

Abstract
GPCRs are a major family of homologous proteins and are key mediators of the effects of numerous endogenous neurotransmitters, hormones, cytokines, therapeutic drugs, and drugs-of-abuse. Despite the enormous amount of research on the pharmacological and biochemical properties of GPCRs, the question as to whether they exist as monomers, dimers, or higher order structures in the body is unanswered. The GPCR dimer field has been dominated by techniques involving recombinant cell lines expressing mutant receptors, often involving the solubilization of the receptors. These techniques cannot be applied in vivo or even to primary cell cultures. This review will focus on a novel approach to exploring the functional properties of homodimers. Studies of the 5-HT7 and 5-HT2A serotonin receptors have revealed that binding of a pseudo-irreversible antagonist (“inactivator”) to one of the orthosteric sites of a homodimer abolishes all receptor activity, and subsequent binding of a competitive antagonist to the orthosteric site of the second protomer releases the inactivator, allowing the receptor to return to an active state. This approach demonstrates allosteric crosstalk between protomers of native GPCR homodimers, indicating that GPCRs do exist and function as homodimers in both recombinant cells and rat primary astrocytes. This technique can be applied universally using intact recombinant or primary cells in culture, membrane homogenate preparations and, potentially, in vivo. The data obtained using the 5-HT7 and 5-HT2A receptors are strongly supportive of a GPCR homodimer structure, with little evidence of monomer involvement in the function of these receptors.
Keywords: GPCR, homodimer, heterodimers, 5-HT7, 5-HT2A, allosterism
1. Introduction
1.1 The importance of studying GPCR dimers
There is a growing sense that G-protein coupled receptors (GPCRs) may be obligatory dimers in vivo (Figure 1). Thus, the commonly depicted seven transmembrane (7TM) structure for a GPCR actually represents a subunit of the overall macromolecular structure. Developing a simple, definitive, relatively non-invasive methodology for detecting homodimer or heterodimer-associated functional effects of native GPCRs in vivo is essential if the question of the prevalence and function of GPCR oligomers is to be answered. Besides the importance, from a basic cell biology viewpoint, of knowing the quaternary structure and function of GPCRs, there are important clinical implications. The processing and positioning of GPCRs have been implicated in disease processes, and strategies for altering these processes have been targeted for novel therapeutic drug development (Rene et al., 2010). Therefore, the processing and positioning of a GPCR oligomer will involve numerous cell biological functions not yet investigated in vivo. Dysfunction in the processing and/or the functioning between the protomers of a GPCR dimer or higher order structure could be responsible for disease states (Wang et al., 2010). As we have no reliable methodologies to investigate GPCR oligomer function in vivo, this is a wide-open area of investigation. Drugs that change the processing and/or function of the GPCR oligomer may prove to be novel therapeutics.
Figure 1. The obligatory dimer GPCR model.

In order for the GPCR to be expressed in a functional state on the plasma membrane, an intracellular dimer must be formed. The dimer may be a homodimer or a heterodimer. Thus, a GPCR may be viewed as containing two “subunits”, each a distinct gene product. This illustration depicts neuronal serotonin (5-HT) receptors, but may be applied to any GPCR.
1.2 Overview of the GPCR dimer field: the recombinant cell line “bottleneck”
The exploration of the molecular properties of the GPCR family of receptors began in the 1970's and resulted in a universally accepted receptor/GTP-binding-protein/adenylate cyclase (or phospholipase C) signal transduction model (Stadel, DeLean, & Lefkowitz, 1980). The techniques dominating the field involved solubilization, purification and reconstitution of the components of this signaling complex, demonstrating the ability to restore most receptor-mediated function in an artificial lipid environment with these three components (Lefkowitz, Cerione, Codina, Birnbaumer, & Caron, 1985). While there was an occasional report that indicated the receptor might exist as a dimer (Fraser & Venter, 1982; Lilly, Fraser, Jung, Seeman, & Venter, 1983), the concept of monomeric GPCRs became accepted. The cloning of the genes for GPCRs did not change the model, only adding more information regarding the basic structure of the receptors as a family, i.e. revealing the now classical seven trans-membrane structure (O'Dowd, Lefkowitz, & Caron, 1989).
Thus, it was a shock to many GPCR researchers when multiple groups convincingly demonstrated that co-expression of GABAB1 and GABAB2 cDNA was required to produce a functional GABAB receptor expressed on the plasma membrane (Jones et al., 1998; Marshall, Jones, Kaupmann, & Bettler, 1999; Kuner et al., 1999; Ng et al., 1999). The in vivo validity of the original work, which involved co-transfection of the two cDNAs into one recombinant cell, developed rapidly as mRNA and immunocytochemical studies showed that both receptors were co-localized in vivo in all tissues that demonstrate properties of the metabotropic GABAB receptor (Jones et al., 1998; Ng et al., 1999; Kaupmann et al., 1998; Marshall et al., 1999). There are two important aspects of the GABAB1/GABAB2 discovery that we wish to emphasize: (A) None of the groups set out to prove the existence of heterodimers: the expectation was that the receptors would be functional when expressed individually. The heterodimer structure was discovered, in large part, through serendipity and went against the conventional model. (B) The in vivo relevance of the recombinant cell line observations was quickly established through various observations, including the co-localization of mRNA for both receptor sub-types in any cell with a metabotropic GABAB receptor response and co-immunoprecipitation studies in brain tissue, supporting the physical interaction of the two receptors.
The discovery of the heterodimeric structure of the GABAB1/GABAB2 receptor led to the publication of numerous reports on the formation of homo- and heterodimers in recombinant cell lines. Although in-depth discussion of these studies is beyond the scope of this review, these studies have been reviewed numerous times (Pin et al., 2007; Ferre et al., 2009; Franco et al., 2007; Satake & Sakai, 2008). A major difference concerning the nature of these studies and the original GABAB1/GABAB2 studies became apparent: rather than a rapid and convincing demonstration of the in vivo relevance of other GPCR dimers observed in recombinant cells, there were very few reports that extended the evidence for dimer formation to an in vivo preparation, and these reports were unconvincing to many GPCR researchers. Thus, by 2007 the GPCR dimer field was a confusing one.
In 2007 and 2009 committees were convened to organize the information and develop a nomenclature for GPCR dimers (Pin et al., 2007; Ferre et al., 2009). The first committee declared that in order to assign a name to a dimer the following questions should be answered, with an emphasis on the first question:
1) Are the properties observed in transfected heterologous expression systems also valid in native tissue in vivo? 2) Do we have to consider such GPCR complexes as a receptor unit or just an association of receptor units? 3) Are the pharmacological properties of homodimers or heterodimers substantially different from those of the monomeric receptor? (Pin et al., 2007)
The committee decided that the critical first question has been answered for the GABAB receptor (GABAB1/GABAB2 heterodimer), the sweet taste receptor (T1R2/T1R3 heterodimer), and the umami taste receptor (T1R1/T1R3 heterodimer) (Pin et al., 2007), as co-expression of both protomers of these heterodimers were required to produce functional receptors. These receptors are all Class C GPCRs, which like all other GPCRs contain seven transmembrane spanning domains but also contain large extracellular ligand binding (venus flytrap) domains, wherein disulfide bonds covalently link the receptor protomers. Recently, another group of GPCR researchers (18 authors) convened as an IUPHAR committee and published a similar review emphasizing the difficulties in determining properties of GPCR heterodimers and homodimers in vivo (Ferre et al., 2009). This second committee declared the following:
The controversy has now moved to the existence and functional significance of receptor heteromers in native tissues. In order to address these questions, we must find evidence for the unique biochemical and functional signatures (different from those of its constituent receptors) that characterize the receptor heteromer. (Ferre et al., 2009)
Just as the 2007 committee concluded, this 2009 committee found that the GABAB receptor, the sweet taste receptor, and umami taste receptor exist as GPCR dimers in vivo (i.e. genetic deletion of one protomer abolishes the heteromeric receptor's function) (Pin et al., 2007; Ferre et al., 2009; Yasuo, Kusuhara, Yasumatsu, & Ninomiya, 2008; Marshall et al., 1999). Subsequent to the 2009 IUPHAR committee meeting, a report was published involving the luteinizing hormone receptor that supports the existence of this receptor as a homodimer in vivo (Rivero-Muller et al., 2010). The experimental design involves a transgenic mouse expressing mutated forms of the native luteinizing receptor. As with many of the studies addressing the issue of homodimers, the applicability of this experimental design is limited in that an appropriate pair of mutant receptors must be designed, tested in vitro, and then become functionally incorporated into a transgenic mouse line. While not detracting from the importance of this report, this approach will clearly not be readily available to a majority of GPCR research programs. In this review, one of the reasons we are enthusiastic about our approach is its applicability to many GPCR research groups using basic, fundamental receptor technologies available in most pharmacological laboratories.
A database has been established (http://data.gpcr-okb.org/gpcr-okb/oligomer/list) attempting to organize the literature on hetero- and homodimers (Skrabanek et al., 2007). Whether a given dimer listed in the database truly fulfils the criteria established through the studies on the Class C heterodimers, including the GABAB1/GABAB2, TR2–TR3 (sweet), or TR1–TR3 (umami) receptors, is up for debate.
1.3 GPCR heterodimers as therapeutic drug targets
A great deal of attention has been focused on GPCR heterodimers (Pin et al., 2007; Ferre et al., 2009). This appears to be due to several factors. As mentioned above, the GABAB1/GABAB2 discovery demonstrates that the process for forming heterodimers exists in vivo. There are numerous reports of the production of GPCR heterodimers in recombinant cell lines. The study of heterodimers lacks several of the technical obstacles that face the study of GPCR homodimers (see below). Heterodimers are composed of two distinctly different protomers that, in many cases, can be discriminated by ligand binding properties, signal transduction mechanisms, and potentially molecular weight and immunoreactivity, all of which make identifying heterodimers in vivo more feasible than identifying homodimers. From a novel drug development perspective, the concept of GPCR heterodimers appears to be alluring (Pin et al., 2007; Ferre et al., 2009; Yasuo et al., 2008; Marshall et al., 1999). The concept includes the idea that monomers (or homodimers) and heterodimers co-exist and that many of the current GPCR-directed drugs interact indiscriminately with both the monomeric (or homodimeric) and heterodimeric forms of these receptors. If one hypothesizes that (A) the heterodimer has an orthosteric (or allosteric) binding site(s) that is different from the monomer, (B) the heterodimer is expressed in vivo, and (C) selectively altering the heterodimer's activity with agonists or antagonists may produce a therapeutic effect, then GPCR heterodimer-directed drugs do represent a potential novel therapeutic strategy. It is clear that finding a way to determine the presence and properties of GPCR heterodimers in vivo is of great importance. It seems logical that before any GPCR heterodimer is proposed to be worthy of attention, the co-localization of the two GPCRs forming the heterodimer should be demonstrated in vivo. This can be done at a gross level using receptor autoradiography; in situ hybridization techniques can accomplish this at the cellular level.
1.4 GPCR homodimers: a difficult field of study
In contrast to GPCR heterodimers, the existence and function of GPCR homodimers in vivo has not been established for any GPCR, with the possible exceptions of rhodopsin (see below), the luteinizing hormone receptor (Rivero-Muller et al., 2010), and our recent publication of r5-HT7 receptors in primary cultures of rat astrocytes (Smith, Toohey, Knight, Klein, & Teitler, 2011), which may be more appropriately regarded as “ex vivo”.
The concept of GPCR homodimers is an intriguing one. As mentioned above, the relevant GPCR heterodimer species are limited to those GPCRs shown to be expressed in the same cells in vivo, and for many GPCR heterodimers studied in recombinant cells, this is not the case. GPCR homodimers do not have this criterion. Any cell expressing a GPCR potentially can produce a homodimeric form of that receptor. Based on this reasoning, it seems likely that GPCR homodimers are more prevalent in vivo than GPCR heterodimers. While there are some examples that GPCRs might exist in a homodimeric structure in vivo, a convincing demonstration of native GPCRs functioning as homodimers in vivo does not exist. Using atomic force microscopy, the rhodopsin receptor has been visualized as a dimer and possibly a tetramer in retinal tissue; however, this technique involves subjecting the retinal tissue to lipid-depleting treatments (Fotiadis et al., 2006). Furthermore, there is dispute amongst biophysicists as to the validity of the procedures used to produce the images indicating dimer pairs for rhodopsin (Chabre, Cone, & Saibil, 2003).
The studies described in the following sections of this review, involving novel receptor functional properties, provide support for the existence of GPCR homodimers in vivo. While they are not direct, structural studies, these studies do identify the biological fingerprints of GPCR homodimeric function ex vivo and potentially in vivo. By the nature of the experimental design, another important aspect of GPCR dimeric pharmacology is addressed: protomer-protomer allosteric crosstalk (i.e. the allosteric modulation of one protomer's binding properties and/or function by the binding of a ligand at the other protomer). Demonstrating protomer-protomer allosteric crosstalk in GPCR homodimers has been another difficult aspect of GPCR pharmacology. Other than the studies described in the following sections, the only other report that clearly addresses protomer-protomer allosteric crosstalk of GPCRs in vivo is based on mutated luteinizing hormone receptors expressed in transgenic mice (Rivero-Muller et al., 2010), wherein mutated receptors that are signaling-deficient can be rescued by the expression of receptors that are ligand binding-deficient. This research strategy requires the use of mutated receptors; thus, the information garnered form this study is not easily translatable to the allosteric interactions of native luteinizing hormone receptor protomers. The studies reviewed below address this issue in native 5-HT7 and 5-HT2A receptors, and in fact, protomer-protomer allosteric crosstalk is an essential part of the experimental design.
The identity of the two protomers in a GPCR homodimer present experimental difficulties not present in study of GPCR heterodimers. If one starts with the simplest structure of a homodimer and proposes that the interaction of a ligand with one orthosteric site produces no allosteric effect on the affinities of ligands at the other orthosteric site, then detecting the dimer structure through purely pharmacological studies will be impossible. The homodimer will appear as a single population of binding sites. Thus, there is an inherent irony in contemplating the study of GPCR homodimers versus studying GPCR heterodimers. Because every cell that expresses a GPCR has the potential to form a homodimer, it appears that demonstrating GPCR homodimer expression and function should be relatively easy compared to finding GPCR heterodimers, where the expression of two gene products must be demonstrated in one cell, leading to the formation of the heterodimer. However, the identity of the two orthosteric binding sites on a GPCR homodimer creates a situation in which discerning a homodimer from a monomer may be very difficult. In this review, we will demonstrate that it is possible to use ligands that perturb the GPCR to induce allosteric changes that reveal the homodimeric structure and function.
1.5 GPCR monomers
The concept that GPCR monomers might not exist on the plasma membrane in vivo is a startling one. As mentioned above, the classical work on GPCRs relied on a receptor monomeric model and there were no significant data to dispute this model until the GABAB1/GABAB2 receptor complex opened up the field of GPCR dimers. Furthermore, many papers in the last decade, attempting to demonstrate dimers or higher order structures, also appear to identify monomers (Hern et al., 2010). Groups have reported that monomeric GPCRs can stimulate G-proteins (Whorton et al., 2007; Chabre & le, 2005). That GPCRs can be expressed exclusively as dimers is clearly shown by the GABAB1/GABAB2 complex. It was quickly recognized that in order for the mature, functional receptor to be released from the endoplasmic reticulum (ER) the heterodimer has to be formed. The GABAB2 monomer appears to allow the release of the GABAB1/GABAB2 heteromer from the ER by occluding the GABAB1 domain that is used to anchor the protein to the endoplasmic reticulum membrane (ER retention signal). This results in only heterodimers expressed at the plasma membrane. The question is whether this is a common phenomenon in the GPCR field or a rare occurrence, possibly unique to the GABAB1/GABAB2 complex. At this point, most GPCR researchers convinced of the prevalence of dimers include monomers at the plasma membrane in their cellular model of GPCR processing (Hern et al., 2010; Whorton et al., 2007). It may well be that monomers, dimers and higher order structures co-exist on the plasma membrane. The data and models presented herein do not appear to support functional monomers (see below); however, these data involve only the 5-HT7 and 5-HT2A receptors, and it is possible that similar experiments on other receptors would reveal a role for monomers in function.
In summary, while the existence of dimeric GPCRs is strongly indicated in recombinant cell lines, the function of the dimeric structure remains a challenging but potentially critical area of GPCR research. “A specific functional property for the heterodimeric receptor will be critical to identify such receptors in native tissue. This could include the identification of a specific pharmacological property such as a positive or negative allosteric interaction between the two binding sites…” (Pin et al., 2007). If this is true for heterodimers, it is certainly true for homodimers, which present a greater challenge due to the identity of the two orthosteric sites involved in the homodimer structure. The remainder of this review involves the examination of four papers we have published that illustrate a novel strategy that provides data strongly indicating the presence of functional, native h5-HT7 homodimers in recombinant cell lines and in primary cell culture, and demonstrates the ligand-dependent protomer-protomer allosteric crosstalk that can occur within the homodimer. We also present similar findings for the 5-HT2A receptor. The potential global importance of these studies is that the experimental design involves the native GPCR, readily measured outputs from the stimulation of the GPCR, and can be applied in vitro and in vivo. Rather than examining the quaternary structure of these receptors though a biophysical approach, these studies identify novel functional properties of native 5-HT7 and 5-HT2A receptors that reveal the homodimeric nature of these receptors. The replication of such studies in a variety of GPCRs would shift the accepted paradigm of a monomeric structure to a homodimeric structure. This, in turn, would enhance the relevance of other GPCR homodimers studied solely in recombinant cell lines up to this point as well as renew research into the implications of GPCR homodimeric structure on cellular function and the role of homodimer dysfunction in disease states. Novel therapeutic strategies to ameliorate dysfunctions due to homodimer processing or function would represent a truly novel avenue of drug development.
2. Inactivation and reactivation of the h5-HT7 receptor: a demonstration of GPCR homodimer protomer-protomer allosteric crosstalk
2.1 Discovery and characterization of h5-HT7 receptor “inactivators”
In 2006 and 2009 we reported that risperidone, 9-OH-risperidone, methiothepin, bromocryptine, lisuride, and metergoline appeared to produce an unusual effect on h5-HT7 receptors stably expressed in HEK-293 cells (HEK-293-h5-HT7 cells)(Smith et al., 2006; Knight, Smith, Toohey, Klein, & Teitler, 2009). These drugs, all of which are classic serotonergic compounds, non-competitivly antagonized h5-HT7-mediated accumulation of cAMP. Brief exposure of the HEK-293-h5-HT7 cells to these drugs at concentrations only 10-fold over their Ki values produced an apparently irreversible inhibition (“inactivation”) of the receptor (Figure 2). This was demonstrated by thoroughly washing out the drugs after the brief exposure to drug and stimulating the cells with 10μM 5-HT, which is 200-fold over the EC50 of 5-HT for the h5-HT7 receptor. Even at this high level of 5-HT, little or no receptor activity was seen, despite the absence of the antagonists from the media. Other potent 5-HT7 receptor antagonists did not produce this effect: clozapine, mesulergine, penfluridol, amperozide, and cinanserin exposure followed by washout had no effect on h5-HT7 receptor activity (Figure 2). Using this drug washout procedure, we characterized the “inactivating antagonist” activity of the drugs and found their potencies as inactivators correlated with their apparent affinities for the h5-HT7 receptor as determined using homogenate radioligand binding assays (Borison, Diamond, Pathiraja, & Meibach, 1994). Of special interest were the actions of risperidone and 9-OH-risperidone. While both these drugs fully inactivated the h5-HT7 receptor (Figure 2), they produced only a 50% loss of receptor binding activity (Figure 3)(Knight et al., 2009). This was a key observation (see below). The other inactivators fully inhibited the h5-HT7 radioligand binding signal in a wash-resistant manner, except for metergoline, which was eventually found to have different properties than the other five inactivators (Knight et al., 2009). Studies utilizing [3H]risperidone to directly measure the risperidone-h5-HT7 receptor complex also revealed pseudo-irreversible binding to 50% of the sites and competitive binding to the remaining 50% of sites. The interpretation of these two reports was that the six inactivating antagonists interact with the h5-HT7 receptor in a pseudo-irreversible manner (i.e. non-covalent and wash-resistant interaction), occluding the orthosteric binding site, thus inactivating the receptor. The effect of risperidone and 9-OH-risperidone in fully inactivating the receptor but only pseudo-irreversibly binding to 50% of the receptors was noted. At the time a cogent interpretation of the 50% effect was not readily apparent. It appeared as though the HEK-293-h5-HT7 receptor cell line was producing two populations of h5-HT7 receptors that could only be discerned by their interaction with risperidone (or 9-OH-risperidone). All other properties of the receptors appeared homogeneous. Thus, the 0% effect appeared inexplicable.
Figure 2. Effect of drug pre-treatment on h5-HT7 receptor-stimulated cAMP production.

HEK-293-h5-HT7 cells were suspended in isotonic buffer and exposed to 10 × Ki concentrations (inactivators and clozapine) or 1μM of drugs for 30 min. Cells were thoroughly washed and assayed for response to 10μM 5-HT. These results are from (Smith et al., 2006) and are the means ± S.E.M. of three independent experiments performed in triplicate (*p<0.005 versus “No drug,” one-way ANOVA with Dunnett's post-test).
Figure 3. Risperidone concentration-response curve for wash-resistant inhibition of specific [3H]5-HT binding to the h5-HT7 receptor.

HEK-293-h5-HT7 cells were suspended in isotonic buffer and exposed to buffer (control) or varying concentrations of risperidone, a drug that displayed full inactivating properties (see Fig. 1). After the drug washout procedure, the cells were incubated with 2 nM [3H]5-HT; nonspecific binding was determined with the addition of 10μM clozapine. These results are from (Knight et al., 2009) and are the means ± S.E.M. of three independent experiments performed in triplicate.
2.2 Reactivation of inactivated h5-HT7 receptors using competitive antagonists
The observation of the 50% maximal pseudo-irreversible binding of risperidone and 9-OH-risperidone to the h5-HT7 receptor led to the second phase of the investigation (Teitler, Toohey, Knight, Klein, & Smith, 2010). We measured the dissociation rate of the fraction of [3H]risperidone that binds competitively to the h5-HT7 receptor using the standard protocol of allowing 1nM [3H]risperidone to equilibrate with the cells, adding a saturating level of competitive antagonist (1μM mesulergine or clozapine) and measuring specific binding at different time points. Surprisingly, rather than observing the dissociation of ~50% of the [3H]risperidone from the receptor, almost all of the specific binding dissociated over 90 minutes. This was not expected as 50% of the [3H]risperidone specific binding had been shown to be pseudo-irreversible; it was expected that, at most, 50% of the specific binding would dissociate. Thus, an apparent anomaly existed: 50% of [3H]risperidone binding to h5-HT7 receptors is “wash resistant”, and yet almost all of the specific binding of [3H]risperidone can be dissociated when a competitive antagonist is added to the [3H]risperidone-labeled HEK-293-h5-HT7 cells. One major difference between the experimental design of showing wash resistance and performing dissociation rate experiments is the inclusion of the competitive antagonist in the dissociation rate experiments. We hypothesized that the dissociation of the wash-resistant fraction of [3H]risperidone binding (50% of 5-HT7 binding sites) is greatly influenced by receptor interaction with competitive antagonist. In other words, the addition of competitive antagonist(s) to an inactivated h5-HT7 receptor somehow “releases” risperidone from the receptor in a concentration-dependent (i.e. receptor-mediated) manner. It was further hypothesized that if that was true, washing out the drugs after the competitive antagonist treatment, should leave the receptors in their original state, i.e. capable of being stimulated by 5-HT. This was shown to be true (Figure 4). The addition of competitive antagonist to the risperidone-bound, inactive h5-HT7 receptors, followed by washout of the drugs results in the reactivation of the receptor. Addition of other inactivators to the risperidone-inactivated h5-HT7 receptor does not reactivate the receptor (Figure 4). The potencies of the competitive antagonists in reactivating the h5-HT7 receptor were determined and found to be highly correlated with, if not identical to, their affinities for the h5-HT7 receptor (Figure 5), indicating the reactivating antagonists mediate their effects through the 5-HT7 orthosteric site despite the presence of risperidone at 50% of the sites.
Figure 4. Reactivation of risperidone-inactivated h5-HT7 receptors.
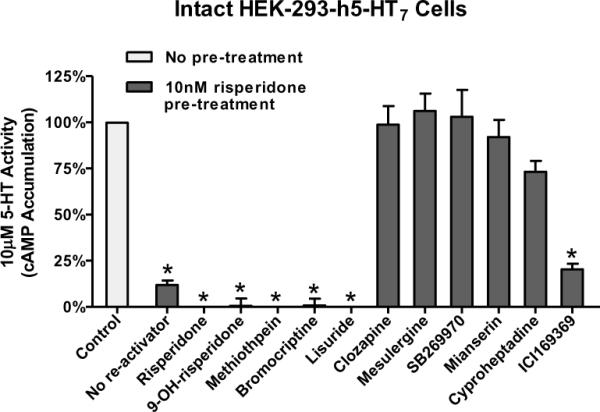
Four inactivating antagonists and six non-inactivating antagonists (competitive antagonists) were tested for their ability to reverse risperidone inactivation of the h5-HT7 receptor. Cells were exposed to risperidone, thoroughly washed, then exposed to 1 μM of the various drugs for 90 min. After removal of drugs, receptor activity was tested with 10 μM 5-HT. The four inactivators (risperidone, 9-OH-risperidone, methiothepin, and bromocriptine) as well as ICI169369 did not reactivate the receptor; all other drugs reactivated the receptor as defined by sensitivity to 10 μM 5-HT. ICI169369 proved to be a low potency reactivator due to its relatively low affinity for the h5-HT7 receptor (Teitler et al., 2010) (Figure 5). These results are from (Teitler et al., 2010) and are the mean±S.E.M. of three experiments performed in triplicate (*p<0.001 vs. control, one-way ANOVA with Dunnett's post-test).
Figure 5. Correlation between the reactivation potencies (log EC50, ordinate) and affinities (log Ki, abscissa) of competitive antagonists at the h5-HT7 receptor.

The high correlation observed (r2=0.95) indicates that the reactivation of risperidone-inactivated h5-HT7 receptors is due to the binding of the reactivating drugs to the orthosteric binding site of the h5-HT7 receptor despite the presence of wash-resistant risperidone bound to the orthosteric site. These results are from (Teitler et al., 2010) and are the mean±S.E.M. of three experiments performed in triplicate.
2.3 A homodimer model explains the reactivation of h5-HT7 receptors
In developing a feasible explanation for these observations, especially the apparent ability of competitive antagonists to bind to fully inactivated receptors, we adhered to two fundamentally accepted pharmacological principles: (1) Drugs bind to empty binding sites on receptors. This is true whether the binding site is the orthosteric binding site or an allosteric binding site. Drugs (and endogenous ligands) are designed to bind to sites unoccupied by other drugs or endogenous substances. The presence of a drug or an endogenous ligand at the binding site eliminates that binding site from interaction with a second ligand, until the first ligand dissociates. As elementary and basic as this principle is in receptor pharmacology, we feel it is important to emphasize this point. (2) Classical allosteric binding sites have very different affinities for drugs compared to the orthosteric site and generally no affinity for the endogenous ligand. While, in theory, an allosteric site on a protein could form a binding domain that is very similar to an orthosteric binding site, this has never been observed. Allosteric drugs tend to have very low affinities for the orthosteric binding site; conversely, orthosteric site-directed drugs have very low affinities for allosteric sites.
Thus, the observations could not be explained within a monomer GPCR paradigm. Competitive antagonists cannot “displace” pseudo-irreversibly bound risperidone from the orthosteric site on a monomer, and yet the unique h5-HT7 pharmacological profile of the reactivating potencies of the drugs indicated the reactivating drugs interact with an available orthosteric site. The most plausible explanation for these observations is that the 5-HT7 receptor functions as a homo-oligomer, the simplest form being a homodimer (Figure 6). In fact, a homodimer structure explained several puzzling observations:
-
(1)
Risperidone pseudo-irreversibly binds only 50% of 5-HT7 receptors yet inactivates all receptor activity This phenomenon can be explained if the binding of risperidone to the orthosteric site of one protomer of a 5-HT7 homodimer allosterically modulates the orthosteric binding properties of the second protomer (i.e. allows the second protomer to bind risperidone reversibly) via protomer-protomer allosteric crosstalk. After washing out excess risperidone, this would leave all 5-HT7 homodimers with one orthosteric site pseudo-irreversibly bound to risperidone and the other orthosteric site unoccupied, the conclusion being that the inactive conformation of the risperidone-bound protomer globally inactivates the homodimer. Hence, agonist binding at the unoccupied second orthosteric site would not be capable of simulating receptor activity. Central to this hypothesis is that the protomers of the 5-HT7 homodimer are capable of allosterically modulating each other's activity and binding properties. This principle, though novel in the GPCR field, is a common observation amongst ligand-gated ion channels where the state of one subunit of the channel impacts the activity of other subunits (Barnard, 1997).
-
(2)
Exposure to competitive antagonists causes pseudo-irreversibly bound [3H]risperidone to dissociate from 5-HT7 receptors and all receptor activity is restored (“reactivation”) A homodimer structure allows for a straight forward explanation for this phenomenon and is consistent with the previous observation: the unoccupied binding site of a [3H]risperidone-inactivated 5-HT7 homodimer allows competitive antagonists to bind the receptor, and via protomer-protomer crosstalk this interaction allosterically modulates the binding properties of the [3H]risperidone-bound protomer. Thus, the [3H]risperidone-bound protomer no longer binds [3H]risperidone pseudo-irreversibly and allows its dissociation. After washing away the competitive antagonist and released [3H]risperidone from the cellular media, both orthosteric sites of a 5-HT7 homodimer can be radiolabeled and are available to bind agonist - full receptor activity is restored. By this reasoning, the observed full reactivation of risperidoneinactivated receptors implied that none of the 5-HT7 receptors existed as monomers. A partial recovery of 5-HT7 receptor activity would have left open the possibility that 5-HT7 monomers co-exist with homodimers and bind risperidone pseudo-irreversibly and are inactivated but fail to be reactivated by competitive antagonists because of the lack of protomer-protomer interactions, but this was not observed.
-
(3)
Exposure to risperidone and 9-OH-risperidone also causes pseudo-irreversibly bound [3H]risperidone to dissociate from 5-HT7 receptors, and yet the receptors remain inactivated This finding can be explained by a homodimer structure and is in fact predicted by the two previous observations: like a competitive antagonist, risperidone binds the unoccupied second orthosteric site of a [3H]risperidone-inactivated 5-HT7 receptor homodimer and allosterically induces the dissociation of [3H]risperidone. However, unlike a competitive antagonist, risperidone remains pseudo-irreversibly bound to the second orthosteric site, thus keeping the 5-HT7 homodimer in an inactivated state. A cycle can be envisioned where risperidone molecules are constantly exchanged at the 5-HT7 receptor homodimer: one pseudo-irreversibly bound risperidone molecule is released but only upon the pseudo-irreversible binding of another risperidone molecule, thus the homodimer is never free of risperidone and remains inactivated. Since the non-radioactive risperidone is present at saturating levels and the total amount of [3H]risperidone is very low, the net effect over time is the apparent loss of radioactive binding.
-
(4)
Unlike risperidone and 9-OH risperidone, 5-HT7 receptors that are inactivated by the other inactivators (methiothepin, bromocryptine, lisuride, and metergoline) cannot be reactivated by competitive antagonists This observation is consistent with a homodimer structure and the three previous observations. Only risperidone and 9-OH-risperidone exhibited the unusual behavior of maximally binding 50% of 5-HT7 receptors pseudo-irreversibly; the other inactivators appeared to bind all sites pseudo-irreversibly. Thus, 5-HT7 homodimers inactivated by saturating concentrations of methiothepin, bromocryptine, lisuride, or metergoline have no unoccupied orthosteric binding sites for competitive antagonists to bind. Unable to interact with competitive antagonists, these homodimers remain inactivated. Whether or not the pseudo-irreversible binding of a single molecule of methiothepin, bromocryptine, lisuride, or metergoline is sufficient to globally inactivate the homodimer is not clear from this early report, as all experiments were conducted with saturating levels of these inactivators, thus occupying all receptor sites. However, later data garnered from primary rat astrocyte 5-HT7 receptors and from 5-HT2A receptors suggests this may be the case and that so long as one orthosteric site on a homodimer remains unoccupied, the homodimer can be reactivated by exposure to competitive antagonists (see Sections 3 & 4).
Figure 6. Schematic presentation of a homodimeric model consistent with the anomalous effects of drugs on the h5-HT7 receptor.

Risperidone (Risp) binds to a protomer of a h5-HT7 receptor homodimer, resulting in a pseudo-irreversible complex between the drug and the protomer, the inactivation of the h5-HT7 receptor, and the inability of the second protomer to bind risperidone irreversibly. The second protomer retains reversible drug binding properties. The binding of a competitive antagonist, such as mesulergine (Mesul) to the second protomer releases the pseudo-irreversibly bound risperidone from the first protomer, leaving a reactivated receptor. Presumably, methiothepin and bromocriptine bind in a wash-resistant manner to both protomers, and when saturating concentrations of these drugs are used, this results in an inactivated receptor that cannot be reactivated (not shown in model: see Figure 9).
3. Applying pharmacological inactivation and reactivation to a primary culture system: demonstration of, native GPCR homodimers
As mentioned in Section 1, major obstacles to expanding our knowledge of the prevalence and properties of GPCR dimers were created by the experimental designs and methodologies used to study dimers. The use of mutated forms of GPCRs to demonstrate heterodimers or homodimers requires transfection and over-expression of these receptors in recombinant cell lines. As described in Section 2, the use of pseudo-irreversible antagonists to inactivate the h5-HT7 receptor and competitive antagonists to reactivate the receptor allows the study of native h5-HT7 receptor homodimer functionality (i.e. protomerprotomer allosteric crosstalk) in a recombinant cell line (Teitler et al., 2010). The properties of the receptor were measured using the most common and most relevant functional assay for Gs-coupled GPCRs: cAMP accumulation. Thus, it appeared likely that this approach could be applied to a primary cell culture that endogenously expresses functional 5-HT7 receptors.
It had been reported that cortical astrocytes express a functional 5-HT7 receptor in primary culture (Hirst, Cheung, Rattray, Price, & Wilkin, 1998; Hirst, Price, Rattray, & Wilkin, 1997). We were able to reproduce these results, and all the 5-HT-stimulated cAMP production in the primary astrocytes cultures appeared to be 5-HT7-receptor mediated. Consistent with previous reports (Hirst et al., 1998; Hirst et al., 1997), we could not detect a substantial radioligand binding signal, indicating the astrocytes express a low level of highly functional 5-HT7 receptors (Smith, Toohey, Klein, Knight, & Teitler, 2010). Nevertheless, the presence of a functional r5-HT7 receptor allowed us to perform the same series of experiments we had performed using HEK-293-h5-HT7 cells described in Section 2. The r5-HT7 receptor endogenously expressed by cortical astrocytes produced very similar results to the h5-HT7 receptor expressed in the HEK-293-h5-HT7 cells.
-
1)
Risperidone, 9-OH-risperidone, methiothepin, and bromocryptine interact with the r5-HT7 orthosteric site to produce a wash-resistant inhibition of r5-HT7 receptor activity, i.e. these drugs are “inactivators” at the r5-HT7 receptor in primary culture.
-
2)
Competitive antagonists reverse the inactivation produced by risperidone, 9-OH-risperidone, and non-saturating levels of methiothepin. The reactivation of methiothepin, though initially surprising, is consistent with the homodimer model: methiothepin, which is capable of binding both protomers pseudo-irreversibly (see Section 2.3), need only bind one protomer to globally inactivate the 5-HT7 homodimer. Non-saturating levels of methiothepin allow for many of these single methiothepin-bound homodimers to accumulate, which can then be reactivated by competitive antagonist interaction with the unoccupied second orthosteric site. This property of GPCR homodimers (i.e. partial inactivation – partial reactivation) was not observed in the HEK-293-h5-HT7 cells because all experiments carried out in this earlier report used saturating levels of inactivators. Thus, methiothepin pseudo-irreversibly occluded both binding sites of h5-HT7 homodimers and prevented reactivation. More extensive studies of partial inactivation are conducted for 5-HT2A receptors (see Section 4) and are consistent with these findings for r5-HT7 receptors in astrocytes.
-
3)
The potencies of clozapine, mesulergine, mianserin, cyproheptadine, and SB269970 as reactivators closely correlate with their potencies as antagonists of r5-HT7 receptor-mediated stimulation of cAMP (Figure 7).
Based on the reasoning applied to the h5-HT7 receptor expressed in HEK-293 cells (Section 2), these observations indicated the r5-HT7 receptor endogenously expressed by rat astrocytes appears to function as a homodimer, which allows competitive antagonists to “release” risperidone and reactivate the homodimer through protomer-protomer allosteric crosstalk. This was the first functional evidence for a native, endogenously expressed GPCR homodimer and the first evidence for allosteric interaction between the protomers of an endogenously expressed GPCR homodimer. This was a major step forward in establishing the in vivo relevance of GPCR homodimers (Smith et al., 2010; Teitler et al., 2010). This work demonstrates that using pseudo-irreversible antagonists (inactivators), the experimental design of pharmacologically inactivating and reactivating native GPCRs can be used to assess the functionality and relevance of other GPCR homodimers ex vivo and potentially in vivo as well.
Figure 7. Correlation between competitive antagonists' reactivation potencies (pEC50, ordinate) and potencies to inhibit 5-HT-stimulated cAMP production (pKb, abscissa) in rat cortical astrocytes.

The correlation is highly significant (r2=0.92, p=0.01); this indicates that the reversal of risperidone's inactivating effect is occurring through the r5-HT7 receptor orthosteric binding site. These results are from (Smith et al., 2010) and are the means±S.E.M. of three independent experiments performed in triplicate.
4. Inactivation and reactivation of the h5-HT2A receptor: quantitative analysis of homodimer protomer-protomer allosteric crosstalk
4.1 Methiothepin and ritanserin: Inactivating antagonists at the h5-HT2A receptor
The strategy of using the inactivation and reactivation of 5-HT7 receptors to provide strong evidence for homodimers in vitro and in primary cell culture was a novel approach. In order to determine if our observations were unique to the 5-HT7 receptor, we decided to apply this approach to other GPCRs. We chose to investigate the possibility of 5-HT2A homodimers for several reasons: (1) The 5-HT2A receptor is a target for antipsychotic drugs and hallucinogens, is highly expressed in the brain, and is involved in control of platelet function (Meltzer, Matsubara, & Lee, 1989; Titeler, Lyon, & Glennon, 1988; Meltzer, 1993; Appel et al., 1990). Thus, the 5-HT2A receptor is of interest to many biomedical researchers. (2) Unlike the 5-HT7, the 5-HT2A receptor is Gq-coupled receptor (stimulation of intracellular inositol phosphate formation), and thus adds diversity of second messenger systems to our investigation of GPCR homodimers. (3) Rat primary cultured astrocytes have been reported to express a highly functional 5-HT2A receptor, which produces a robust stimulation of inositol phosphates (Hansson, Simonsson, & Alling, 1987). Thus, we could examine our strategy of inactivating and reactivating the 5-HT2A receptors in a recombinant cell line and a primary cell culture in order to substantiate the utility of this approach to the question of the prevalence and properties of GPCR homodimers.
We were able to reproduce the 5-HT2A receptor-mediated stimulation of inositol phosphate production: the use of 5-HT2A-selective antagonists indicated the stimulation was solely due to 5-HT2A receptor activity (data not shown). We screened 12 drugs for inactivating activity and two drugs, methiothepin and ritanserin, proved to be potent inactivators of the h5-HT2A receptor (Figure 8). While methiothepin's inactivating properties at the 5-HT2A receptor are not entirely surprising, as it has shown this activity at the h5-HT7 receptor (Figure 4) (Smith et al., 2010; Teitler et al., 2010), ritanserin was a novel finding. Subsequent to observing the inactivating properties of ritanserin, we found two reports of “non-competitive antagonism” of the 5-HT2A receptor by ritanserin (Leysen et al., 1985; Leysen, Van, Gommeren, & Laduron, 1987). However, we did not find a drug with the properties that risperidone displayed in regulating 5-HT7 receptor activity, i.e. the ability to fully inactivate all receptors while pseudo-irreversibly binding only 50% of sites after washout. As mentioned above, this property allowed us to use risperidone, at saturating concentrations, to fully inactivate the h5-HT7 receptor and fully reactivate the receptor, as every inactive dimer contained an unoccupied protomer after washout (Figure 6). Thus, we needed to modify our strategy in order to study 5-HT2A receptor inactivation and reactivation.
Figure 8. Concentration-response curves for inactivation of h5-HT2A receptor stimulated IP3 production in HEK-293-h5-HT2A cells.

Cells were exposed to varying concentrations of methiothepin or ritanserin for 30 min. After the drug washout procedure, the cells were exposed to 100μM 5-HT for 30 min and intracellular IP3 levels were determined. The results are the means ±S.E.M. of three independent experiments performed in triplicate.
4.2 Reactivation of partially inactivated h5-HT2A receptors
To produce inactive h5-HT2A receptors that could be reactivated, we made a slight alteration to the experimental design. Instead of saturating the h5-HT2A receptors with the inactivator ritanserin, we exposed the HEK-293-h5-HT2A cells to non-saturating concentration of ritanserin (10nM). As shown in Figure 9, if the h5-HT2A receptor is a homodimer with properties similar to that of the h5-HT7 receptor, a non-saturating concentration of an inactivator like ritanserin should produce a mixed population of homodimers. This mode of inactivation and reactivation makes the fewest new assumptions about GPCR homodimer function:
-
1)
Protomers on the unoccupied homodimer are equally likely to interact with methiothepin
-
2)
The binding of methiothepin to one protomer does not change the likelihood of the second protomer interacting with methiothepin
-
3)
A homodimer with one protomer occupied by methiothepin is inactive.
-
4)
The binding of a competitive antagonist to a homodimer occupied by a single inactivator results in the release of the inactivator; washing out the drugs returns the homodimer to a fully activatable state (“reactivation”).
Figure 9. Schematic depiction of a homodimer structure for the 5-HT2A receptor and the effects of inactivating antagonist at 50% fractional occupancy.

The binding of ritanserin to the dimer produces a pseudo-irreversible protomer-drug complex. The binding of one inactivator produces an inactive dimer. Washing out ritanserin leaves three populations of 5-HT2A receptors: doublets (two inactivators bound), singlets (one inactivator bound), and empty receptors (no inactivator bound). Only the singlets can be reactivated by the subsequent exposure of the receptor to a competitive antagonist such as spiperone. The binding of a competitive antagonist to the unoccupied protomer of the singlets produces an allosteric affect on the inactivator-bound protomer, dissociating the inactivator from that protomer. Washing out drugs at this point leaves the singlet receptors unoccupied and thus “reactivated”. The experimental design involves native receptors and thus can be utilized in cell culture, other types of tissue preparations, and, conceivably, in vivo.
When using an inactivator that can pseudo-irreversibly bind both protomers of a dimer, the model (Figure 9) predicts that after exposure to non-saturating levels of inactivator, dimers will exist as “doublets” (inactive and non-recoverable dimer), “singlets” (inactive but recoverable dimer), and “empties” (no protomers are occupied and the dimer remains active). Hence, a partially inactivated population of dimers results in a situation where some, but not all, receptor activity can be recovered: reactivation is limited by the amount of doublets present. This is demonstrated in Figure 10, where HEK-293-h5-HT2A cells are exposed to a non-saturating concentration of inactivator (10nM ritanserin); only the empty receptors remain active. Following reactivation by competitive antagonist (1μM spiperone), receptor activity is only partially restored; the fraction of receptor activity that has been restored should equal the fraction of dimers that were singlets before the reactivation step. The fraction of receptor activity that cannot be reactivated is due to the presence of doublets. With knowledge of the proportion of dimers that are doublets, singlets, and empties, a quantitative and predictive model of dimer inactivation/reactivation can be established.
Figure 10. Partial inactivation and partial reactivation of ritanserin-inactivated h5-HT2A receptors.

Intact HEK-293-h5-HT2Acells were treated with buffer (control) or 10nM ritanserin, thoroughly washed, then exposed to buffer (“Ritan/Veh”) or 1μM spiperone (“Ritan/Spip”). The competitive antagonist spiperone partially reactivates the ritanserin-inactivated 5-HT2A receptors. It is important to note that unlike risperidone's effect at the h5-HT7 receptor, ritanserin appears capable of inhibiting the binding of 100% of the h5-HT2A receptors in a wash-resistant manner (data not shown). Thus, the use of ritanserin as an inactivator presents unique challenges. The model presented in Figure 9 predicts partial reactivation using a drug such as ritanserin. Data are means±s.e.m. from three experiments performed in triplicate. *p<0.001 compared to “Veh/Veh,” #p<0.001 compared to “Ritan/Veh,” one-way ANOVA with Bonferroni corrected t-test.
4.3 A predictive model for inactivation and reactivation of h5-HT2A homodimers
The proportions of doublet, singlet, and empty dimers resulting from exposure to any concentration of inactivator can be defined by simple binomial distribution. The model illustrated in Figure 9 is the simplest and most straight forward example of this: if all receptors are dimers and the fractional occupancy of the inactivator is 50% (i.e. there is a (1/2)1 probability that any one protomer is occupied by inactivator), then the probability that a dimer is double occupied is 25% (i.e. there is a (1/2)2 probability that any two protomers are occupied by inactivator). Hence, to determine the number of doublets, one need only raise the fractional occupancy of the inactivator (“FOi”) to a power equal to the number of “trials” of a binomial probability test. In this case “trials” is synonymous with the number of interacting protomers, and for a homodimer this number is 2. Conversely, to find the proportion of empty dimers, the fractional occupancy must be subtracted from 1 (“1 − FOi”) then raised to a power equal to the number of “trials” (interacting protomers). This is very simple for the situation illustrated in Figure 9, where at 50% fractional occupancy, 25% of dimers are entirely unoccupied (i.e. there is a (1 − 1/2)2 probability that any two protomers are left unoccupied by inactivator). The population of dimers remaining after subtracting doublets (25%) and empties (25%) are the singlets (50%) or, in the case of higher order oligomers, the population of receptors that are inactivated (bound to at least one molecule of inactivator) but have at least one binding site available to interact with reactivating antagonist. These relationships are governed by binomial distribution and represented in equation 1, where the term “Oligomer” is used to represent the number of “trials” (interacting protomers) and receptor activity is solved for by determining the proportion of empties (“(1 − FOi)Oligomer”) plus the level of reactivated singlets (“(1 − (FOi)Oligomer − (1 − FOi)Oligomer”), which is governed directly by the fractional occupancy of the reactivator (FOr). Equations 2 and 3 describe the fractional occupancy of the inactivator and the reactivator, respectively.
| eq.1 |
| eq.2 |
| eq.3 |
Equation 1 is the quantitation of a rather straight forward concept: as inactivator concentration varies, the maximal level of reactivation will vary by a predictable degree. At low concentrations of inactivator, much of the inactivation is due to the formation of singlets and very few doublets are formed, thus most of the inactivation is reversible. At high concentrations of inactivator, more doublets are formed and the population of singlets dwindles, thus much of the receptor activity is non-recoverable. The maximal level of reactivation is at the EC50 of the inactivator's fractional occupancy curve, wherein the maximal amount of singlets is formed. The total population of reactivatable receptors is dictated by the number of interacting protomers per receptor oligomer: higher order oligomers, if capable of being inactivated by a single molecule of inactivator and reactivated by a single molecule of reactivator, would allow for more reactivatable receptors to form over a wider range of inactivator concentrations. This concept is demonstrated in Figure 11A, where the inactivation concentration-response curve of ritanserin is simulated based on equation 1 for monomers, dimers, tetramers, and octomers of 5-HT2A receptors. The solid lines indicate receptor activity immediately after inactivation: only the empty receptors remain active. The dashed lines indicate the receptor activity after reactivation with a saturating concentration of reactivator. When the Oligomer value equals 1 (i.e. a monomer: no allosterism between protomers), no reactivation is possible: non-reactivated and reactivated curves overlap. When the Oligomer value equals 2 (i.e. dimer: allosteric interaction between two protomers), reactivation is possible: the non-reactivated curve is negatively inflected and appears leftward shifted, the reactivated curve is positively inflected and appears rightward shifted. As the oligomer value increases, the separation of the non-reactivated and reactivated curves increases, indicative of the potential for the formation of more singlets (or inactivated but recoverable oligomers). We have hypothesized that the h5-HT2A receptor functions as a homodimer, and supporting this is Figure 11B, which displays the inactivation concentration-response curve for ritanserin before (solid line) and after (dashed line) reactivation by a saturating concentration of spiperone. These results closely match the predicted properties of h5-HT2A receptor homodimers (blue curves of Figure 11A) The Oligomer value best-fit to these data is 2.2±0.2 (95% confidence interval=1.936 to 2.464), which is not significantly different from 2 (p > 0.05, F-test), but is significantly different from 1,3 or any larger integer (p > 0.05, F-test). This supports the hypothesis that h5-HT2A receptors function as dimers.
Figure 11. Simulation (A) and actual effect at h5-HT2A receptors (B) of ritanserin inactivation concentration-response and reactivation by a competitive antagonist.

A) At different levels of inactivation, different levels of maximal reactivation are predicted based on the reactivation model presented in Figure 9. Data are simulated for monomers, dimers, tetramers and octomers (Oligomer values equal 1, 2, 4, and 8, respectively). B) Reactivation results appear to be consistent with the simulated behavior of a dimer (i.e. Oligomer value is not significantly different from 2). The effect of the competitive antagonist spiperone (at a saturating concentration of 1μM) is the apparent rightward shift of the ritanserin inactivation concentration-response curve; the degree of this shift is limited by the number of interacting protomers. Arrows emphasize the partial reactivation of partially inactivated receptors. Data are mean±S.E.M. three independent experiments performed in triplicate.
This method of probing the dimer-related functionality of GPCRs is extremely sensitive, but it requires knowledge of the reactivator's potency in order to reliably generate Oligomer values. Thus, the effects of various concentrations of the reactivator should be assessed, i.e. there are two independent variables: (1) inactivator concentration and (2) reactivator concentration. The most robust means of analyzing data of this experimental design is three-dimensional nonlinear regression (i.e. surface fitting). Figure 12 displays the results of such an experiment; surface fitting to these data produces an Oligomer value of 2.14±0.07, which is not significantly different from 2 (p > 0.05, F-test) and supports the homodimer hypothesis. Although all data points were included in the surface fitting analysis, the data points pertaining to the reactivation concentration-response curve at a single concentration of ritanserin are highlighted as red spheres and a best-fit curve is displayed to illustrate the concentration-dependency and high potency of spiperone's reactivating abilities. Other inactivating and reactivating drugs must be examined in this fashion in order to codify these observations as a characteristic of the native h5-HT2A homodimers and not a drug-dependent phenomenon. Preliminary experiments with other drugs have produced results similar to those displayed in Figure 12 (unpublished observations).
Figure 12. Three-dimensional depiction of inactivation and reactivation of the h5-HT2A receptor.

The inactivation and reactivation of a GPCR involves the effects of two drugs, each with a concentration-dependent effect. Thus, the dependent variable (receptor activity), plotted on the z-axis, is a function of two independent variables, plotted on the x-axis (ritanserin, the inactivator) and y-axis (spiperone, the reactivator). The effects on receptor activity of varying the concentrations of the inactivator and reactivator can be surface-fit to equation 8, yielding a globally best-fit determination for the Oligomer value (2.2±0.2, not significantly different from 2, p > 0.05, F-test). The spiperone reactivation concentration-response curve for a single concentration of ritanserin is highlighted (points are red spheres, red sigmoidal curve), depicting a more conventional concentration-response curve.
The model used to calculate Oligomer values (eq. 1) and predict the reactivation of partially inactivated receptors (Figures 11 & 12) assumes that all singlets (inactivated oligomers with at least one unoccupied orthosteric site) can be reactivated upon binding of competitive antagonist at the available orthosteric site. The Oligomer value is directly associated with the magnitude of the “rightward shift” of the inactivation CRC caused by reactivation. The presence of inactivated monomeric receptors, which are incapable of binding a second orthosteric ligand and cannot be reactivated, would lessen the magnitude of the “rightward shift” and decrease the Oligomer value. Thus, an Oligomer value significantly less than 2 would indicate the presence of inactivated monomeric receptors. If all receptors exist as monomers, no reactivation is possible (i.e. no “shift” in the inactivation CRC), yielding an Oligomer value of 1. If a mixture of functional monomeric and dimeric receptors is expressed on the plasma membrane, the Oligomer value would range from 1 to 2, dependent on the proportion of functional receptors that exist as monomers and dimers. For scenarios where functional monomers, dimers, and higher order oligomers all coexist on the plasma membrane, the Oligomer value will be difficult to interpret, as it is possible that some combination of oligomers and monomers would allow for the exact magnitude “rightward shift” necessary to produce an Oligomer value of 2. However, Oligomer values have been determined at different receptors (5-HT7 and 5-HT2A), in multiple preparations (recombinant cell lines and primary cell culture), and using multiple combinations of inactivators and reactivators, and as yet, no Oligomer value has been found to significantly differ from 2. This is an unlikely occurrence if monomers, dimers, and higher order oligomers are all expressed on the plasma membrane.
In summary, methiothepin and ritanserin were identified as inactivators at h5-HT2A receptors and reactivation of partially inactivated populations of h5-HT2A receptors has demonstrated the homodimeric functionality of these receptors. Although no h5-HT2A receptor inactivating drugs have been identified that have properties similar to risperidone at 5-HT7 receptors, the mode in which methiothepin and ritanserin inactivate the receptor (i.e. pseudo-irreversibly binding both sites of a h5-HT2A homodimer) is likely to be the more common mechanism of inactivation when inactivators are identified at other GPCRs. Thus, the quantitative method that we developed for assessing the dimeric functionality of GPCRs via inactivation and reactivation will likely be indispensable in future investigations of this type. Much work remains to be done for h5-HT2A homodimers, including assessment of homodimers in astrocytes and in vivo, but the evidence presented herein strongly indicates that native h5-HT2A receptors function as homodimers.
5. Conclusions and Perspectives
The GPCR is a fundamental component of intercellular communication, GPCR signaling and GPCR dysfunctions are involved in various pathologies, and drugs that target these receptors are a major class of therapeutics. A tremendous amount of research has gone into uncovering the basic structure and function of GPCRs, and yet the prevalence of GPCR homodimers and heterodimers in their native environment is unknown. The techniques available to study GPCR dimers generally could not be employed outside of recombinant cell lines, thus little insight into the prevalence of GPCR homodimers and heterodimers in vivo has been achieved. New techniques were clearly needed.
We have discovered that GPCRs can bind certain antagonists (inactivators) in a pseudo-irreversible manner that is reversible through the binding of other competitive antagonists to what appears to be the same orthosteric binding site. This property of a GPCR can best be interpreted as being due to a homodimer structure in which, initially, one protomer binds the inactivator and, subsequently, the second protomer binds the competitive antagonist. An allosteric interaction between the protomers (i.e. protomer-protomer allosteric crosstalk) releases the inactivator and subsequent washout of the drugs restores the GPCR to an active state. This drug-induced inactivation/reactivation procedure, besides revealing novel drug-receptor interactions in recombinant cell lines, can also be utilized in primary culture, as demonstrated with the r5-HT7 receptor in astrocyte cultures. Thus, this approach to GPCR pharmacology allows the investigation of the prevalence and functionality of GPCR homodimers ex vivo, and possibly in vivo. This procedure employs basic pharmacological techniques, which are available to many laboratories, and should allow a determination as to whether the results presented herein concerning the 5-HT7 and 5-HT2A receptors are unusual or are commonly observed when inactivators and competitive antagonists are available for the GPCR being studied.
Pseudo-irreversible binding of ligands to the CCR2/CCR5 heterodimer, and the allosteric acceleration of dissociation of the ligands by drugs binding to the other protomer in the heterodimer has been reported (Springael, Urizar, Costagliola, Vassart, & Parmentier, 2007; Springael et al., 2006). The authors correctly concluded their results were unambiguous evidence for protomer-protomer crosstalk in heterodimers (Springael et al., 2006). Unfortunately this work was dismissed by the IUPHAR committee: “…one cannot so far rule out the possibility that such negative interaction may also result from indirect cross-talk.” As these studies were performed in homogenate preparations, indirect effects seem highly unlikely. It should be emphasized that while the CCR2/CCR5 studies were performed in membrane homogenates and involved radioligand binding studies the studies presented herein were mainly performed in intact cells and involved functional measurements. Our experimental design may more easily be adapted to the in vivo situation.
We propose that most GPCRs are expressed and function as homodimers on the plasma membrane of cells. GPCRs do not function as monomers; rather they exist either as homodimers or heterodimers (e.g. the GABAB1/GABAB2 complex). Our reasoning is that we have studied two receptors using the inactivation/reactivation strategy: both receptors reveal properties best interpreted as due to homodimer structure and function. The dimer-related properties of the 5-HT7 receptor were discovered serendipitously; to determine if these properties would be present in other another GPCR; the 5-HT2A receptor was the first receptor we chose to investigate. If the dimeric structure is rare, we have certainly been very lucky when choosing which receptors to study (2 positive hits out of an n of 2). It seems more likely that GPCRs prefer the homodimer structure for functional reasons not yet known, and this will become evident as research proceeds using the inactivation/reactivation experimental design (or other designs not yet developed).
One can organize the various views on the structure of GPCRs as a continuum of complexity. The simplest is that GPCRs are generally monomers. This has been the conventional opinion, and for decades there was no significant literature disputing this viewpoint. The most complex view that has been expressed is the dynamic system of monomers, dimers, and higher order structures co-existing on the plasma membrane and, furthermore, drugs change the proportion of these forms of the receptors. Our sense, based on the two receptors we have studied and the existing literature on GPCR structure, is that GPCRs function as homodimers except for the few that function as heterodimers. In the context of the classical monomer model and the chaotic oligomeric model, the homodimer model may be viewed as less than radical, but rather a somewhat conservative interpretation, given our results.
If most GPCRS exist as a dimeric structure, there is presumably a functional reason for this. It is known that the protomers of the GABAB1/GABAB2 receptor protomers must be co-expressed so that the complex can exit the endoplasmic reticulum (ER) and translocate to the plasma membrane. However, the view that this is the purpose of the dimeric structure, i.e. to cover the ER retention signal of the GABAB1 receptor, may be only part of the story. We feel that this mechanism is a cellular check to ensure the expression of a dimeric GPCR structure on the plasma membrane. Why plasma membrane expression of GPCR dimers is needed is unknown but may involve mechanisms for rapid regulation of GPCR signaling, i.e. the protomer-protomer interaction may be critical for maintaining proper levels of GPCR activity. If so, there may be many instances of disease states due to improper GPCR dimer formation. The role(s) of such dysfunctions cannot be addressed until the basic questions of GPCR dimer prevalence and function in vivo are settled.
It is our sense that our experimental design can be used to study most GPCRs. This is dependent on the availability of drugs that form a pseudo-irreversible complex with the GPCR, inactivating the GPCR. We have now studied three receptors, the 5-HT7, 5-HT2A, and D2 dopamine receptors (Teitler, Smith, Toohey, & Klein, 2011), and in all three cases, within only a few weeks of low throughput screening we found several pseudo-irreversible antagonists. This implies that inactivating antagonists are common and that if several dozen antagonists of a GPCR are tested, at least several inactivating antagonists will be found. Of course this requires the development of a sizable group of antagonists for the GPCR. Medicinal chemists have generally been able to develop numerous antagonists for GPCRs in a short period of time. GPCRs for which antagonists cannot be developed would not be subject to the experimental design outlined in this review. Once inactivators are identified, the experimental design described in this review can be attempted. Thus, it likely that GPCR protomer-protomer allosteric crosstalk can be studied using inactivating antagonists for many, if not a majority, of GPCRs.
If our experimental design using inactivators and competitive antagonists to expose the presence and properties of GPCR homodimers is exploited and reveals that this is the preferred form of GPCRs, a paradoxical situation would arise. We have emphasized the limitations of using mutated forms of GPCRs in recombinant cell lines to establish the basic question as to the structure of GPCRs in vivo (see sections 1.2&1.4). However, if GPCRs do form dimers in vivo, then the work in recombinant cells during the last decade will have added significance. At the present time, one may choose to dismiss these studies, many of which are brilliant in design and results, as not relevant to the in vivo situation. If our experimental design, or some other approach that allows the demonstration of GPCR structure and function, does indicate that GPCRs are dimers in vivo, then the work in recombinant cell lines will be viewed far less skeptically and appreciated far more fully. We suspect that our work on native GPCR homodimers will, in the end, substantiate many of the studies in recombinant cell lines and lead to many more experiments well-justified by results indicating GPCR oligomers are relevant in vivo.
Acknowledgments
Funding source Supported by the National Institutes of Health, National Institute of Mental Health [Grant MH56650] (M.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Appel NM, Mitchell WM, Garlick RK, Glennon RA, Teitler M, De Souza EB. Autoradiographic characterization of (+−)-1-(2,5-dimethoxy-4-[125I] iodophenyl)-2-aminopropane ([125I]DOI) binding to 5-HT2 and 5-HT1c receptors in rat brain. J.Pharmacol.Exp.Ther. 1990;255:843–857. [PubMed] [Google Scholar]
- Barnard EA. Ionotropic glutamate receptors: new types and new concepts. Trends Pharmacol.Sci. 1997;18:141–148. doi: 10.1016/s0165-6147(97)01053-5. [DOI] [PubMed] [Google Scholar]
- Borison RL, Diamond B, Pathiraja A, Meibach RC. Pharmacokinetics of risperidone in chronic schizophrenic patients. Psychopharmacol.Bull. 1994;30:193–197. [PubMed] [Google Scholar]
- Chabre M, Cone R, Saibil H. Biophysics: is rhodopsin dimeric in native retinal rods? Nature. 2003;426:30–31. doi: 10.1038/426030b. [DOI] [PubMed] [Google Scholar]
- Chabre M, le MM. Monomeric G-protein-coupled receptor as a functional unit. Biochemistry. 2005;44:9395–9403. doi: 10.1021/bi050720o. [DOI] [PubMed] [Google Scholar]
- Ferre S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, et al. Building a new conceptual framework for receptor heteromers. Nat.Chem.Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Jastrzebska B, Philippsen A, Muller DJ, Palczewski K, Engel A. Structure of the rhodopsin dimer: a working model for G-protein-coupled receptors. Curr.Opin.Struct.Biol. 2006;16:252–259. doi: 10.1016/j.sbi.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Franco R, Casado V, Cortes A, Ferrada C, Mallol J, Woods A, et al. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. ScientificWorldJournal. 2007;7:48–57. doi: 10.1100/tsw.2007.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Venter JC. The size of the mammalian lung beta 2-adrenergic receptor as determined by target size analysis and immunoaffinity chromatography. Biochem.Biophys.Res.Commun. 1982;109:21–29. doi: 10.1016/0006-291x(82)91560-1. [DOI] [PubMed] [Google Scholar]
- Hansson E, Simonsson P, Alling C. 5-Hydroxytryptamine stimulates the formation of inositol phosphate in astrocytes from different regions of the brain. Neuropharmacology. 1987;26:1377–1382. doi: 10.1016/0028-3908(87)90102-x. [DOI] [PubMed] [Google Scholar]
- Hern JA, Baig AH, Mashanov GI, Birdsall B, Corrie JE, Lazareno S, et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc.Natl.Acad.Sci.U.S.A. 2010;107:2693–2698. doi: 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst WD, Cheung NY, Rattray M, Price GW, Wilkin GP. Cultured astrocytes express messenger RNA for multiple serotonin receptor subtypes, without functional coupling of 5-HT1 receptor subtypes to adenylyl cyclase. Brain Res.Mol.Brain Res. 1998;61:90–99. doi: 10.1016/s0169-328x(98)00206-x. [DOI] [PubMed] [Google Scholar]
- Hirst WD, Price GW, Rattray M, Wilkin GP. Identification of 5-hydroxytryptamine receptors positively coupled to adenylyl cyclase in rat cultured astrocytes. Br.J.Pharmacol. 1997;120:509–515. doi: 10.1038/sj.bjp.0700921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Knight JA, Smith C, Toohey N, Klein MT, Teitler M. Pharmacological analysis of the novel, rapid, and potent inactivation of the human 5-Hydroxytryptamine7 receptor by risperidone, 9-OH-Risperidone, and other inactivating antagonists. Mol.Pharmacol. 2009;75:374–380. doi: 10.1124/mol.108.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R, Kohr G, Grunewald S, Eisenhardt G, Bach A, Kornau HC. Role of heteromer formation in GABAB receptor function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Cerione RA, Codina J, Birnbaumer L, Caron MG. Reconstitution of the beta-adrenergic receptor. J.Membr.Biol. 1985;87:1–12. doi: 10.1007/BF01870694. [DOI] [PubMed] [Google Scholar]
- Leysen JE, Gommeren W, Van GP, Wynants J, Janssen PF, Laduron PM. Receptor-binding properties in vitro and in vivo of ritanserin: A very potent and long acting serotonin-S2 antagonist. Mol.Pharmacol. 1985;27:600–611. [PubMed] [Google Scholar]
- Leysen JE, Van GP, Gommeren W, Laduron PM. Differential regulation of dopamine-D2 and serotonin-S2 receptors by chronic treatment with the serotonin-S2 antagonists, ritanserin, and setoperone. Psychopharmacol.Ser. 1987;3:214–224. doi: 10.1007/978-3-642-71288-3_25. [DOI] [PubMed] [Google Scholar]
- Lilly L, Fraser CM, Jung CY, Seeman P, Venter JC. Molecular size of the canine and human brain D2 dopamine receptor as determined by radiation inactivation. Mol.Pharmacol. 1983;24:10–14. [PubMed] [Google Scholar]
- Marshall FH, Jones KA, Kaupmann K, Bettler B. GABAB receptors - the first 7TM heterodimers. Trends Pharmacol.Sci. 1999;20:396–399. doi: 10.1016/s0165-6147(99)01383-8. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. Serotonin receptors and antipsychotic drug action. Psychopharmacol.Ser. 1993;10:70–81. doi: 10.1007/978-3-642-78010-3_7. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J.Pharmacol.Exp.Ther. 1989;251:238–246. [PubMed] [Google Scholar]
- Ng GY, Clark J, Coulombe N, Ethier N, Hebert TE, Sullivan R, et al. Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J.Biol.Chem. 1999;274:7607–7610. doi: 10.1074/jbc.274.12.7607. [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Lefkowitz RJ, Caron MG. Structure of the adrenergic and related receptors. Annu.Rev.Neurosci. 1989;12:67–83. doi: 10.1146/annurev.ne.12.030189.000435. [DOI] [PubMed] [Google Scholar]
- Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, et al. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol.Rev. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]
- Rene P, Le Gouill C, Pogozheva ID, Lee G, Mosberg HI, Farooqi IS, et al. Pharmacological chaperones restore function to MC4R mutants responsible for severe early-onset obesity. J.Pharmacol.Exp.Ther. 2010;335:520–532. doi: 10.1124/jpet.110.172098. [DOI] [PubMed] [Google Scholar]
- Rivero-Muller A, Chou YY, Ji I, Lajic S, Hanyaloglu AC, Jonas K, et al. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc.Natl.Acad.Sci.U.S.A. 2010;107:2319–2324. doi: 10.1073/pnas.0906695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake H, Sakai T. Recent advances and perceptions in studies of heterodimerization between G protein-coupled receptors. Protein Pept.Lett. 2008;15:300–308. doi: 10.2174/092986608783744207. [DOI] [PubMed] [Google Scholar]
- Skrabanek L, Murcia M, Bouvier M, Devi L, George SR, Lohse MJ, et al. Requirements and ontology for a G protein-coupled receptor oligomerization knowledge base. BMC.Bioinformatics. 2007;8:177. doi: 10.1186/1471-2105-8-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M. Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor. Mol.Pharmacol. 2006;70:1264–1270. doi: 10.1124/mol.106.024612. [DOI] [PubMed] [Google Scholar]
- Smith C, Toohey N, Klein MT, Knight JA, Teitler M. Risperidone-induced inactivation and clozapine-induced re-activation of rat cortical astrocyte 5-HT7 receptors: evidence for in situ GPCR homodimer protomer cross-talk. Mol.Pharmacol. 2010 doi: 10.1124/mol.110.069278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Toohey N, Knight JA, Klein MT, Teitler M. Risperidone-induced inactivation and clozapine-induced reactivation of rat cortical astrocyte 5-hydroxytryptamine7 receptors: evidence for in situ g protein-coupled receptor homodimer protomer cross-talk. Mol.Pharmacol. 2011;79:318–325. doi: 10.1124/mol.110.069278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springael JY, Le Minh PN, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric modulation of binding properties between units of chemokine receptor homo- and heterooligomers. Mol.Pharmacol. 2006;69:1652–1661. doi: 10.1124/mol.105.019414. [DOI] [PubMed] [Google Scholar]
- Springael JY, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric properties of G protein-coupled receptor oligomers. Pharmacol.Ther. 2007;115:410–418. doi: 10.1016/j.pharmthera.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Stadel JM, DeLean A, Lefkowitz RJ. A high affinity agonist. beta-adrenergic receptor complex is an intermediate for catecholamine stimulation of adenylate cyclase in turkey and frog erythrocyte membranes. J.Biol.Chem. 1980;255:1436–1441. [PubMed] [Google Scholar]
- Teitler M, Smith C, Toohey N, Klein MT. Fluphenazine potently and rapidly inactivates the human D2 dopamine receptor. Society for Neuroscience Abstracts 41. 2011 [Google Scholar]; Ref Type: Abstract
- Teitler M, Toohey N, Knight JA, Klein MT, Smith C. Clozapine and other competitive antagonists reactivate risperidone-inactivated h5-HT7 receptors: radioligand binding and functional evidence for GPCR homodimer protomer interactions. Psychopharmacology (Berl) 2010;212:687–697. doi: 10.1007/s00213-010-2001-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titeler M, Lyon RA, Glennon RA. Radioligand binding evidence implicates the brain 5-HT2 receptor as a site of action for LSD and phenylisopropylamine hallucinogens. Psychopharmacology (Berl) 1988;94:213–216. doi: 10.1007/BF00176847. [DOI] [PubMed] [Google Scholar]
- Wang M, Pei L, Fletcher PJ, Kapur S, Seeman P, Liu F. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: increased dopamine D2 receptor dimerization. Mol.Brain. 2010;3:25. doi: 10.1186/1756-6606-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc.Natl.Acad.Sci.U.S.A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuo T, Kusuhara Y, Yasumatsu K, Ninomiya Y. Multiple receptor systems for glutamate detection in the taste organ. Biol.Pharm.Bull. 2008;31:1833–1837. doi: 10.1248/bpb.31.1833. [DOI] [PubMed] [Google Scholar]