

Abstract
Post-operative cognitive dysfunction (POCD) is a clinical phenomenon that has drawn significant attention from the public and scientific community. Age is a risk factor for POCD. However, the contribution of general anesthesia/anesthetics to POCD and the underlying neuropathology are not clear. Here, we showed that 18-month-old male Fisher 344 rats exposed to 1.2% isoflurane, a general anesthetic, for 2 h had significant learning and memory impairments assessed at 2 to 4 weeks after isoflurane exposure. These isoflurane effects were attenuated by intravenous lidocaine (1.5 mg/kg as a bolus and then 2 mg/kg/h during isoflurane exposure), a local anesthetic that has neuroprotective effect. Exposure to isoflurane or isoflurane plus lidocaine did not change the neuronal and synaptic density as well as the expression of NeuN (a neuronal protein), drebrin (a dendritic spine protein), synaptophysin (a synaptic protein), activated caspase 3 and caspase-activated DNase in the hippocampus at 29 days after isoflurane exposure when cognitive impairment was present. Isoflurane and lidocaine did not affect the amount of β-amyloid peptide, total tau and phospho-tau in the cerebral cortex as well as interleukin-1β and tumor necrosis factor-α in the hippocampus at 29 days after isoflurane exposure. Thus, isoflurane induces learning and memory impairment in old rats. Lidocaine attenuates these isoflurane effects. Isoflurane may not cause long-lasting neuropathological changes.
Keywords: aged rats, β-amyloid peptide, cognitive functions, isoflurane
1. Introduction
Post-operative cognitive dysfunction (POCD) is a fairly well-documented clinical phenomenon [1]. Studies have shown short-term and long-term POCD after cardiac and non-cardiac surgeries [2-4]. Advancing age has been consistently identified as a risk factor for POCD [2,3]. About 10% of elderly (60 years or older) patients have POCD 3 months after non-cardiac surgery [3]. In addition to affecting patients’ daily activities due to cognitive impairment, POCD has been found to be associated with an increased mortality [3,5].
It is estimated that more than 200 million people each year undergo surgery in the world. Most patients have their surgeries under general anesthesia. Among them, ~80% patients will receive volatile anesthetics as their primary anesthetics due to many features of volatile anesthetics [6]. Although patients often regain consciousness quickly after general anesthesia with volatile anesthetics, there is a growing concern of volatile anesthetic safety in recent years. Cognitive impairment in rats can be detected at least 3 weeks after being exposed to volatile anesthetics [7,8], although improvement of cognitive functions after volatile anesthetic exposure in rodents also has been reported [7,9,10]. Volatile anesthetics increase activated caspase 3 expression and β-amyloid peptide (Aβ) production in mouse brains [11]. Those anesthetics at clinically relevant concentrations can induce Aβ oligomerization in vitro [12]. Aβ overproduction, oligomerization and accumulation as well as accumulation of hyperphosphorylated tau in the brain are considered as underlying neuropathology for Alzheimer's disease (AD) [13], the most common form of dementia in elderly patients. However, it is not known yet whether these neuropathological changes contribute to the cognitive impairment after anesthetic exposure.
Very little is known about how to attenuate volatile anesthetic-induced cognitive impairment. Neuroinflammation can impair cognitive functions [14]. We have shown that isoflurane increases the expression of inducible nitric oxide synthase, a protein that contributes to inflammatory response, in rat brains [15]. Isoflurane has been shown to increase activated caspase 3 [11], a key enzyme to lead to cell apoptosis, in mouse brains. Neuroinflammation, Aβ accumulation, neurodegeneration and hyperphosphorylated tau accumulation are key pathological features of AD brains [13,16]. Lidocaine, a local anesthetic, decreases required concentrations of general anesthetics to maintain anesthesia [17] and has neuroprotective and anti-inflammatory effects when given systemically [18-20]. Thus, we hypothesize that isoflurane induces cognitive impairment and AD-like neuropathology and that lidocaine attenuates these changes in aged animals. To test our hypothesis, we exposed 18-month old Fisher 344 rats to isoflurane in the presence or absence of lidocaine. Cognitive functions, brain Aβ and phosphorylated tau levels, neuroinflammation as reflected by the levels of the proinflammatory cytokines interleukin 1β (IL-1β) and tumor necrosis factor-α (TNF-α), and brain cell death were evaluated after the exposure to anesthetics.
2. Materials and Methods
The animal protocol was approved by the institutional Animal Care and Use Committee of the University of Virginia (Charlottesville, VA). All animal experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publications number 80-23) revised in 1996.
2.1. Animal Groups
Eighteen-month-old male Fisher 344 rats weighing 470 – 550 g from the National Institutes of Health, Bethesda, MD, were divided into three groups: control, isoflurane, and isoflurane plus lidocaine. Animals in the isoflurane and isoflurane plus lidocaine groups were exposed to 1.2% isoflurane for 2 h. Animals in the isoflurane plus lidocaine group received intravenous lidocaine (1.5 mg/kg as a bolus and then 2 mg/kg/h during the 2-h isoflurane exposure). Since isoflurane was carried by 100% O2, rats in the control group were kept in a chamber that was gassed with 100% O2 for 2 h.
2.2. Isoflurane exposure
As we described before [21], anesthesia was induced by placing rats in a chamber gassed with 3% isoflurane in oxygen. Rats were intubated with a 14-gauge catheter. They were mechanically ventilated with isoflurane carried by 100% O2 to maintain end tidal isoflurane concentration at 1.2%. The inhaled and exhaled gas concentrations were monitored continuously with a DatexTM infrared analyzer (Capnomac, Helsinki, Finland). The ventilator settings usually were 2 ml as the tidal volume at a rate of 60 breaths/min and were adjusted to maintain the end tidal CO2 at ~32 mm Hg. Rectal temperature was maintained at 37°C ± 0.5°C. Heart rate and pulse oximeter oxygen saturation (SpO2) were measured continuously during anesthesia with a MouseOx™ Pulse Oximeter (Harvard Apparatus, Holliston, MA). Non-invasive blood pressure was measured by using a CODA Monitor (Kent Scientific Corp., Torrington, CT). The isoflurane exposure was for 2 h. Rats were extubated when they were responsive. Animals in the isoflurane plus lidocaine group received lidocaine via a tail vein. Lidocaine was dissolved in normal saline at 8 mg/ml. The rats in the isoflurane only group were given the same volume of saline. All animals were recovered for 20 min in a chamber gassed with 100% O2 and at 37°C. They then were placed in their home cages.
2.3. Barnes maze
Two weeks after isoflurane exposure, animals were subjected to Barnes maze. Barnes maze is designed to test spatial learning and memory. Animals were placed in the middle of a circular platform with 20 equally spaced holes (SD Instruments, San Diego, CA). One of the holes was connected to a dark chamber that was called target box. Animals were encouraged to find this box by aversive noise (85 dB) and bright light (200 W) shed on the platform. Animals went through a spatial acquisition phase that took 4 days with 3 min per trial, 2 trials per day and 15 min between each trial. Animals then went through the reference memory phase to test the short-term retention on day 5 and long-term retention on day 12. One trial on each of these two days was performed. No test was performed during the period from day 5 to day 12. The latency and number of errors to find the target box during each trial were recorded with the assistance of ANY-Maze video tracking system (SD Instruments). The numbers of pokes in each hole during the reference memory phase also were recorded.
2.4. Fear conditioning
One day after Barnes maze test, rats were subjected to fear conditioning test. Fear conditioning is a very sensitive and non-effort-dependent test of learning and memory [22]. Each animal was placed in a test chamber wiped with 70% alcohol and subjected to 3 tone-foot shock pairings (tone: 2000 Hz, 85 db, 30 s; foot shock: 1 mA, 2 s) with an intertrial interval 1 min in a relatively dark room. Animal was removed from this test chamber 30 s after the conditioning training. The animal was placed back to the chamber 24 h later for 8 min in the absence of tone and shock. The amount of time with freezing behavior was recorded in an 8 s interval. The animal was placed 2 h later in a test chamber that had different context and smell from the first test chamber (this second chamber was wiped with 1% acetic acid) in a relatively light room. Freezing was recorded for 3 min without the auditory conditioning stimulus. The auditory stimulus then was turned on for 3 cycles, each cycle for 30 s followed by 1-min inter-cycle interval (4.5 min in total). The freezing behavior in the 4.5 min was recorded. The order of the context and tone fear conditioning tests was arranged in a way so that half of the animals in each group were tested first by contextual test and the other half by tone test. Freezing behavior recorded in the video was scored by an observer who was blind to group assignment. These tests test hippocampus-dependent (context-related) and hippocampus-independent (tone-related) learning and memory functions [22].
2.5. Brain tissue harvest
Thirty minutes after fear conditioning test, rats were deeply anesthetized with isoflurane and perfused transcardially with saline. Brains were removed. The right hippocampi were dissected out immediately for the Western blotting of various proteins and enzyme-linked immunosorbent assay (ELISA) of IL-1β and TNF-α. The left cerebral hemisphere at Bregma -2.5 to -3.5 and -3.5 to -4.5 was used for preparing sections for electronic microscopic examination and Nissl staining, respectively. Cerebral cortex from Bregma -1 to -3 was harvested for ELISA of Aβ and Western blotting of phospho-tau.
2.6. Nissl staining
The cerebral block containing hippocampus was fixed in 10% neutral buffered formalin overnight and then paraffin embedded. Coronal 8 μm sections were prepared and subjected to Nissl staining [15]. These sections were examined by an observer blinded to the group assignment of the sections. The number of positive cells (neurons) in a high magnification field (x 400, ~0.2 mm2) in the CA1 and CA3 regions was counted. Three determinations, each on different locations in these two brain regions, were performed for each section. Three sections were used for each rat. These 9 determinations were averaged to yield a single number (density of the neurons) for each brain region of individual rat.
2.7. Western blotting
Hippocampal tissues were homogenated in RIPA buffer (catalogue number: 89900; Thermo Scientific, Worcester, MA) containing protease inhibitor cocktail (catalogue number: P2714; Sigma, St Louis, MO) and protease inhibitor mixture (catalogue number: 1697498; Roche Applied Science, Indianapolis, IN). Homogenates were centrifuged at 13,000 g at 4°C for 30 min. The supernatant was saved and its protein concentration was determined by Bradford assay. To detect phospho-tau, total protein lysates from cerebral cortex were prepared in a lysis buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitors (Protease Inhibitor Cocktail; Sigma Aldrich, St Louis, MO) and phosphatase inhibitors (phosSTOP Phosphatase Inhibitor Cocktail Tablets; Roche, Nutley, NJ).
Fifty microgram proteins per lane were electrophoresed on a polyacrylamide gel and then blotted onto a polyvinylidene difluoride membrane. After being blocked with Protein-Free T20 Blocking Buffer (catalogue number: 37573; Thermo Scientific, Lot LB141635), membranes were incubated with the following primary antibodies: mouse monoclonal anti-synaptophysin antibody (1:15000; catalogue number: MAB329; Millipore Cor. Billerica, MA), rabbit polyclonal anti-drebrin antibody (1:1000; catalogue number: D3816; Sigma), mouse monoclonal anti-NeuN, clone A06 antibody (1:1000; catalogue number: MAB377; Millipore Cor.), mouse monoclonal anti-caspase-activated DNase (CAD) antibody (1:1000; catalogue number: sc-56035; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit monoclonal anti-cleaved caspase-3 antibody (1:1000; catalogue number: 9664; Cell Signaling Technology, Inc., Danvers, MA), rabbit polyclonal anti-tau [pS199/202] phosphospecific antibody (1:1000; catalogue number: 44768G; Invitrogen, Camarillo, CA); rabbit polyclonal anti-tau [pS396] phosphospecific antibody (1:1000; catalogue number: 4137; Invitrogen); mouse monoclonal anti-tau (tau5) antibody (1:1000; catalogue number: MA512805; Millipore Cor.); and mouse monoclonal anti-β-actin polyclonal antibody (1:2000; catalogue number: A2228; Sigma). Appropriate secondary antibodies were used. Proteins were visualized using a Genomic and Proteomic Gel Documentation (Gel Doc) Systems from Syngene (Frederick, MD). The protein band intensities of NeuN, caspase 3, CAD, synaptophysin and drebrin were normalized by the corresponding band intensities of β-actin from the same samples. The results from animals under various experimental conditions were then normalized by those of the corresponding control animals.
2.8. Quantification of IL-1β, TNFα and Aβ
Brain tissues were homogenized on ice in 20 mM Tris–HCl buffer (pH = 7.3) containing protease inhibitors (10 μg/ml aproteinin, 5 μg/ml peptastin, 5 μg/ml leupeptin, and 1 mM phenylmethanesulfonylfluoride). Homogenates were centrifuged at 10,000 g for 10 min at 4°C. The supernatant was then ultracentrifuged at 150,000 g for 2 h 4°C. Bradford protein assay of the supernatant was performed for each sample. ELISA kits for measuring rat IL-1β and TNFα (catalogue number: DY501 and RTA00, respectively; R&D Systems, Minneapolis, MN) were used to quantify the contents of these cytokines in the samples according to the manufacturer’ instructions. The quantity of IL-1β and TNFα in each brain sample was standardized to the protein contents.
Aβ was extracted from brain tissues as described before [23]. Briefly, parietal cerebral cortex was homogenized in 8 x volume of 70% glass distilled formic acid. The homogenates were kept at 4°C for 15 min and then centrifuged at 100,000 g for 1 h. The formic acid extract layer between a thin overlying lipid layer and a small pellet was removed and used for Aβ1-42 quantification by a Aβ1-42 ELISA kit (catalog number KMB3441; Invitrogen, Carlsbad, CA, USA). The level of the Aβ1-42 in each brain sample was standardized to the brain tissue weight and expressed as picogram of Aβ1-42 per gram brain tissues.
2.9. Synaptic density quantification
Coronary brain slices were fixed in 0.1 M cacodylate buffer containing 4% paraformaldehyde and 2.5% glutaraldehyde for at least 24 h. Vibratome sections of 400 μm were obtained and re-fixed in the above fixative solution overnight. Hippocampus was isolated and post-fixed with 1% OsO4 in 0.1 M cacodylate buffer for 1 h. Thick sections were obtained and stained with toludine blue. Stratum radiatum was identified, embedded in epoxy resin and used to prepare 85-nm ultra-thin sections. The sections were stained with 2% uranyl acetate and 0.25% lead citrate and observed under a JEOL 1230 transmission electron microscope (Japan Electron Optics Limited, Tokyo, Japan). Synapses in 40 randomly selected fields (1752 μm2) per animal were counted by an investigator who is blind to the group assignment of the animals [24]. This value then was used to calculate synaptic density in mm2. Both CA1 and CA3 are involved in fear conditioning [25-27]. We choose the stratum radiatum because it is in CA1 region and contains mossy fiber to CA3 and Shaeffer collaterals from CA3 to CA1.
2.10. Statistical analysis
Parametric results, such as physiological parameters and results from behavioral testing, western blotting and ELISA, are presented as means ± S.D. (n ≥ 4) and were analyzed by one way analysis of variance followed by the Tukey test after confirmation of normal distribution of the data, by Kruskal-Wallis analysis of variance on ranks followed by the Tukey or the Dunn's test when the data are not normally distributed, or by Student's t test as appropriate. The data of training sessions of Barnes maze were analyzed by one way repeated measures analysis of variance followed by the Tukey test. A P ≤ 0.05 was accepted as significant. All statistical analyses were performed with the SigmaStat (Systat Software, Inc., Point Richmond, CA, USA).
3. Results
Animals in the isoflurane and isoflurane plus lidocaine groups did not have any episodes of hypoxia (defined as SpO2 < 90%) during the exposure to isoflurane. The SpO2 was 98 ± 1% and 97 ± 1%, respectively, for the isoflurane and isoflurane plus lidocaine groups (n = 6 for each group, P = 0.142). The mean arterial blood pressures were 110 ± 6, 109 ± 20 and 114 ± 29 mmHg (n = 4 – 6, P = 0.912), respectively, for the control, isoflurane and isoflurane plus lidocaine groups. The corresponding heart rates were 373 ± 43, 352 ± 46 and 388 ± 17 beats/min (n = 4 – 6, P = 0.385). There was no difference in the mean arterial blood pressure and heart rates among the different groups. These measurements were obtained by non-invasive methods and performed when animals were awake (control group) or at ~1 h after the initiation of the isoflurane exposure for the isoflurane alone and isoflurane plus lidocaine groups.
All three groups of rats trended to take less time to find the target hole over the time during training sessions. Statistical results for the training sessions were F(7, 5) = 5.576 and P < 0.001 for the control group, F(7, 5) = 3.679 and P = 0.004 for the isoflurane exposure group, and F(7, 5) = 2.342 and P = 0.045 for the isoflurane plus lidocaine group. The latency to find the target hole in the last training session on the fourth day was significantly shorter than that in the first training session on the first day (Fig. 1), suggesting that animals in all three groups developed spatial learning. However, animals exposed to isoflurane took much longer to find the target hole and searched more non-target holes prior to identification of the target hole than control rats during the reference memory tests occurred 1 day or 8 days after the last training session. Animals in the isoflurane plus lidocaine group were not significantly different from control rats in these tests (Fig. 2). Statistical results were H(2) = 12.063 and P = 0.002 for the time to identify the target hole at 1 day after the last training session and F(2, 5) = 3.838 and P = 0.047 for the time to identify the target hole at 8 days after the last training session. Similar to these results, rats in the isoflurane group had less freezing behavior in the contextual fear conditioning test than control rats and this reduction was abolished by the use of lidocaine during isoflurane exposure. Statistical results were F(2, 5) = 20.175 and P < 0.001 for the comparisons. However, there was no difference in the freezing behavior among the three groups of rats during the tone-related fear conditioning test (Fig. 3). No animals from the three groups had a freezing event in the 3-min period prior to the application of the auditory conditioning stimulus during the tone-related fear conditioning test.
Fig. 1. Performance on Barnes maze in the training sessions.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. They were subjected to Barnes maze training sessions 2 weeks later. Results are mean ± S.D. (n = 6). * P < 0.05 compared with the corresponding data in the first trial on day 1.
Fig. 2. Performance on Barnes maze in the reference memory sessions.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. They were subjected to Barnes maze for testing reference memory 19 days (short-term memory, panels A and B) and 26 days (long-term memory, panels C and D) later. Results are mean ± S.D. (n = 6). * P < 0.05 compared with the control group. ^ P < 0.05 compared with isoflurane alone group.
Fig. 3. Performance on fear conditioning test.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. They were subjected to fear conditioning test for measuring contextual (panel A) and tone-related (panel B) fear conditioning. Results are mean ± S.D. (n = 5 - 6). * P < 0.05 compared with the control group. ^ P < 0.05 compared with isoflurane alone group.
There was no difference in the Aβ1-42 levels in the parietal cerebral cortex among the three groups of animals at 29 days after isoflurane exposure. There was also no difference in the IL-1β and TNFα concentrations in the hippocampus among the animals (Fig. 4). Compared with control, exposure to isoflurane or isoflurane plus lidocaine did not increase the expression of CAD and activated caspase 3 in the hippocampus (Fig. 5). Two caspase 3 fragments at 20 KDa and 17 KDa were detected. These fragments are subunits of activated caspase 3 [28]. CAD has a molecular weight ~40 KDa. Two prominent bands at ~39 and 41 KDa were detected by the monoclonal anti-CAD antibody and might represent different post-translational modifications. The final results presented in the bar graphs of figure 5 were the sum of the two fragments of caspase 3 and two protein bands for CAD.
Fig. 4. Expression of β amyloid peptide (Aβ), interleukin (IL) 1β and tumor necrosis factor (TNF)-α in rat brains.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. Aβ in the parietal cerebral cortex (panel A) and IL-1β (panel B) and TNFα (panel C) in the hippocampus were measured 29 days after the isoflurane exposure. Results are mean ± S.D. (n = 4 - 5).
Fig. 5. Expression of caspase-activated DNase (CAD) and activated caspase 3 in rat brains.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. The hippocampus was harvested at 29 days after the isoflurane exposure for Western analysis. A representative Western blot is shown in panel A and the graphic presentation of the CAD and activated caspase 3 protein abundance quantified by integrating the volume of autoradiograms from 4 to 5 animals is shown in panel B. Values in graphs are expressed as fold changes over the mean values of control animals and are presented as the means ± S.D.
Tau is a family of closely related proteins at 55 – 62 KDa [29]. At least three protein bands in this molecular weight range were detected with antibodies that were developed to specifically bind to tau or phosphorylated tau (Fig. 6). The quantification results described in this report were the sum of all tau protein bands. There was no significant difference in the total tau and tau phosphorylated at S199/202 or S396 in the cerebral cortex of the three groups of animals at 29 days after isoflurane exposure (Fig. 6).
Fig. 6. Expression of phosphorylated tau and total tau in rat cerebral cortex.

A representative Western blot is shown in panel A and the graphic presentation of protein abundance quantified by integrating the volume of autoradiograms from 4 rats for each experimental condition is shown in panel B. In panel B, the results of phosphorylated tau and total tau were normalized by the corresponding actin data and then expressed as fold change over the mean value of the control rats. Results are means ± S.D.
Isoflurane exposure significantly increased the activated caspase 3 expression (mostly the 20 KDa fragment) in the hippocampus at 6 h after the exposure. This increase was attenuated by lidocaine application. Statistical results for these comparisons were F(2, 3) = 8.658 and P = 0.008. However, the activated caspase 3 expression in the cerebral cortex was not different among the three groups of animals at this time point (Fig. 7).
Fig. 7. Effects of isoflurane on the expression of activated caspase 3 in rat brains.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. The hippocampus was harvested at 6 h after the isoflurane exposure for Western analysis. A representative Western blot is shown in panel A and the graphic presentation of the activated caspase 3 protein abundance quantified by integrating the volume of autoradiograms from 4 animals is shown in panel B. Values in graphs are expressed as fold changes over the mean values of control animals and are presented as the means ± S.D. * P < 0.05 compared with the control group. ^ P < 0.05 compared with isoflurane alone group.
All three groups of rats had similar neuronal density in the CA1 and CA3 regions at 29 days after isoflurane exposure (Fig. 8). Protein abundance of NeuN (a neuronal protein), drebrin (a dendritic spine protein) and synaptophysin (a synaptic protein) in the hippocampus of all three groups of rats were similar (Fig. 9). The synaptic density in the hippocampal stratum radiatum was not different among the three groups of animals (Fig. 10).
Fig. 8. Neuronal density in the hippocampus.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. The hippocampus was harvested at 29 days after the isoflurane exposure for Nissl staining to determine neuronal density in the CA1 (panel A) and CA3 (panel B) regions. Results are mean ± S.D. (n = 6).
Fig. 9. Expression of NeuN, drebrin and synaptophysin in rat brains.

Eighteen-month-old Fisher 344 rats were exposed to or not exposed to 1.2% isoflurane in the presence or absence of lidocaine for 2 h. The hippocampus was harvested at 29 days after the isoflurane exposure for Western analysis. A representative Western blot is shown in panel A and the graphic presentation of the NeuN, drebrin and synaptophysin protein abundance quantified by integrating the volume of autoradiograms from 4 to 5 animals is shown in panel B. Values in graphs are expressed as fold changes over the mean values of control animals and are presented as the means ± S.D.
Fig. 10. Synaptic density in the hippocampus.
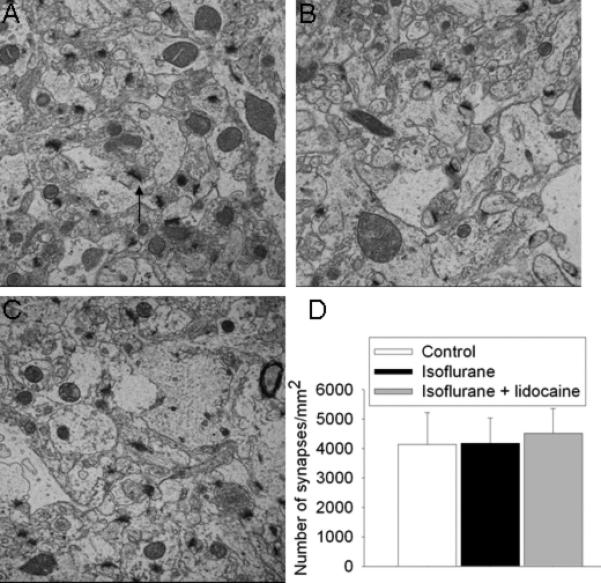
Electron microscopic images of the stratum radiatum from 18-month-old Fisher 344 rats that were control (A) and 29 days after isoflurane exposure (B) or isoflurane plus lidocaine exposure (C). An arrow is placed in panel A to indicate a typical synapse. Other synapses are not marked. Panel D is the quantitative results in means ± S.D. (n = 4 - 6).
4. Discussion
POCD can occur after cardiac and non-cardiac surgeries. Multiple studies have shown that aging is a risk factor for POCD [2,3,30,31]. To make things worse, elderly patients also have higher chances of having surgery than younger patients [32]. Since most patients have surgeries under general anesthesia, general anesthetics have been suspected to contribute to the development of POCD. Up till now, there is no clear clinical evidence for this suspicion because patients under general anesthesia for surgery appear to have a similar rate of POCD to patients under regional/neuraxial blockade [33,34]. However, patients under regional/neuraxial anesthesia often receive sedatives during surgery, which may reduce the differences between general anesthesia and regional/neuraxial anesthesia. In recent years, there is an increasing concern that general anesthesia/anesthetics may contribute to POCD because laboratory research has shown that animals exposed to general anesthetics develop learning and memory impairments and neuropathology [7,11,35], although volatile anesthetics also have been reported to improve cognitive functions of young adult rodents [7,9,10].
We used 18-month old rats in this study. Rats at this age may be considered as in their early elderly stage or late phase of middle age. Exposure of these rats to 1.2% isoflurane for 2 h resulted in a poorer performance on spatial reference memory tasks in Barnes maze test. Consistent with our results, previous studies have shown that exposure of rats to general anesthetics impairs their spatial memory [7,8,35]. Our study also showed that animal exposed to isoflurane had less freezing behavior in the contextual fear conditioning test. Since contextual fear conditioning is considered to be hippocampus-dependent [22], our results suggest that isoflurane impairs hippocampus-dependent learning and memory in rats. Interestingly, intravenous lidocaine attenuated isoflurane-induced learning and memory impairments. This attenuation was very significant in the contextual fear conditioning results and when the reference memory was tested at 1 day after the training sessions in the Barnes maze test. The attenuation did not reach statistical significance when reference memory was tested at 8 days after the training sessions in the Barnes maze. Lidocaine is a commonly used local anesthetic. Intravenous lidocaine infusion has been used in clinical practice for many years to reduce the required concentrations of general anesthetics during surgery [17]. The novel lidocaine effect identified in this study can be readily applied to clinical practice if this effect is confirmed in humans.
We designed our study to be clinically relevant. One minimum alveolar concentration (MAC, the concentration at which 50% of animals do not have a motor response to painful stimuli) of isoflurane in rats is ~1.5% [36]. Concentrations between 0.5 to 1.3 MACs are commonly used in the clinical practice. We started to test cognitive functions of the animals at 2 weeks after isoflurane exposure. Many patients may have been discharged home by this time. Normal cognitive functions are very important for the daily activity of patients at home.
The mechanisms for volatile anesthetic-induced cognitive impairment are not known. One school of thought is that this cognitive impairment may be due to Aβ accumulation in the brain and its subsequent neurotoxicity because these mechanisms have been hypothesized for the development of AD [13]. A few lines of evidence support this possibility. Volatile anesthetics at clinically relevant concentrations can induce Aβ oligomerization in vitro [12]. Volatile anesthetics increase Aβ formation in cell cultures [37]. A recent study showed that exposure of mice (the age of the mice was not specified in the paper) to 1.4% isoflurane for 2 h increased the expression of Aβ in their brains (cerebral cortex) assessed at 24 h after isoflurane exposure [11]. However, previous studies have not provided evidence to support the connection between Aβ accumulation and cognitive impairment because no study has measured cognitive impairments and Aβ production and accumulation in the brains of the same wild-type animals to determine the correlation between them after anesthetic exposure. Our study showed that isoflurane impaired the learning and memory functions but did not affect the Aβ concentrations in the cerebral cortex. We measured Aβ concentrations at a time when animals had significant cognitive impairments. Thus, our results do not indicate a role of Aβ production and accumulation in isoflurane-induced cognitive impairments in the elderly rats. Consistent with this suggestion, our results showed that the Aβ1-42 is in picomolar levels in the brains of the Fisher 344 rats, which is similar to those measured in nontransgenic mouse brains [38,39] and is much lower than the concentrations used in the in vitro studies showing Aβ cytotoxicity (often in micromolar levels) [12,40].
Associated with the Aβ theory, it has been proposed that brain cell death after anesthetic exposure may contribute to the brain functional changes [1]. Isoflurane has been shown to increase activation of caspase 3, a key enzyme to induce apoptosis, in both cultured cells and mouse brains [11,37]. This increased activated caspase 3 may increase Aβ expression, which then causes further activation of caspase 3 [37]. This vicious cycle may ultimately result in brain cell death. In our study, isoflurane exposure increased activated caspase 3 expression in the hippocampus at 6 h after the exposure and lidocaine attenuated this increase. Interestingly, there was no difference in the activated caspase 3 expression in the cerebral cortices of the three groups of rats. Similar to our situation, toxic effects of drugs administrated systemically on specific brain regions and cell types have been reported before [41]. Since the behavioral data of our study indicate that isoflurane impairs hippocampus-dependent learning and memory and lidocaine attenuated this impairment, our results suggest that the possible cell injury after isoflurane exposure may contribute to its effects on learning and memory.
Accumulation of hyperphosphorylated tau that forms intracellular neurofibrillary tangles is a pathological feature of AD brains [42]. A previous study showed that adult mice after isoflurane anesthesia had increased phospho-tau in their brains. However, this increased phospho-tau was found to be due to anesthesia-induced hypothermia, not due to direct anesthetic effects [43]. Consistent with this finding, we showed here that the phospho-tau was not increased after isoflurane or isoflurane plus lidocaine exposure.
Our current study showed that isoflurane did not change the neuronal density of CA1 and CA3 regions and the expression of hippocampal NeuN, drebrin and synaptophysin assessed at 29 days after isoflurane exposure. These negative results may not be due to the insensitivity of these measurements because we have detected a significant reduction of neuronal density in the hippocampi of young adult rats at 29 days after isoflurane exposure [44]. NeuN is a protein specifically expressed in neurons. Drebrin is a dendritic spine protein and synaptophysin is a synaptic protein. Consistent with these biochemical data, isoflurane exposure did not change the synaptic density in the hippocampus. These results suggest that isoflurane does not cause a significant change in the hippocampal neuronal structures. Our study also showed that isoflurane exposure did not increase cell injury at 29 days after isoflurane exposure because the expression of activated caspase 3 and CAD in animals exposed to isoflurane was not different from that in control animals at this time point. These results suggest that the possible acute cell injury detected at 6 h after isoflurane exposure may not last for a long time and may not significantly change the hippocampal neuronal structures.
In addition to being a local anesthetic, lidocaine has anti-inflammatory property [19]. Since lidocaine attenuated isoflurane-induced cognitive impairment, it is possible that isoflurane induces neuroinflammation that then leads to cognitive dysfunction. Consistent with this idea, it is known that neuroinflammation can cause cognitive impairments [14]. A recent study showed that tibial surgery under isoflurane anesthesia induced a rapid increase of IL-1β in the brain and hippocampal inflammation within 3 days after the surgery in young adult mice. These animals also had significant cognitive impairments assessed at 3 days after the surgery and anesthesia [45]. However, isoflurane anesthesia alone did not increase IL-1β level and cause inflammation in the hippocampus in the same study [45]. Our study showed that isoflurane anesthesia did not change IL-1β and TNFα concentrations in the hippocampus at a time point when animals had significant hippocampus-dependent learning and memory impairments. Thus, neuroinflammation may not be a mechanism for isoflurane-induced cognitive impairment detected in a delayed phase in our study.
Of note, significant cell death was not shown in the brains of 16-month old rats at 16 h after isoflurane exposure [36,46]. Isoflurane did not affect the rats’ learning and memory functions assessed at 4 months after the exposure in the same study. These findings seem to be different from our results and findings of others [7,8,11,35]. The reasons for this discrepancy are not known but may be due to the assessment of cell injury and cognitive functions at different times.
Our study has limitations. First, since intravenous lidocaine reduces the required concentrations of general anesthetics for maintaining anesthesia, animals in the isoflurane group may have lighter general anesthesia than animals in the isoflurane plus lidocaine group. This difference in the depth of general anesthesia may have contributed to the different cognitive functions of these two groups of animals. However, evidence for the influence of depth of general anesthesia in POCD is inconsistent. There is at least one study showing that patients under lighter general anesthesia have somewhat worse cognitive functions [47]. Opposite results are shown in another study [48]. There are more studies showing that there is no clear relationship between the depths of general anesthesia and POCD [49,50]. Since one of our goals for this study was to determine whether lidocaine reduced isoflurane-induced cognitive impairment, the same concentration of isoflurane was used for animals in the isoflurane or isoflurane plus lidocaine groups. Second, we did not include a group of rats receiving lidocaine only. Intravenous infusion of lidocaine for 2 h in animals will be difficult to perform unless the animals are anesthetized, physically restrained or implanted with an infusion pump under anesthesia. This requirement will invalidate the usefulness of this group of animals. The lack of lidocaine only group may affect the interpretation of possible mechanisms for the observed lidocaine protection. However, there is no evidence yet that lidocaine can improve cognitive functions of control animals in the literature. Also, it is unlikely that attenuation of cell death or IL-1β and TNFα production plays a role in the lidocaine-induced improvement of cognitive functions of 18-month old rats after isoflurane anesthesia because there is no difference in the levels of IL-1β, TNFα and cell death among the three groups of rats at a time when cognitive impairments are present. These results also suggest that lidocaine does not affect the baseline level of cytokines and markers of cell injury, which indicates a non-critical need for having the lidocaine alone group in our study. Also, identifying a method to reduce isoflurane-induced cognitive impairment is one of the major goals for this study. Thus, we did not perform the study on lidocaine alone group. Finally, we detected significant isoflurane-induced cognitive impairment and lidocaine protection against this impairment. However, we did not found any changes in the parameters measuring neuropathological or neurochemical status. There were at least 4 animals per group in these studies. If there are three groups, each group has 4 animals and the difference in means among groups is 50% with an expected standard deviation of 20%, the power to detect a significant difference (α level at 0.05) is 0.765. Thus, one possible reason for no detectable changes in the neuropathological and neurochemical parameters is that our study is underpowered because the effects size in these parameters is smaller than 50%. However, our results on these parameters did not suggest a trend for the changes that are consistent with the changes detected in the cognitive functions among the three groups of rats. Nevertheless, our study may be underpowered to detect the lidocaine protection in the long-term memory experiment of Barnes maze.
In conclusion, we have shown that isoflurane at a clinically-relevant concentration can induce learning and memory impairment in old rats. This detrimental effect is attenuated by lidocaine co-administrated during the isoflurane exposure. If these findings are confirmed in humans, lidocaine could become a useful drug to reduce cognitive impairment in patients after anesthesia and surgery.
Research highlight.
The volatile anesthetic isoflurane impairs cognitive function of 18-month old rats.
Lidocaine, a local anesthetic, attenuates the above isoflurane effects
Isoflurane may not cause significant long-term neuropathological changes
Acknowledgement
This study was supported by a grant (R01 GM065211 to Z Zuo) from the National Institutes of Health, Bethesda, Maryland, by a grant from the International Anesthesia Research Society (2007 Frontiers in Anesthesia Research Award to Z Zuo), Cleveland, Ohio, by a Grant-in-Aid from the American Heart Association Mid-Atlantic Affiliate (10GRNT3900019 to Z Zuo), Baltimore, Maryland and the Robert M. Epstein Professorship endowment, University of Virginia.
Abbreviations
- Aβ
β-amyloid peptide
- AD
Alzheimer's disease
- CAD
caspase-activated DNase
- ELISA
enzyme-linked immunosorbent assay
- IL-1β
interleukin 1β
- POCD
post-operative cognitive dysfunction
- SpO2
pulse oximeter oxygen saturation
- TNF-α
tumor necrosis factor-α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baranov D, Bickler PE, Crosby GJ, Culley DJ, Eckenhoff MF, Eckenhoff RG, et al. Consensus statement: First International Workshop on Anesthetics and Alzheimer's disease. Anesth Analg. 2009;108:1627–30. doi: 10.1213/ane.0b013e318199dc72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, et al. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet. 1998;351:857–61. doi: 10.1016/s0140-6736(97)07382-0. [DOI] [PubMed] [Google Scholar]
- 3.Monk TG, Weldon BC, Garvan CW, Dede DE, van der Aa MT, Heilman KM, et al. Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology. 2008;108:18–30. doi: 10.1097/01.anes.0000296071.19434.1e. [DOI] [PubMed] [Google Scholar]
- 4.Newman MF, Kirchner JL, Phillips-Bute B, Gaver V, Grocott H, Jones RH, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med. 2001;344:395–402. doi: 10.1056/NEJM200102083440601. [DOI] [PubMed] [Google Scholar]
- 5.Steinmetz J, Christensen KB, Lund T, Lohse N, Rasmussen LS. Long-term consequences of postoperative cognitive dysfunction. Anesthesiology. 2009;110:548–55. doi: 10.1097/ALN.0b013e318195b569. [DOI] [PubMed] [Google Scholar]
- 6.Clergue F, Auroy Y, Pequignot F, Jougla E, Lienhart A, Laxenaire MC. French survey of anesthesia in 1996. Anesthesiology. 1999;91:1509–20. doi: 10.1097/00000542-199911000-00045. [DOI] [PubMed] [Google Scholar]
- 7.Culley DJ, Baxter M, Yukhananov R, Crosby G. The memory effects of general anesthesia persist for weeks in young and aged rats. Anesth Analg. 2003;96:1004–9. doi: 10.1213/01.ANE.0000052712.67573.12. [DOI] [PubMed] [Google Scholar]
- 8.Culley DJ, Baxter MG, Crosby CA, Yukhananov R, Crosby G. Impaired acquisition of spatial memory 2 weeks after isoflurane and isoflurane-nitrous oxide anesthesia in aged rats. Anesth Analg. 2004a;99:1393–7. doi: 10.1213/01.ANE.0000135408.14319.CC. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu H, Nogaya J, Anabuki D, Yokono S, Kinoshita H, Shirakawa Y, et al. Memory facilitation by posttraining exposure to halothane, enflurane, and isoflurane in ddN mice. Anesth Analg. 1993;76:609–12. doi: 10.1213/00000539-199303000-00028. [DOI] [PubMed] [Google Scholar]
- 10.Rammes G, Starker LK, Haseneder R, Berkmann J, Plack A, Zieglgansberger W, et al. Isoflurane anaesthesia reversibly improves cognitive function and long-term potentiation (LTP) via an up-regulation in NMDA receptor 2B subunit expression. Neuropharmacology. 2009;56:626–36. doi: 10.1016/j.neuropharm.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Xie Z, Culley DJ, Dong Y, Zhang G, Zhang B, Moir RD, et al. The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann Neurol. 2008;64:618–27. doi: 10.1002/ana.21548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, et al. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703–9. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–38. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- 14.Sanderson DJ, Cunningham C, Deacon RM, Bannerman DM, Perry VH, Rawlins JN. A double dissociation between the effects of sub-pyrogenic systemic inflammation and hippocampal lesions on learning. Behav Brain Res. 2009;201:103–11. doi: 10.1016/j.bbr.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 15.Zhao P, Zuo Z. Isoflurane preconditioning induces neuroprotection that is inducible nitric oxide synthase-dependent in the neonatal rats. Anesthesiology. 2004;101:695–702. doi: 10.1097/00000542-200409000-00018. [DOI] [PubMed] [Google Scholar]
- 16.Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. 2001;22:903–8. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 17.Himes RS, Jr., DiFazio CA, Burney RG. Effects of lidocaine on the anesthetic requirements for nitrous oxide and halothane. Anesthesiology. 1977;47:437–40. doi: 10.1097/00000542-197711000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Flondor M, Listle H, Kemming GI, Zwissler B, Hofstetter C. Effect of inhaled and intravenous lidocaine on inflammatory reaction in endotoxaemic rats. Eur J Anaesthesiol. 2010;27:53–60. doi: 10.1097/EJA.0b013e32832b8a70. [DOI] [PubMed] [Google Scholar]
- 19.Caracas HC, Maciel JV, Martins PM, de Souza MM, Maia LC. The use of lidocaine as an anti-inflammatory substance: a systematic review. J Dent. 2009;37:93–7. doi: 10.1016/j.jdent.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Lei B, Cottrell JE, Kass IS. Neuroprotective effect of low-dose lidocaine in a rat model of transient focal cerebral ischemia. Anesthesiology. 2001;95:445–51. doi: 10.1097/00000542-200108000-00029. [DOI] [PubMed] [Google Scholar]
- 21.Lee JJ, Li L, Jung H-H, Zuo Z. Postconditioning with isoflurane reduced ischemia-induced brain injury in rats. Anesthesiology. 2008;108:1055–62. doi: 10.1097/ALN.0b013e3181730257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–7. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- 23.Gravina S, Ho L, Eckman C, Long K, Otvos L, Jr, Younkin L, et al. Amyloid beta protein in Alzheimer's disease brain. J Biol Chem. 1995;270:7013, 7–16. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 24.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunsaker MR, Tran GT, Kesner RP. A behavioral analysis of the role of CA3 and CA1 subcortical efferents during classical fear conditioning. Behav Neurosci. 2009;123:624–30. doi: 10.1037/a0015455. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Zhang S, Zhu Y, Fu Q, Gong Y, Ohtsu H, et al. Improved learning and memory of contextual fear conditioning and hippocampal CA1 long-term potentiation in histidine decarboxylase knock-out mice. Hippocampus. 2007;17:634–41. doi: 10.1002/hipo.20305. [DOI] [PubMed] [Google Scholar]
- 27.Rogers JL, Hunsaker MR, Kesner RP. Effects of ventral and dorsal CA1 subregional lesions on trace fear conditioning. Neurobiol Learn Mem. 2006;86:72–81. doi: 10.1016/j.nlm.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 29.Hirokawa N, Shiomura Y, Okabe S. Tau proteins: the molecular structure and mode of binding on microtubules. J Cell Biol. 1988;107:1449–59. doi: 10.1083/jcb.107.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ancelin ML, de Roquefeuil G, Ledesert B, Bonnel F, Cheminal JC, Ritchie K. Exposure to anaesthetic agents, cognitive functioning and depressive symptomatology in the elderly. Br J Psychiatry. 2001;178:360–6. doi: 10.1192/bjp.178.4.360. [DOI] [PubMed] [Google Scholar]
- 31.Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, et al. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology. 2002;96:1351–7. doi: 10.1097/00000542-200206000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Hall MJ, Kozak LJ. Ambulatory and inpatient surgery: national patterns for the elderly. Stat Bull Metrop Insur Co. 1999;80:22–31. [PubMed] [Google Scholar]
- 33.Williams-Russo P, Sharrock NE, Mattis S, Szatrowski TP, Charlson ME. Cognitive effects after epidural vs general anesthesia in older adults. A randomized trial. JAMA. 1995;274:44–50. [PubMed] [Google Scholar]
- 34.Nielson WR, Gelb AW, Casey JE, Penny FJ, Merchant RN, Manninen PH. Long-term cognitive and social sequelae of general versus regional anesthesia during arthroplasty in the elderly. Anesthesiology. 1990;73:1103–9. doi: 10.1097/00000542-199012000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Culley DJ, Baxter MG, Yukhananov R, Crosby G. Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology. 2004b;100:309–14. doi: 10.1097/00000542-200402000-00020. [DOI] [PubMed] [Google Scholar]
- 36.Stratmann G, Sall JW, Bell JS, Alvi RS, May LV, Ku B, et al. Isoflurane does not affect brain cell death, hippocampal neurogenesis, or long-term neurocognitive outcome in aged rats. Anesthesiology. 2010;112:305–15. doi: 10.1097/ALN.0b013e3181ca33a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Z, Dong Y, Maeda U, Moir RD, Xia W, Culley DJ, et al. The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J Neurosci. 2007;27:1247–54. doi: 10.1523/JNEUROSCI.5320-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–53. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu J, Igarashi A, Kamata M, Nakagawa H. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta ); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–8. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- 41.Noguchi KK, Walls KC, Wozniak DF, Olney JW, Roth KA, Farber NB. Acute neonatal glucocorticoid exposure produces selective and rapid cerebellar neural progenitor cell apoptotic death. Cell Death Differ. 2008;15:1582–92. doi: 10.1038/cdd.2008.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 43.Planel E, Bretteville A, Liu L, Virag L, Du AL, Yu WH, et al. Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB J. 2009;23:2595–604. doi: 10.1096/fj.08-122424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin D, Zuo Z. Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology. 2011;61:1354–9. doi: 10.1016/j.neuropharm.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, et al. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68:360–8. doi: 10.1002/ana.22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stratmann G, Sall JW, May LD, Bell JS, Magnusson KR, Rau V, et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 2009;110:834–48. doi: 10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 47.Farag E, Chelune GJ, Schubert A, Mascha EJ. Is depth of anesthesia, as assessed by the Bispectral Index, related to postoperative cognitive dysfunction and recovery? Anesth Analg. 2006;103:633–40. doi: 10.1213/01.ane.0000228870.48028.b5. [DOI] [PubMed] [Google Scholar]
- 48.Jildenstal PK, Hallen JL, Rawal N, Gupta A, Berggren L. Effect of Auditory Evoked Potential-Guided Anaesthesia on Consumption of Anaesthetics and Early Postoperative Cognitive Dysfunction: a randomised controlled trial. Eur J Anaesthesiol. 2011;28:213–9. doi: 10.1097/EJA.0b013e328340dbb9. [DOI] [PubMed] [Google Scholar]
- 49.Steinmetz J, Funder KS, Dahl BT, Rasmussen LS. Depth of anaesthesia and post-operative cognitive dysfunction. Acta Anaesthesiol Scand. 2010;54:162–8. doi: 10.1111/j.1399-6576.2009.02098.x. [DOI] [PubMed] [Google Scholar]
- 50.Wong J, Song D, Blanshard H, Grady D, Chung F. Titration of isoflurane using BIS index improves early recovery of elderly patients undergoing orthopedic surgeries. Can J Anaesth. 2002;49:13–8. doi: 10.1007/BF03020413. [DOI] [PubMed] [Google Scholar]