or any other suitable optic is used to create a focus and the sample is placed in the beam diverging from it. The resolution of the resulting image, as with other forms of CDI and crystallography, is limited by the maximum angle at which diffraction is observed (not by the properties of the optics producing the diverging beam) and it has been established that this configuration displays rather more stable and consistent image recovery characteristics [30]. This configuration, in turn, allowed the finite object to be replaced by the finite illumination of an extended object [31] and so eliminating the need for the sample to be isolated. Scanning methods also borrowed from electron microscopy [32] and known diversely as Wigner phase space deconvolution [33], ptychography [34] and scanning diffraction microscopy [35] have also enabled diffractive imaging of extended objects with high resolution. The different names really refer to the analysis methods for the data, with the latter two forms relying on iterative methods, and the Wigner phase-space method using a deterministic Fourier transform based deconvolution method.
As argued in this paper, modern high-resolution microscopy also needs the capacity to image in three dimensions and this has also been convincingly demonstrated for non-biological objects [36] at a spatial resolution of around 50nm. The application of CDI to biological samples has been developing rapidly. The first convincing demonstration was of a yeast cell in a form that allowed for imaging of both the phase and the amplitude [37] at a resolution of 30nm. Images have also been published of the malaria-infected red blood cell [38] and samples of a human chromosome [39]. In all these cases, the samples were dried prior to imaging.
Realistic biological imaging will require that the sample be prepared in a frozen hydrated state and results of such experiments have been reported very recently for X-rays in the water window obtained at the Advanced Light Source [40] where a resolution of better than 25 nm was reported for a yeast (S. cerevisiae) cell (Figure 6; cryo X-ray tomography of S. cerevisiae is shown in Figure 2 for comparison). A frozen hydrated Deinococcus radiodurans bacteria was simultaneously reported from the ESRF [41] where 8keV X-rays were used to image at a spatial resolution of around ~30nm.
Figure 6.

Optical DIC image (left) and X-ray CDI image (right) of a Saccharomyces cerevisiae yeast cell. The arrow in the left indicates the assumed direction of the incident X-ray beam. The red arrow indicates the possible location of a mitochondrion. Reproduced with permission from ref [40].
A recent paper by Nelson et al reports high-resolution molecular localization in a specifically labeled cell, and describes an approach to obtain some depth resolution using the phase information to enable “focusing” through the sample using digital methods [42]. In this manuscript, the authors show that similar images of a yeast cell are obtained using CDI, STXM and SEM. However, none of these images shows the vast array of subcellular organelles that are seen in images of S. cerevisiae obtained with transmission electron tomography [43] or X-ray tomography (Figure 2), indicating that there remains considerable scope for the further development of CDI in bio-imaging.
It has been proposed that using a short pulse of x-rays from a free electron laser could mitigate the effects of radiation damage, with the image being formed from scattered radiation before the specimen is destroyed [44]; Chapman and colleagues have shown that it is indeed possible to obtain useable diffraction patterns from objects that are in the process of disintegrating [45]. Unfortunately the damage caused by the extremely powerful laser pulse rules out the use of tomography to retrieve 3D information, as tomography requires many images of the same cell. The remaining option is to average data from many specimens. While this method could, in principal, work with identical particles such as single molecules, it is unlikely to be effective for cell imaging since each cell is unique. Moreover, recent data has shown that the core electrons are removed from light atoms such as carbon and nitrogen in a few femtoseconds, creating hollow atoms [46] suggesting that the rapid electronic damage effects of the incident pulse must also be considered – the nature of the sample in the Chapman et al paper [45] was such that it could not test the consequences of electronic damage. However, future experiments will no doubt soon clarify whether high-flux femtosecond pulse imaging can mitigate the effects of radiation damage accompanying biological x-ray imaging.
In summary, CDI has a great deal further to go in order to compete with the state-of-the-art three-dimensional X-ray microscopy, and the available resolution of X-ray zone plates will continue to improve and so the requirements for CDI to be truly competitive in biological X-ray microscopy will continue to become ever more demanding. The image reconstruction algorithms will continue to develop and, in the opinion of the authors, the uncertainty as to whether a given reconstruction is correct in every detail – the problem being that the algorithm might become trapped in a spurious solution (ie. a local minimum in solution space) – remains an issue worthy of further examination. However the ptychography-related methods [33-35] and curved beam methods [29,31] seem to be more consistent. However CDI does offer very high spatial resolution imaging that is sensitive to both phase and amplitude and so offers a quantitative form of imaging that has considerable potential for biological imaging.
Conclusions and outlook
Significant advances have been made in X-ray imaging techniques during the past decade. In this article, we have limited our discussion to those X-ray techniques that examine subcellular structures in whole cells at high resolution (better than 50nm). One of these techniques, soft X-ray tomography, has generated remarkable images of a variety of different cell types. The multi-disciplinary team of scientists at the National Center for X-ray Tomography (NCXT) in Berkeley constructed a world-class microscope dedicated to cellular imaging and moved this technology from proof-of-principle demonstrations to a robust and easy to use biological research instrument. A similar multi-purpose microscope (designed for both physical and biological sciences) in Berlin is advancing spectral and spatial resolution capabilities of zone plate microscopy [47].
In the immediate future we can expect to see many new X-ray imaging methods mature into techniques that can be easily applied to a range of biological applications. One powerful new approach will be the routine use of correlated fluorescence and X-ray tomography [48]. This approach will put molecular information into context by overlaying three-dimensional data obtained from super resolution fluorescence microscopy onto precision structural information, obtained from the same cell, using soft X-ray tomography. Continued advances in zone plate optics will also improve the resolution achievable with X-ray tomography.
The method of coherent diffractive imaging is at a much earlier stage of development but the field is progressing rapidly. It is safe to say that, from the initial demonstration of Miao et al, it has evolved into a suite of reliable high-resolution imaging methods. It is also true that new ideas continue to emerge rapidly.
The ultimate role of CDI in the development of X-ray microscopy is not yet clear. One can be certain that at some point the technical limits on the spatial resolution achievable using X-ray optics will be met and that CDI offers a pathway beyond this obstacle, wherever it may lie. CDI also offers access to the quantitative amplitude and phase information that are not easily available through other forms of X-ray imaging. Either or both of these features may turn out to be very important. For example, the access to phase information opens up a new contrast mechanism that may make it easier to explore a wider range of imaging wavelengths and to push for truly wavelength-limited resolution at these shorter wavelengths. Phase contrast does not require energy to be deposited in the sample and so perhaps some strategies that mitigate radiation damage will emerge. More penetrating radiation could facilitate advances such as the use of diffraction reconstruction strategies for thick specimen tissue tomography with the more penetrating multi-kilovolt X-rays.
Biological X-ray microscopy is at an exciting stage. Zone plate-based X-ray tomographic imaging has now reached maturity and truly spectacular images are being obtained. CDI is poised to take over as zone plates reach their limits and together these will ensure that X-ray microscopy grows and develops for many years to come.
Acknowledgments
The authors thank Dr. Mark Le Gros for his assistance with this manuscript and Dr. Maho Uchida for help with preparation of the Figures. CAL acknowledges the support of the National Center for Research Resources (RR019664) and the National Institute of General Medical Sciences (GM070445), both of the National Institutes of Health, and the US Department of Energy, Office of Biological and Environmental Research (Contract No. DE-AC02-05CH11231). KAN acknowledges the support of the Australian Research Council through its Centres of Excellence and Federation Fellowships programs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang B. Super-resolution optical microscopy: multiple choices. Current Opinion in Chemical Biology. 2010;14:10–14. doi: 10.1016/j.cbpa.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Patterson GH. Fluorescence microscopy below the diffraction limit. Seminars in Cell & Developmental Biology. 2009;20:886–893. doi: 10.1016/j.semcdb.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumeister W, Grimm R, Walz J. Electron tomography of molecules and cells. Trends in Cell Biology. 1999;9:81–85. doi: 10.1016/s0962-8924(98)01423-8. [DOI] [PubMed] [Google Scholar]
- 4.Marko M, Hsieh C, Schalek R, Frank J, Mannella C. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nature Methods. 2007;4:215–217. doi: 10.1038/nmeth1014. [DOI] [PubMed] [Google Scholar]
- 5.Meyer-Ilse W, Hamamoto D, Nair A, Lelievre SA, Denbeaux G, Johnson L, Pearson AL, Yager D, Legros MA, Larabell CA. High resolution protein localization using soft X-ray microscopy. J Microsc. 2001;201:395–403. doi: 10.1046/j.1365-2818.2001.00845.x. [DOI] [PubMed] [Google Scholar]
- 6.Schneider G. Cryo x-ray microscopy with high spatial resolution in amplitude and phase contrast. Ultramicroscopy. 1998;75:85–104. doi: 10.1016/s0304-3991(98)00054-0. [DOI] [PubMed] [Google Scholar]
- 7.Le Gros MA, McDermott G, Larabell CA. X-ray tomography of whole cells. Current Opinion in Structural Biology. 2005;15:593–600. doi: 10.1016/j.sbi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 8.McDermott G, Le Gros MA, Knoechel CG, Uchida M, Larabell CA. Soft X-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging. Trends in Cell Biology. 2009;19:587–595. doi: 10.1016/j.tcb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larabell CA, Le Gros MA. X-ray tomography generates 3-D reconstructions of the yeast, Saccharomyces cerevisiae, at 60-nm resolution. Molecular Biology of the Cell. 2004;15:957–962. doi: 10.1091/mbc.E03-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkinson DY, McDermott G, Etkin LD, Le Gros MA, Larabell CA. Quantitative 3-D imaging of eukaryotic cells using soft X-ray tomography. Journal of Structural Biology. 2008;162:380–386. doi: 10.1016/j.jsb.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida M, McDermott G, Wetzler M, Le Gros MA, Myllys M, Knoechel C, Barron AE, Larabell CA. Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19375–19380. doi: 10.1073/pnas.0906145106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weiss D, Schneider G, Niemann B, Guttmann P, Rudolph D, Schmahl G. Computed tomography of cryogenic biological specimens based on X-ray microscopic images. Ultramicroscopy. 2000;84:185–197. doi: 10.1016/s0304-3991(00)00034-6. [DOI] [PubMed] [Google Scholar]
- 13.Chao WL, Harteneck BD, Liddle JA, Anderson EH, Attwood DT. Soft X-ray microscopy at a spatial resolution better than 15nm. Nature. 2005;435:1210–1213. doi: 10.1038/nature03719. [DOI] [PubMed] [Google Scholar]
- 14.Kirkpatrick P, Baez AV. Formation of optical images by x-rays. Journal Of The Optical Society Of America. 1948;38:766–774. doi: 10.1364/josa.38.000766. [DOI] [PubMed] [Google Scholar]
- 15.Andersen EH, Harteneck B, Olynick D, Meyer-IIse W, Attwood D, editors. Nanofabrication of X-ray ZonePlates with the Nanowriter Electron-Beam Lithography System. Berkeley, CA: American Institute of Physics; 1999. [Google Scholar]
- 16.Attwood DT. Soft x-rays and extreme ultraviolet radioation: principles and applications. Cambridge, New York: Cambridge University Press; 1999. [Google Scholar]
- 17.Schmahl G, Rudolph D, Niemann B, Guttmann P, Thieme J, Schneider G, David C, Diehl M, Wilhein T. X-ray microscopy studies. Optik. 1993;93:95–102. [Google Scholar]
- 18.Schmahl G, Rudolph D, Niemann B, Guttmann P, Thieme J, Schneider G. X-ray microscopy. Naturwissenschaften. 1996;83:61–70. [PubMed] [Google Scholar]
- 19.Schneider G, Niemann B, Guttmann P, Rudolph D, Schmahl G. Cryo X-ray microscopy. Synchrotron Radiation News. 1995;8:19–28. [Google Scholar]
- 20.Spitta EJ. Microscopy - the Construction, theory and Use of the Microscope. New York, NY: E.P Dutton; 1907. [Google Scholar]
- 21.Natterer F, Wübbeling F. Mathematical Methods in Image Reconstruction. Cambridge, New York: Cambridge University Press; 2001. [Google Scholar]
- 22.Le Gros M, Larabell CA. Cryotomography X-ray Microscope Stage. US Patent. 2009
- 23.Weiss D, Schneider G, Vogt S, Guttmann P, Niemann B, Rudolph D, Schmahl G. Tomographic imaging of biological specimens with the cryo transmission X-ray microscope. Nuclear Instruments & Methods in Physics Research Section A-Accelerators Spectrometers Detectors & Associated Equipment. 2001;467:1308–1311. [Google Scholar]
- 24.Gu W, Etkin LD, Gros MAL, Larabell CA. X-ray tomography of Schizosaccharomyces pombe. DIFFERENTIATION. 2007;75:529–535. doi: 10.1111/j.1432-0436.2007.00180.x. [DOI] [PubMed] [Google Scholar]
- 25.Sayre D. Imaging Processes and Coherence in Physics. In: Schlenker M, Fink M, Goedgebuer JP, Malgrange C, Vienot J Ch, Wade RH, editors. Lecture Notes in Physics. Vol. 112 Springer-Verlag; Heidelberg: 1980. pp. 229–235. [Google Scholar]
- 26.Bates RHT. Fourier Phase Problems Are Uniquely Solvable In More Than One Dimension.1. Underlying Theory. Optik. 1982;61:247–262. [Google Scholar]
- 27.Fienup JR. Reconstruction Of A Complex-Valued Object From The Modulus Of Its Fourier-Transform Using A Support Constraint. Journal of the Optical Society of America A-Optics Image Science and Vision. 1987;4:118–123. [Google Scholar]
- 28.Miao JW, Charalambous P, Kirz J, Sayre D. Extending the methodology of X-ray crystallography to allow imaging of micrometre-sized non-crystalline specimens. Nature. 1999;400:342–344. [Google Scholar]
- 29.Williams GJ, Quiney HM, Dhal BB, Tran CQ, Nugent KA, Peele AG, Paterson D, de Jonge MD. Fresnel coherent diffractive imaging. Physical Review Letters. 2006;97:025506. doi: 10.1103/PhysRevLett.97.025506. [DOI] [PubMed] [Google Scholar]
- 30.Quiney HM, Nugent KA, Peele AG. Iterative image reconstruction algorithms using wave-front intensity and phase variation. Optics Letters. 2005;30:1638–1640. doi: 10.1364/ol.30.001638. [DOI] [PubMed] [Google Scholar]
- 31.Abbey B, Nugent KA, Williams GJ, Clark JN, Peele AG, Pfeifer MA, De Jonge M, McNulty I. Keyhole coherent diffractive imaging. Nature Physics. 2008;4:394–398. [Google Scholar]
- 32.Hegerl R, Hoppe W. Dynamic Theory Of Crystalline Structure Analysis By Electron Diffraction In Inhomogeneous Primary Wave Field. Berichte Der Bunsen-Gesellschaft Fur Physikalische Chemie. 1970;74:1148. [Google Scholar]
- 33.Chapman HN. Phase-retrieval X-ray microscopy by Wigner-distribution deconvolution. Ultramicroscopy. 1996;66:153–172. [Google Scholar]
- 34.Rodenburg JM, Hurst AC, Cullis AG, Dobson BR, Pfeiffer F, Bunk O, David C, Jefimovs K, Johnson I. Hard-x-ray lensless imaging of extended objects. Physical Review Letters. 2007;98:034801. doi: 10.1103/PhysRevLett.98.034801. [DOI] [PubMed] [Google Scholar]
- 35.Thibault P, Dierolf M, Menzel A, Bunk O, David C, Pfeiffer F. High-resolution scanning x-ray diffraction microscopy. Science. 2008;321:379–382. doi: 10.1126/science.1158573. [DOI] [PubMed] [Google Scholar]
- 36.Chapman HN, Barty A, Marchesini S, Noy A, Hau-Riege SR, Cui C, Howells MR, Rosen R, He H, Spence JCH, et al. High-resolution ab initio three-dimensional x-ray diffraction microscopy. Journal of the Optical Society of America A-Optics Image Science and Vision. 2006;23:1179–1200. doi: 10.1364/josaa.23.001179. [DOI] [PubMed] [Google Scholar]
- 37.Shapiro D, Thibault P, Beetz T, Elser V, Howells M, Jacobsen C, Kirz J, Lima E, Miao H, Neiman AM, et al. Biological imaging by soft x-ray diffraction microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15343–15346. doi: 10.1073/pnas.0503305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams GJ, Hanssen E, Peele AG, Pfeifer MA, Clark J, Abbey B, Cadenazzi G, de Jonge MD, Vogt S, Tilley L, et al. High-resolution X-ray imaging of Plasmodium falciparum-infected red blood cells. Cytometry Part A. 2008;73A:949–957. doi: 10.1002/cyto.a.20616. [DOI] [PubMed] [Google Scholar]
- 39.Nishino Y, Takahashi Y, Imamoto N, Ishikawa T, Maeshima K. Three-Dimensional Visualization of a Human Chromosome Using Coherent X-Ray Diffraction. Physical Review Letters. 2009;102:018101. doi: 10.1103/PhysRevLett.102.018101. [DOI] [PubMed] [Google Scholar]
- 40.Huang XJ, Nelson J, Kirz J, Lima E, Marchesini S, Miao HJ, Neiman AM, Shapiro D, Steinbrener J, Stewart A, et al. Soft X-Ray Diffraction Microscopy of a Frozen Hydrated Yeast Cell. Physical Review Letters. 2009;103:198101. doi: 10.1103/PhysRevLett.103.198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giewekemeyer K, Thibault P, Kalbfleisch S, Beerlink A, Kewish CM, Dierolf M, Pfeiffer F, Salditt T. Quantitative biological imaging by ptychographic x-ray diffraction microscopy. Proceedings of the National Academy of Sciences of the United States of America. 107:529–534. doi: 10.1073/pnas.0905846107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson J, Huang XJ, Steinbrener J, Shapiro D, Kirz J, Marchesini S, Neiman AM, Turner JJ, Jacobsen C. High-resolution x-ray diffraction microscopy of specifically labeled yeast cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7235–7239. doi: 10.1073/pnas.0910874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Toole ET, Winey M, McIntosh JR, Mastronarde DN. Electron tomography of yeast cells. Guide to Yeast Genetics and Molecular and Cell Biology, Pt C. 2002;351:81–95. doi: 10.1016/s0076-6879(02)51842-5. Methods in Enzymology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature. 2000;406:752–757. doi: 10.1038/35021099. [DOI] [PubMed] [Google Scholar]
- 45.Chapman HN, Barty A, Bogan MJ, Boutet S, Frank M, Hau-Riege SP, Marchesini S, Woods BW, Bajt S, Benner H, et al. Femtosecond diffractive imaging with a soft-X-ray free-electron laser. Nature Physics. 2006;2:839–843. [Google Scholar]
- 46.Young L, Kanter EP, Krassig B, Li Y, March AM, Pratt ST, Santra R, Southworth SH, Rohringer N, DiMauro LF, et al. Femtosecond electronic response of atoms to ultra-intense X-rays. Nature. 2010;466:56–61. doi: 10.1038/nature09177. [DOI] [PubMed] [Google Scholar]
- 47.Carrascosa JL, Chichon FJ, Pereiro E, Rodriguez MJ, Fernandez JJ, Esteban M, Heim S, Guttmann P, Schneider G. Cryo-X-ray tomography of vaccinia virus membranes and inner compartments. Journal of Structural Biology. 2009;168:234–239. doi: 10.1016/j.jsb.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 48.Le Gros MA, McDermott G, Uchida M, Knoechel CG, Larabell CA. High-aperture cryogenic light microscopy. Journal of Microscopy-Oxford. 2009;235:1–8. doi: 10.1111/j.1365-2818.2009.03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]