

Abstract
There is an increasing awareness that diabetes has an impact on the CNS and that diabetes is a risk factor for Alzheimer’s disease (AD). Links between AD and diabetes point to impaired insulin signaling as a common mechanism leading to defects in the brain. However, diabetes is predominantly characterized by peripheral, rather than central, neuropathy and despite the common central mechanisms linking AD and diabetes, little is known about the effect of AD on the PNS. In this study, we compared indices of peripheral neuropathy and investigated insulin signaling in the sciatic nerve of insulin-deficient mice and APP overexpressing transgenic mice. Insulin-deficient and APP transgenic mice displayed similar patterns of peripheral neuropathy with decreased motor nerve conduction velocity, thermal hypoalgesia and loss of tactile sensitivity. Phosphorylation of the insulin receptor and GSK3β were similarly affected in insulin-deficient and APP transgenic mice despite significantly different blood glucose and plasma insulin levels and nerve of both models showed accumulation of Aβ-immunoreactive protein. Although diabetes and AD have different primary etiologies, both diseases share many abnormalities in both the brain and the PNS. Our data point to common deficits in the insulin-signaling pathway in both neurodegenerative diseases and support the idea that AD may cause disorders outside the higher CNS.
Keywords: Alzheimer’s disease, Amyloid precursor protein, Diabetes, Insulin signaling, Sciatic nerve, diabetic neuropathy
Alzheimer’s disease (AD) is characterized clinically by progressive cognitive impairment, and pathologically by the presence of extracellular senile plaques and intracellular neurofibrillary tangles in the brain (Terry, 2006). Senile plaques are largely composed of amyloid β (Aβ) derived from the amyloid precursor protein (APP) while neurofibrillary tangles consist mostly of hyperphosphorylated tau. Numerous murine models of AD are available that are essentially based on mutated genes detected in the human form of the disease. APP is one of the favored genes used to generate murine models of AD and mice overexpressing APP develop cognitive deficits and amyloid plaques in the brain (for review see McGowan et al., 2006).
Diabetes mellitus is characterized by disturbances of the insulin-signaling pathway, which result from hypoinsulinemia in type 1 diabetes, or from insulin resistance and impaired insulin secretion in type 2 diabetes. There is an increasing awareness that diabetes has an impact on the central nervous system (CNS), with reports of impaired learning, memory, mental flexibility and problem solving being more common in both type 1 and type 2 diabetic subjects than in the general population (Ryan et al., 1985, Reaven et al., 1990, Ryan and Williams, 1993, McCarthy et al., 2002, Desrocher and Rovet, 2004, Cukierman et al., 2005, Manschot et al., 2006, Biessels et al., 2008). We have previously shown that mice with systemic insulin deficiency display evidences of reduced insulin-signaling pathway activity in the brain that are associated with biochemical and behavioral features of AD, including increased Aβ protein and tau phosphorylation levels, and concurrent learning deficits (Jolivalt et al., 2008). More recently, we demonstrated that type 1 diabetes exacerbates the development of AD features in the brain of an AD mouse model (Jolivalt et al., 2010), confirming the increasing number of studies demonstrating diabetes as a risk factor for AD (Yoshitake et al., 1995, Ott et al., 1996, Leibson et al., 1997, Peila et al., 2002, Ronnemaa et al., 2009, Xu et al., 2009, Valente et al., 2010, Velayudhan et al., 2010).
However, the most common neuropathy resulting from diabetes is peripheral neuropathy, described as a distal symmetrical polyneuropathy (Thomas, 1997). In diabetic patients, indices of peripheral neuropathy range from hyperalgesia and allodynia to progressive hypoalgesia and loss of sensation while the most widely studied functional disorder is nerve conduction velocity slowing (Greene et al., 1999). Diabetic peripheral neuropathy can be modeled in rodents (Calcutt, 2004). Insulin-deficient diabetic mice develop hypoalgesia (Beiswenger et al., 2008, McGuire et al., 2009) and nerve conduction velocity slowing (McGuire et al., 2009, Obrosova, 2009). While the impact of diabetes across all regions of the nervous system is becoming accepted, little is known about potential effects of AD on the peripheral nervous system (PNS) other than occasional reports describing pain in AD patients that is usually underdiagnosed (Scherder and van Manen, 2005, Scherder et al., 2005, Belin and Gatt, 2006). Since the brains of diabetic and AD mice show some similar pathological features (Jolivalt et al., 2008), we have now extended our comparative study of diabetes and AD to the PNS. Similar patterns of nerve injury were detected in all the parameters measured in 12-week insulin-deficient diabetic mice and age-matched AD mice suggesting that common pathogenic mechanisms may affect the PNS in mouse models of diabetes and AD.
Experimental Procedures
1.1. Animals
Animals used in this study were concurrently assessed for learning deficits and brain biochemistry as previously described (Jolivalt et al., 2010), while PNS changes are described here. Briefly, we used hAPP transgenic mice that express mutated (London V717I and Swedish K670M/N671L) hAPP751 under the control of the neuronal murine (m)Thy-1 promoter (Rockenstein et al., 2001a) and their non-transgenic littermates (wild type: WT). Animals were housed 4-5 per cage with free access to food and water and maintained in a vivarium approved by the American Association for the Accreditation of Laboratory Animal Care. All animal studies were carried out according to protocols approved by the Institutional Animal Care and Use Committee of the University of California San Diego. Six to 8 mice were used per group.
1.2. Induction of diabetes
Insulin-deficient diabetes was induced in 4-month-old wild type mice by intraperitoneal (i.p.) injection of 90 mg/kg streptozotocin (STZ, Sigma. St, Louis, MO, dissolved in 0.9% sterile saline) on 2 successive days following an overnight fast. Hyperglycemia was confirmed using a strip-operated reflectance meter in a blood sample obtained by tail prick 4 days after STZ injection and in another sample collected at the conclusion of the study. Plasma insulin levels were also measured at the end of the study using a mouse ultrasensitive insulin ELISA (Mercodia, Winston-Salem, NC, USA).
1.3. Tactile response threshold
Tactile allodynia was assessed using von Frey filaments as described in detail elsewhere (Calcutt and Chaplan, 1997). Briefly, a series of filaments with a range of buckling forces were sequentially applied to the plantar surface of the mouse hindpaw following the up and down method of (Dixon, 1980). Data are represented as 50% paw withdrawal thresholds (PWT) (Chaplan et al., 1994).
1.4. Paw thermal response latency
Mice were placed in an observation chamber on top of the thermal testing apparatus (UARD, San Diego, CA, USA) and allowed to acclimate to the warmed glass surface (30°C) and surroundings for 30 minutes. The mobile heat source was placed below the center of the hind paw and turned on, activating a timer and locally warming the glass surface. Paw withdrawal triggered movement sensors that stopped the timer and turned off the heat source. Both paws were measured and the mean used as a composite score for each mouse. To avoid tissue damage, the upper cut-off limit was set at 20 seconds. The response latency was converted to the response temperature using a withdrawal time to floor temperature calibration curve (Calcutt, 2004).
1.5. Nerve conduction velocity
Under isoflurane anesthesia, core temperature was maintained at 37 °C using a heated pad. The sciatic nerve was stimulated (5 V, 0.05 ms pulse, square wave) with needle electrodes at the sciatic notch or ankle. Evoked early (M wave) responses were recorded from interosseous muscles of the foot with fine needle electrodes, amplified and displayed on an oscilloscope. The difference in response latencies of the M wave after stimulation at the sciatic notch and ankle was recorded as the time required for the motor nerve conduction between those 2 sites. The distance between stimulation sites was measured on fully extended hind limb and divided by the M-wave latencies difference to calculate sciatic motor nerve conduction velocity (MNCV).
1.6. Tissue preparation for western blot analysis
Sciatic nerves were homogenized in buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 0.5% Triton X, 1 mM EDTA, protease inhibitor cocktail). Homogenates were centrifuged at 13,000 g for 30 min and supernatants stored in aliquots at -80°C. Protein concentration was assessed using the bicinchoninic acid method (BCA protein assay kit, Pierce, Rockford, IL, USA).
1.7. Western blotting
Aliquots of the sciatic nerve homogenates were boiled in Laemmli LDS sample buffer (Invitrogen, Carlsbad, CA, USA). Up to twenty μg of total extract protein were separated on 4-12% SDS-PAGE Bis-Tris gels (Novex, Invitrogen, Carlsbad, CA, USA) and immunoblotted on nitrocellulose. To maximize the number of proteins analyzed by Western blot, membranes were cut along the molecular markers into strips containing the proteins of interest. Blot strips were incubated with antibodies against phospho-insulin receptor (phosphorylated Ser 972, 1/1200, Upstate, Temecula, CA, USA), insulin receptor (1/200, Chemicon International, Temecula, CA, USA), phospho-GSK3β (phospho-Ser9; 1/1000, Cell Signaling technology, USA), GSK3α/β (1/5000, Chemicon International, Temecula, CA, USA), full-length APP (FL-APP, mouse monoclonal, clone 22C11, 1/10,000, Chemicon International, Temecula, CA, USA) or Aβ (mouse monoclonal, clone 6E10, 1/1000, Covance/Signet Laboratories, Berkeley, CA, USA) followed by secondary antibodies tagged with horseradish peroxidase (HRP, 1/10000, Santa Cruz Biotechnology, Inc., CA, USA). Blots were developed as previously described (Jolivalt et al., 2008). For sequential analysis of Western blot membranes, previously bound antibodies were removed with Restore Stripping buffer (Pierce, Rockford, Il, USA). Quantification of immunoreactivity was performed by densitometric scanning using Quantity One software (BioRad, San Diego, CA, USA). For each animal, band intensities were normalized by calculating the ratio of the intensity of the band corresponding to primary antigen of interest to the intensity of the band corresponding to actin. The limited amount of sciatic nerve available from each mouse meant that we loaded variable amounts of total protein to each lane to maximize signal. This lead to variable actin signal per lane but did not impede our capacity to normalize other proteins to the amount of actin present for any given sample.
1.8. PGP9.5 staining
Foot skin, obtained from the region where thermal withdrawal latency measurements were made, was fixed for 24 h in 4% paraformyldehyde in 0.1 M phosphate buffer. Samples were embedded in paraffin and cut at a thickness of 6 μm. Sections were collected onto glass slides and incubated with an antibody against the pan-neuronal marker PGP9.5 (1/1000, Biogenesis Ltd., UK) to visualize innervation of the skin. Using a light microscope (400× magnification), intraepidermal nerve fiber (IENF) and subepidermal nerve plexus (SNP) immunoreactive profiles were counted using slides coded to obscure group identity. Epidermal length was determined by drawing a line along the stratum granulosum of the epidermis and recording its length in millimeters. Epidermal thickness was measured from the stratum basale to the stratum granulosum every 0.2-0.3 mm along the length of the section. The number of IENF profiles was normalized to epidermal area to limit any contribution of variations in epidermis thickness caused by diabetes (Beiswenger et al., 2008b) while number of SNP profiles was normalized to epidermal length.
1.9. Statistical analysis
Data are presented as group mean+SEM for figures 1, 2 and 4 and as scatter plots of ratio of intensities for individual animals in figures 5, 6 and 7. Differences between groups were analyzed using one-way ANOVA followed by Dunnett’s post-hoc test.
Figure 1.
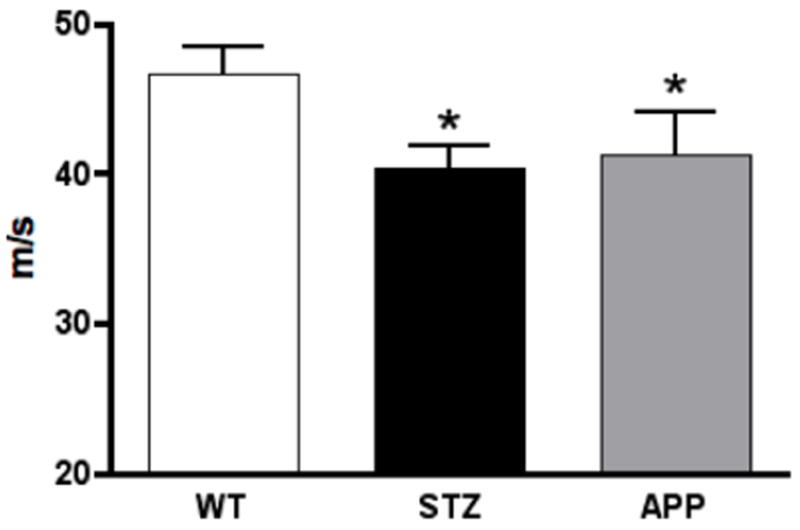
Motor nerve conduction velocity of the mouse sciatic nerve of 7-8 months old APP transgenic (APP) mice, or of age-matched mice after12 weeks of diabetes (STZ) compared to aged-matched wild type mice (WT). Data represent mean+SEM, N= 8 mice per WT and STZ groups, N=5 mice per APP group, *p<0.05 vs WT by one-way ANOVA followed by Dunnett’s post hoc test.
Figure 2.
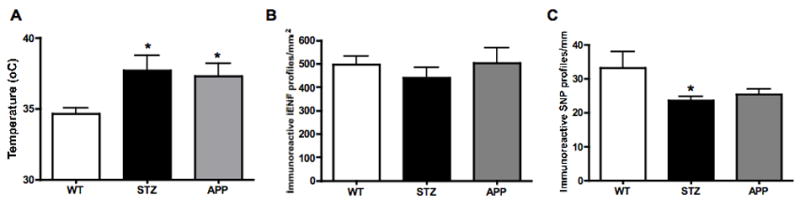
Paw thermal responses (A), IENF (B) and SNP profile (C) counts in 7-8 months old APP transgenic (APP) mice or in age-matched mice after12 weeks of diabetes (STZ) compared to aged-matched wild type mice (WT). Data represent mean+SEM, N= 8 mice per WT and STZ groups, N=5 mice per APP group, *p<0.05 vs WT by one-way ANOVA followed by Dunnett’s post hoc test.
Figure 4.
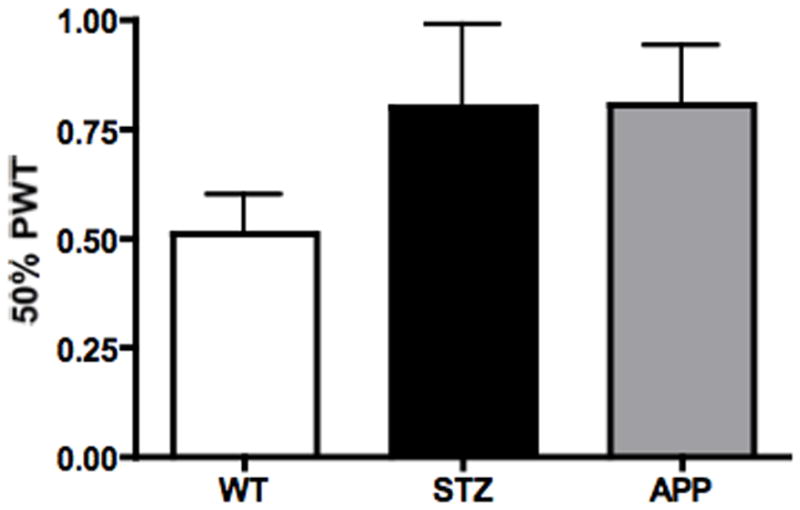
Tactile responses of 7-8 months old APP transgenic (APP) mice or of age-matched mice after12 weeks of diabetes (STZ) compared to aged-matched wild type mice (WT). Data represent mean+SEM. N= 8 mice per WT and STZ groups, N=5 mice per APP group.
Figure 5.
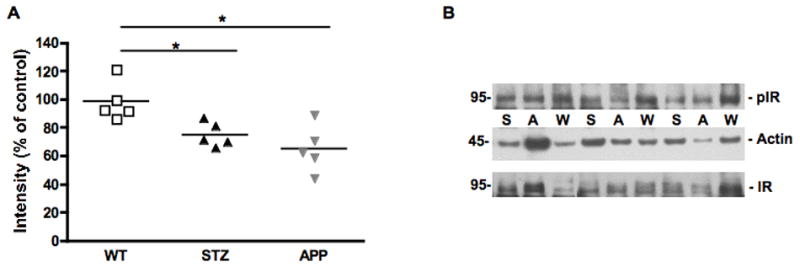
Phosphorylation of insulin receptor in mouse sciatic nerve. Scatter plot representing the individual ratio of intensity of pIR/actin (A) in mouse sciatic nerve of 7-8 months old APP transgenic (APP) mice, or of age-matched mice after 12 weeks of diabetes (STZ) compared to aged-matched wild type mice (WT). N=5 mice per group, *p<0.05 vs WT by one-way ANOVA followed by Dunnett’s post hoc test. Representative western blot of phosphorylated insulin receptor and actin (B) in mouse sciatic nerve of 7-8 months old APP transgenic (A) mice, or of age-matched mice after 12 weeks of diabetes (S) compared to aged-matched wild type mice (W).
Figure 6.
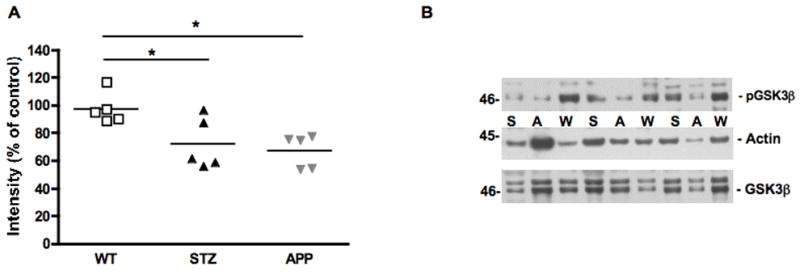
Phosphorylation of GSK3β in mouse sciatic nerve. Scatter plot representing the individual ratio of intensity of pGSK3β/actin (A) in mouse sciatic nerve of 7-8 months old APP transgenic (APP) mice, or of age-matched mice after 12 weeks of diabetes (STZ) compared to aged-matched wild type mice (WT). N=5 mice per group, *p<0.05 vs WT by one-way ANOVA followed by Dunnett’s post hoc test. Representative western blot of phosphorylated GSK3β and actin (B) in mouse sciatic nerve of 7-8 months old APP transgenic (A) mice, or of age-matched mice after 12 weeks of diabetes (S) compared to aged-matched wild type mice (W).
Figure 7.
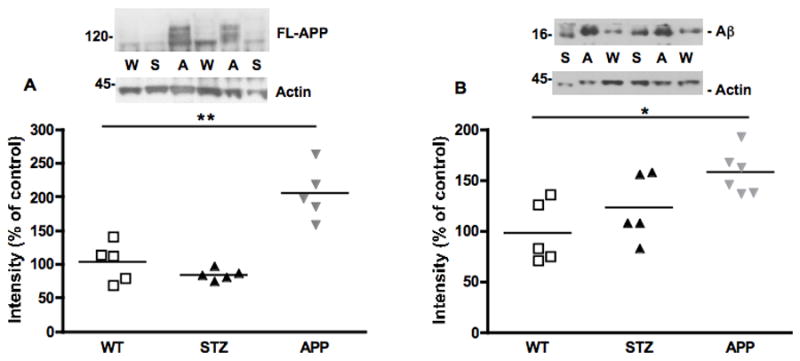
Western blots and scatter plots representing the intensity of band corresponding to FL-APP (A) and amyloid β (16kDa, B) in the sciatic nerve of WT, STZ or APP transgenic mice, normalized against actin. N=5 mice per group, *p<0.05, **p< 0.01 vs WT by one-way ANOVA followed by Dunnett’s post hoc test.
Results
2.1. Diabetes
Before the initiation of the study, the weight of 4-5 months old APP mice was significantly (p<0.05, t test) lower than that of wild-type (WT) littermates (WT: 33.4 ±1.2g, APP: 29.0 ±1.6g). Mice injected with STZ exhibited hyperglycemia (blood sugar >270 mg/dl) 4 days after STZ injection. Twelve weeks later, blood glucose levels for diabetic mice were significantly (p<0.05) higher than for non-diabetic WT and APP transgenic mice (Table 1 and Jolivalt, et al., 2010). Mice were sacrificed at 7-8 months of age, after 3 months of diabetes. Plasma insulin levels were significantly (p<0.01) decreased in STZ-diabetic mice while in APP transgenic mice insulin levels were not different from those of WT mice (Table 1). The study was initiated with 8 WT, 8 STZ and 6 APP mice. One mouse from the APP transgenic group died prior to testing.
Table 1.
Physiological parameters of wild type (WT), diabetic (STZ) and APP transgenic (APP) mice at 7-8 months of age, at the end of the study.
| Body weight (g) | Blood glucose (mg/dl) | Plasma insulin (μg/l) | |
|---|---|---|---|
| WT (N=8) | 35.9±1.3 | 158±7 | 1.28±0.11 |
| STZ (N=8) | 33.4±2.6 | 506±37* | 0.06±0.01** |
| APP (N=5) | 29.1±1.6 | 141±10 | 1.35±0.11 |
Data are represented as group mean±SEM.
p<0.05,
p<0.01 vs WT by one-way ANOVA followed by Dunnett’s post hoc test.
2.2. Nerve conduction velocity
At 4 months of age, and before induction of diabetes, baseline MNCV was similar for WT and APP transgenic mice (WT: 39.3 ± 1.2 m/s, APP: 39.0 ± 2.2 m/s). At 7 months of age and after 3 months of diabetes, MNCV failed to increase over time in aged-matched STZ and APP transgenic mice and consequently was significantly decreased (p<0.05, F(2, 18)=3.556) in both APP transgenic and STZ mice when compared to WT mice (Fig. 1). MNCV of STZ and age-matched APP transgenic mice was not significantly different.
2.3. Paw thermal response
At 4 months of age and before induction of diabetes, the baseline temperature eliciting paw withdrawal was similar for WT and APP transgenic mice (WT: 36.3 ± 0.4 °C, APP: 35.7 ± 0.9 °C). Paw response temperatures were significantly increased (p<0.05, F(2,18)=4.932) in both 7-8 months old APP transgenic mice and age-matched STZ mice after 3 months of diabetes compared to age-matched WT mice (Fig. 2A), while no significant difference between age-matched STZ and APP transgenic mice was evident. Epidermis thickness was similar in all 3 groups (WT: 37.1±2.9 μm, STZ: 36.8±3.0 μm, APP: 44.1±4.5 μm). Numbers of PGP9.5-immunoreactive intraepidermal nerve fiber (IENF) profiles were similar in all 3 groups when expressed by area (Fig. 2B) or by length of epidermis (WT: 18.6±0.9, STZ: 17.4 ±1.8, APP: 19.8±1.7 immunoreactive profiles/mm). However, subepidermal nerve plexus (SNP) profiles were significantly (p<0.05, F(2,15)=3.663) decreased in STZ-diabetic mice compared to age-matched WT mice (Fig. 2C, Fig. 3). Although not significant, SNP profiles were reduced in APP transgenic mice to a level similar to that of STZ mice (Fig. 2C, Fig. 3).
Figure 3.
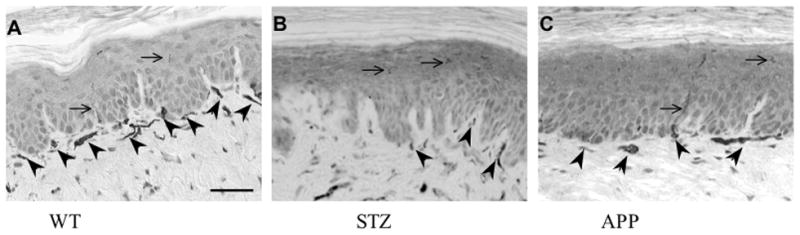
PGP9.5-immunostained epidermal (arrows) and sub-epidermal (arrowheads) nerves fiber profiles in the skin of the hind paw of wild type (A), STZ (B) and APP transgenic (C) mice. Bar=40μm
2.4. Tactile response
Seven to eight month old APP transgenic mice and age-matched STZ mice after 3 months of diabetes tended to be less responsive to von Frey filaments than age-matched WT mice (Fig. 4).
2.5. Insulin receptor phosphorylation
When compared to age-matched WT mice, phosphorylated insulin receptor levels were significantly reduced (p<0.05, F(2,12)=8.279) in the sciatic nerve of both STZ and APP transgenic mice (Fig. 5).
2.6. GSK3β phosphorylation
Phosphorylation of GSK3β at the recognized inactivating site (ser 9) was significantly (p<0.05, F(2, 12)=6.045) reduced in age-matched STZ and APP transgenic mice when compared to age-matched WT mice (Fig. 6).
2.7. Full-length APP and amyloid β protein levels
FL-APP protein levels were significantly (p<0.01, F(2,12)=25.92) increased in APP transgenic mice when compared to levels in sciatic nerve from either WT or STZ mice (Fig. 7A). Protein levels of FL-APP were similar in sciatic nerve from WT and STZ. When analyzing the western blots for Aβ protein, a 16kDa band was detected, possibly representing a tetramer of Aβ. Putative tetramer levels were significantly increased (p< 0.05, F(2,13)=6.137) in the sciatic nerve of APP transgenic mice, while a non-significant increase of 30% was observed in sciatic nerve from STZ diabetic mice (Fig. 7B).
Discussion
The observation that insulin-deficient diabetic mice develop features similar to AD in the CNS (Jolivalt et al., 2008) prompted us to also examine the PNS of diabetic and AD mice. Diabetic peripheral neuropathy is a distal symmetrical polyneuropathy that affects both small and large fibers, resulting in symptoms including loss of sensation, chronic numbness, tingling or pain (Greene et al., 1999), some of which can be modeled in diabetic rodents (for review see Calcutt, 2004, Obrosova, 2009). Our STZ-diabetic mice developed the established neuropathy phenotype of altered nociception accompanied by NCV slowing (Christianson et al., 2003, Christianson et al., 2007, Toth et al., 2010). Thermal hypoalgesia has been associated with loss of C fibers in paw skin (Johnson et al., 2008) but may also incorporate other components as dissociation between thermal responses and IENF in diabetic mice has been reported, with thermal hypoalgesia occurring in the absence of IENF loss (Beiswenger et al., 2008). In the present study, STZ mice displayed hypoalgesia while IENF profiles counts were similar to those of WT mice. Interestingly, SNP counts were decreased in the foot skin of STZ-diabetic mice - a finding that is consistent with decreased dermal myelinated fibers in foot skin of diabetic mice (Christianson et al., 2007). It is possible that early retraction of IENF and SNP has been followed by collateral sprouting of remaining IENF that are non-responsive to heat but contribute to IENF counts.
APP transgenic mice showed disorders of both small and large peripheral nerve fibers that were notably similar to those of STZ-diabetic mice, including sciatic MNCV slowing, paw thermal hypoalgesia and depletion of SNP but not IENF profiles. Unfortunately, our interest in collecting sciatic nerve for biochemical assays (see below) precluded fixation of nerve for structural analysis, so we do not yet know whether the MNCV slowing has a metabolic or structural etiology. We are not aware of prior reports of peripheral nerve dysfunction in mouse models of AD. The co-incidence of similar disorders of the CNS (Jolivalt et al., 2010) and now the PNS in STZ-diabetic and APP makes it tempting to speculate that these two neurodegenerative diseases may share converging pathogenic mechanisms while recognizing that the primary etiologies are distinct.
Peripheral nerve dysfunction during diabetes results in part from loss of neurotrophic support, including the neurotrophic properties of insulin (reviewed in Calcutt et al., 2008) and altered cellular signaling (review in Yasuda et al., 2003). To begin to explore mechanisms leading to a common peripheral neuropathy phenotype in STZ-diabetic and APP transgenic mice, we focused on the insulin-signaling pathway in order to parallel our previous observations in the CNS (Jolivalt et al., 2010). In the PNS, insulin receptors are present in Schwann cells, endothelial cells, pericytes (Sugimoto et al., 2000) and neurons, particularly sensory neurons (Sugimoto et al., 2002, Brussee et al., 2004). Insulin activates the PI3K/AKT pathway (Huang et al., 2005) in both adult primary sensory neurons in culture and ex vivo rat sciatic nerve (Jolivalt, unpublished) and promotes neurite outgrowth (Fernyhough et al., 1993) and sprouting of IENF (Guo et al., 2011). Disruption of insulin signaling in the brain of insulin-deficient diabetic mice is associated with increased GSK3 activity, tau phosphorylation and Aβ levels (Jolivalt et al., 2008). In our present study, phopshorylation of the insulin receptor was significantly decreased after 12 weeks of diabetes, along with decreased phosphorylation of GSK3β, which is indicative of increased GSK3β activity. These findings are consistent with systemic insulin deficiency in the STZ-diabetes model and parallel our prior work in the CNS of diabetic mice (Jolivalt et al. 2008). Similar changes were also present in the sciatic nerves of APP mice, despite plasma glucose and insulin levels similar to those of WT mice. Such changes in phosphorylation status are indicative of impaired insulin signaling and suggest a possible state of insulin resistance in the nerve of APP transgenic mice. Systemic insulin resistance has been reported in aged TG2576 mouse model of AD (Pedersen and Flynn, 2004). Whether our APP mice show insulin resistance in tissues other than peripheral nerve remains to be determined. Nevertheless, impaired insulin signaling in the PNS offers a potential pathogenic mechanism for peripheral neuropathy that coexists in both diabetic and AD models.
Potential downstream consequences of impaired insulin signaling and the subsequent increase in GSK3 activity that may contribute to peripheral neuropathy include abnormal phosphorylation of cytoskeletal proteins such as neurofilament and tau and the modulation of Aβ production from APP (Jope and Johnson, 2004). At present, we have focused our studies on Aβ and the small amount of sciatic nerve available from mice precluded analysis of neurofilament and tau by Western blot, histology or electron microscopy of large fibers in this study. APP is expressed and axonally transported in normal sciatic nerve (Sisodia et al., 1993, Papp et al., 2002). When Western blotting for Aβ, we detected a 16kDa band in the sciatic nerve from all 3 groups that may represent Aβ tetramers. The intensity of this 16kDa band was significantly higher in APP transgenic mice compared to controls, which is consistent with both increased APP protein expression driven by a neuronal promoter (Rockenstein et al., 2001b) and the measured increase in nerve GSK3 activity. Diabetic mice also showed both increased GSK3 activity and a trend (30% increase) towards increased expression of the presumed Aβ tetramer. The accumulation of Aβ in peripheral nerve of both AD and diabetic mouse models offers a number of potential neurotoxic mechanisms that may converge to produce a common neuropathy phenotype. For example, Aβ has been shown to disrupt peripheral axonal transport in vivo (Kasa et al., 2000) and Aβ neurotoxicity and subsequent disruption of APP transport is regulated by GSK3 and tau phosphorylation (Takashima et al., 1995), so that Aβ accumulation may contribute to axonal transport defects reported in both AD and diabetes. Accumulation of Aβ may also contribute to the reduction of insulin receptor phopshorylation as Aβ induced dephosphorylation of proteins via increased activation of calcineurin in cultured cells and a mouse model of AD (Dineley et al., 2007, Dineley et al., 2010, Li et al., 2010). This presents the possibility of a feedback loop where impaired insulin signaling induces Aβ accumulation, which in turn further suppresses insulin signaling. This mechanism remains to be investigated in the sciatic nerve.
In summary, we have shown that APP transgenic and insulin-deficient diabetic mice share similar indices of peripheral neuropathy and potentially convergent neurotoxic mechanisms. Both animal models displayed MNCV slowing, decreased tactile and thermal responses and reduced insulin signaling that were associated with increased activity of GSK3 and increased levels of Aβ protein in the sciatic nerve. As blood glucose and insulin levels are altered by diabetes but not AD, it appears that decreased insulin signaling in nerve is due to insulin deficiency in diabetic mice and possibly to tissue-specific insulin resistance and/or Aβ accumulation in the APP transgenic mice. Although, it is tempting to link the peripheral neuropathy phenotype of both diabetic and AD mice to impaired insulin signaling, further studies at the structural and pharmacological levels need to be performed to test this apparent association and to determine if insulin signaling is a primary insult, as in diabetic mice, or a contributor to PNS neuropathy in APP mice. While both diseases have different primary etiologies, diabetes and AD share many pathological features, both in the brain and the PNS. Our data highlight common deficits in the insulin-signaling pathway in both neurodegenerative diseases and support the idea that AD may be considered a systemic disorder (Joachim et al., 1989).
Highlights.
Similar indices of peripheral neuropathy despite different levels of glucose and insulin.
Decreased NCV, tactile and thermal sensitivity in diabetic and AD mouse models.
Decreased phosphorylation of insulin receptor and GSk3β in diabetic and AD mice sciatic nerve.
Increased Aβ levels in the sciatic nerve of AD mice
Acknowledgments
This work was supported by a Career Development Award from the Juvenile Diabetes Research Foundation (CGJ) and NIH grants AG5131 (EM) and DK057629 (NAC). The authors would like to thank Alex Marquez, Nick Anderson and Mario Brock for technical assistance and Dr. Andrew P. Mizisin for critical review of the manuscript.
Abbreviations
- Aβ
amyloid β
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CNS
central nervous system
- IENF
intraepidermal nerve fiber
- MNCV
motor nerve conduction velocity
- PNS
peripheral nervous system
- PWT
paw withdrawal thresholds
- SNP
subepidermal nerve plexus
- STZ
streptozotocin
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beiswenger KK, Calcutt NA, Mizisin AP. Dissociation of thermal hypoalgesia and epidermal denervation in streptozotocin-diabetic mice. Neurosci Lett. 2008;442:267–272. doi: 10.1016/j.neulet.2008.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin C, Gatt MT. Pain and dementia. Psychol Neuropsychiatr Vieil. 2006;4:247–254. [PubMed] [Google Scholar]
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a lifespan perspective. Lancet Neurol. 2008;7:184–190. doi: 10.1016/S1474-4422(08)70021-8. [DOI] [PubMed] [Google Scholar]
- Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53:1824–1830. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- Calcutt NA. Modeling diabetic sensory neuropathy in rats. Methods Mol Med. 2004;99:55–65. doi: 10.1385/1-59259-770-x:225. [DOI] [PubMed] [Google Scholar]
- Calcutt NA, Chaplan SR. Spinal pharmacology of tactile allodynia in diabetic rats. Br J Pharmacol. 1997;122:1478–1482. doi: 10.1038/sj.bjp.0701538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcutt NA, Jolivalt CG, Fernyhough P. Growth factors as therapeutics for diabetic neuropathy. Curr Drug Targets. 2008;9:47–59. doi: 10.2174/138945008783431727. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience. 2007;145:303–313. doi: 10.1016/j.neuroscience.2006.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JA, Ryals JM, McCarson KE, Wright DE. Beneficial actions of neurotrophin treatment on diabetes-induced hypoalgesia in mice. J Pain. 2003;4:493–504. doi: 10.1016/j.jpain.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- Desrocher M, Rovet J. Neurocognitive correlates of type 1 diabetes mellitus in childhood. Child Neuropsychol. 2004;10:36–52. doi: 10.1076/chin.10.1.36.26241. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Willars GB, Lindsay RM, Tomlinson DR. Insulin and insulin-like growth factor I enhance regeneration in cultured adult rat sensory neurones. Brain Res. 1993;607:117–124. doi: 10.1016/0006-8993(93)91496-f. [DOI] [PubMed] [Google Scholar]
- Greene DA, Stevens MJ, Feldman EL. Diabetic neuropathy: scope of the syndrome. Am J Med. 1999;107:2S–8S. doi: 10.1016/s0002-9343(99)00007-8. [DOI] [PubMed] [Google Scholar]
- Guo G, Kan M, Martinez JA, Zochodne DW. Local insulin and the rapid regrowth of diabetic epidermal axons. Neurobiol Dis. 2011;43:414–421. doi: 10.1016/j.nbd.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Huang TJ, Verkhratsky A, Fernyhough P. Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol Cell Neurosci. 2005;28:42–54. doi: 10.1016/j.mcn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Mori H, Selkoe DJ. Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature. 1989;341:226–230. doi: 10.1038/341226a0. [DOI] [PubMed] [Google Scholar]
- Johnson MS, Ryals JM, Wright DE. Early loss of peptidergic intraepidermal nerve fibers in an STZ-induced mouse model of insensate diabetic neuropathy. Pain. 2008;140:35–47. doi: 10.1016/j.pain.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol. 2010;223:422–431. doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Kasa P, Papp H, Kovacs I, Forgon M, Penke B, Yamaguchi H. Human amyloid-beta1-42 applied in vivo inhibits the fast axonal transport of proteins in the sciatic nerve of rat. Neurosci Lett. 2000;278:117–119. doi: 10.1016/s0304-3940(99)00863-0. [DOI] [PubMed] [Google Scholar]
- Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- Li Q, Fang J, Yang M, Wu D, Zhang L, Zhang Y. Galantamine inhibits calpain-calcineurin signaling activated by beta-amyloid in human neuroblastoma SH-SY5Y cells. Neurosci Lett. 2010;480:173–177. doi: 10.1016/j.neulet.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Manschot SM, Brands AM, van der Grond J, Kessels RP, Algra A, Kappelle LJ, Biessels GJ. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–1113. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- McCarthy AM, Lindgren S, Mengeling MA, Tsalikian E, Engvall JC. Effects of diabetes on learning in children. Pediatrics. 2002;109:E9. doi: 10.1542/peds.109.1.e9. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- McGuire JF, Rouen S, Siegfreid E, Wright DE, Dobrowsky RT. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes. 2009;58:2677–2686. doi: 10.2337/db09-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrosova IG. Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics. 2009;6:638–647. doi: 10.1016/j.nurt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- Papp H, Pakaski M, Kasa P. Presenilin-1 and the amyloid precursor protein are transported bidirectionally in the sciatic nerve of adult rat. Neurochem Int. 2002;41:429–435. doi: 10.1016/s0197-0186(02)00014-1. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol Dis. 2004;17:500–506. doi: 10.1016/j.nbd.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes. 2002;51:1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Thompson LW, Nahum D, Haskins E. Relationship between hyperglycemia and cognitive function in older NIDDM patients. Diabetes Care. 1990;13:16–21. doi: 10.2337/diacare.13.1.16. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Ab1-42. J neurosci Res. 2001a;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001b;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, Degerman-Gunnarsson M, Lannfelt L, Berne C, Kilander L. Glucose metabolism and the risk of Alzheimer’s disease and dementia: a population-based 12 year follow-up study in 71-year-old men. Diabetologia. 2009;52:1504–1510. doi: 10.1007/s00125-009-1393-9. [DOI] [PubMed] [Google Scholar]
- Ryan C, Vega A, Drash A. Cognitive deficits in adolescents who developed diabetes early in life. Pediatrics. 1985;75:921–927. [PubMed] [Google Scholar]
- Ryan CM, Williams TM. Effects of insulin-dependent diabetes on learning and memory efficiency in adults. J Clin Exp Neuropsychol. 1993;15:685–700. doi: 10.1080/01688639308402589. [DOI] [PubMed] [Google Scholar]
- Scherder E, van Manen F. Pain in Alzheimer’s disease: nursing assistants’ and patients’ evaluations. J Adv Nurs. 2005;52:151–158. doi: 10.1111/j.1365-2648.2005.03577.x. [DOI] [PubMed] [Google Scholar]
- Scherder EJ, Oosterman JM, Ooms ME, Ribbe MW, Swaab DF. Chronic pain in dementia and in disorders with a high risk for congnitive impairment. Tijdschr Gerontol Geriatr. 2005;36:116–121. [PubMed] [Google Scholar]
- Sisodia SS, Koo EH, Hoffman PN, Perry G, Price DL. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J Neurosci. 1993;13:3136–3142. doi: 10.1523/JNEUROSCI.13-07-03136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Murakawa Y, Sima AA. Expression and localization of insulin receptor in rat dorsal root ganglion and spinal cord. J Peripher Nerv Syst. 2002;7:44–53. doi: 10.1046/j.1529-8027.2002.02005.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto K, Murakawa Y, Zhang W, Xu G, Sima AA. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes Metab Res Rev. 2000;16:354–363. doi: 10.1002/1520-7560(200009/10)16:5<354::aid-dmrr149>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Takashima A, Yamaguchi H, Noguchi K, Michel G, Ishiguro K, Sato K, Hoshino T, Hoshi M, Imahori K. Amyloid beta peptide induces cytoplasmic accumulation of amyloid protein precursor via tau protein kinase I/glycogen synthase kinase-3 beta in rat hippocampal neurons. Neurosci Lett. 1995;198:83–86. doi: 10.1016/0304-3940(95)11964-x. [DOI] [PubMed] [Google Scholar]
- Terry RD. Alzheimer’s disease and the aging brain. J Geriatr Psychiatry Neurol. 2006;19:125–128. doi: 10.1177/0891988706291079. [DOI] [PubMed] [Google Scholar]
- Thomas PK. Classification, differential diagnosis, and staging of diabetic peripheral neuropathy. Diabetes. 1997;46(Suppl 2):S54–57. doi: 10.2337/diab.46.2.s54. [DOI] [PubMed] [Google Scholar]
- Toth CC, Jedrzejewski NM, Ellis CL, Frey WH., 2nd Cannabinoid-mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain. 2010;6:16. doi: 10.1186/1744-8069-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente T, Gella A, Fernandez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol Dis. 2010;37:67–76. doi: 10.1016/j.nbd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L. Uncontrolled diabetes increases the risk of Alzheimer’s disease: a population-based cohort study. Diabetologia. 2009;52:1031–1039. doi: 10.1007/s00125-009-1323-x. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Terada M, Maeda K, Kogawa S, Sanada M, Haneda M, Kashiwagi A, Kikkawa R. Diabetic neuropathy and nerve regeneration. Prog Neurobiol. 2003;69:229–285. doi: 10.1016/s0301-0082(03)00034-0. [DOI] [PubMed] [Google Scholar]
- Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K, et al. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology. 1995;45:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]