

Abstract
Vein graft intimal hyperplasia remains the leading cause of graft failure, despite many pharmacological approaches that have failed to translate to human therapy. We investigated whether local suppression of inflammation and fibrosis with MMI-0100, a novel peptide inhibitor of Mitogen Activated Protein Kinase Activated Protein Kinase II (MK2), would be an alternative strategy to reduce cell proliferation and intimal hyperplasia. The cell permeable peptide MMI-0100 was synthesized using standard Fmoc chemistry. Pharmacological doses of MMI-0100 induced minimal human endothelial and smooth muscle cell proliferation (30% and 12% respectively). MMI-0100 suppressed IL-6 expression to control levels, without effect on IL-8 expression. MMI-0100 caused sodium nitroprusside induced smooth muscle cell relaxation and inhibited intimal thickening in human saphenous vein rings in a dose-dependent fashion. In a murine aortic bypass model, MMI-0100 reduced intimal thickness in vein grafts by 72%, and there were fewer F4/80-reactive cells in vein grafts treated with MMI-0100. MMI-0100 prevents vein graft intimal thickening ex vivo and in vivo. These results suggest that inhibition of MK2 with the cell-permeant peptide MMI-0100 may be a novel strategy to suppress fibrotic processes such as vein graft disease.
Keywords: intimal hyperplasia, vein graft, MMI-0100, MAPKAP kinase II, signal transduction
1.0 Introduction
There are >1 million coronary bypass procedures a year worldwide, with human greater saphenous vein remaining the most commonly used conduit. However, less than half of these grafts remain patent after 12 years [Motwani 1998], with more recent data from the PREVENT IV trial demonstrating 42% graft occlusion within 18 months [Alexander 2005]. Graft failure typically leads to myocardial infarction and death, the need for repeated coronary bypass procedures and, consequently, substantial costs to the healthcare system. Thus, approaches to decrease vein graft failure rates would improve outcomes after arterial bypass procedures, yielding significant clinical and health economic benefits.
The leading cause of bypass graft failure is intimal hyperplasia of the vein conduit [Clowes 1991]. While its causes are as yet incompletely understood, intimal hyperplasia results from a cascade of events triggered by the tissue response to mechanical injury associated with surgical vein harvest and conduit preparation; in addition, the damage induced by mechanical dilation used to “break” vessel spasm is refractory to current vasodilators and other pharmacologic approaches [Dashwood 2004, Dashwood 2007].
On a cellular-molecular level, intimal hyperplasia is mediated by a sequence of events, including inflammatory processes in response to vessel trauma, resulting in vascular smooth muscle proliferation, migration, and extracellular matrix production [Allaire 1997]. This is associated with a phenotypic modulation of smooth muscle cells from a contractile to a synthetic phenotype, with “synthetic” cells secreting extracellular matrix proteins [Mosse 1985]. Graft functional responses are also impaired, leading to abnormal vasorelaxation [Klyashkin 1993, Lorusso 2007]. All of these processes lead to pathologic narrowing of the vessel lumen, graft stenosis, and ultimately graft failure [LoGerfo 1983].
Although a number of drugs aiming to reduce development of intimal hyperplasia have been tested in clinical trials, these products have failed. Antithrombotic and antiplatelet agents such as warfarin, clopidogrel and aspirin have little or no effect on intimal hyperplasia [Kent 2004]. Two large clinical trials for the prevention of coronary and peripheral vascular vein graft failure using an E2F decoy to prevent smooth muscle proliferation also failed in their primary endpoint [Alexander 2005; Conte 2006]. Accordingly, availability of novel therapeutic approaches to improve graft patency remains an unmet need.
Recently, Epstein, et al. demonstrated that suppression of the innate immune response in the context of vascular injury dramatically down-regulated the degree of intimal hyperplasia [Danenberg 2002, Epstein 2008]. These results suggest that inflammation plays a major role in intimal thickening and that peri-procedural suppression of inflammation could decrease intimal hyperplasia by a clinically meaningful degree. However, immune suppression on a systemic level during surgical procedures and the post-operative recovery period can increase infection risk, and as such is not clinically feasible. Therefore, we investigated whether local suppression of inflammation, via ex vivo vein graft treatment with MMI-0100, a peptide inhibitor of MAPKAP kinase II (MK2), would be a novel alternative strategy to reduce intimal thickening following vein bypass surgery.
Mitogen Activated Protein Kinase Activated Protein Kinase II (MAPKAP Kinase II, MK2) is an intracellular kinase activated by the p38 Mitogen Activated Protein Kinase (MAPK) [Rouse 1994] that, in turn, phosphorylates transcription factors tristetraprolin (TTP) [Sandler 2008] and hnRNPA0 [Rousseau 2002]. TTP and hnRNPA0 are known to interact with AU-rich regions of mRNA to control mRNA stability and expression. Importantly, studies show that suppression of MK2 activity results in down-regulation of inflammatory cytokine expression, including TNF-α, IL-1β, and IL-6 [Kotlyarov 1999, Winzen 1999, Lehner 2002, Neininger 2002, Wang 2002, Thomas 2008, Funding 2009].
We recently developed a cell-permeant MK2 inhibitor peptide [Lopes 2009] that was based on a peptide designed by Hayess and Bendorff [Hayess 1997]. However, further work with this peptide demonstrated that it was relatively nonselective and toxic, which led to development of significantly more specific inhibitor peptides, including MMI-0100 [Ward 2009]. In an animal model of abdominal adhesions, i.e. rat bowel anastomosis, we reported that a single dose of MMI-0100 applied locally at the time of surgery reduces both number and severity of abdominal adhesions without impairing normal intestinal healing, as determined by hydroxyproline content and burst pressure of the colonic anastomosis [Ward 2011].
These results suggest that inhibition of MK2 with MMI-0100 inhibits inflammatory responses leading to excess extracellular matrix deposition and formation of scars and adhesions. Given the role of inflammation in the development of intimal hyperplasia, we investigated whether MMI-0100 could similarly reduce this clinically relevant vascular process and perhaps ultimately vein graft failure. Therefore, we tested whether MMI-0100 affected vascular cell proliferation and reduced intimal hyperplasia ex vivo and in vivo.
2.0 Material and Methods
2.1 Cell culture
Primary human aortic endothelial cells (HAEC) were obtained from Invitrogen; HAEC were cultured in Medium 200 supplemented with LSGS (Low Serum Growth Supplement), containing FBS (2% v/v), hydrocortisone (1 μg/mL), human epidermal growth factor (EGF, 10 ng/mL), Basic Fibroblast Growth Factor (bFGF, 3 ng/mL), gentamycin/amphotericin (GA) and heparin (10 μg/mL). Primary human aortic smooth muscle cells (HASMC) were obtained from Invitrogen; HASMC were cultured in EGM Bullet Kit – EBM-2 Endothelial Basal Medium 2 supplemented with hEGF (10ng/ml), hydrocortisone (1.0 μg/ml), GA (50 μg/ml), FBS (5%), VEGF, hFGF-B, R3-IGF-1, and ascorbic acid. Primary human coronary artery endothelial cells (HCAEC) were obtained from Lonza; HCAEC were cultured in Medium 231 supplemented with SMGS (Smooth Muscle Growth Supplement), containing FBS (4.9% v/v), bFGF (2ng/ml), hEGF (0.5ng/ml), heparin (5ng/ml), insulin (5 μg/ml), BSA (0.2 μg/ml), and GA.
All cultures were maintained in 25cm2 polystyrene tissue culture flasks in a 37°C, 5% CO2/95% air environment, with cell culture media refreshed every other day. All cells were seeded at a density of 20,000~30,000 cells/cm2, as required by the specific experiment, and allowed to grow to 80–90% confluence before being harvested/passaged. Only cells from early passages (numbers 2–8) were utilized in experiments.
Primary cultures of mouse lung endothelial cells (MLEC) were isolated as previously described [Ackah 2005, Muto 2011]. After immunoselection with magnetic beads, endothelial cells were immortalized with polyoma middle T-antigen. Isolated MLEC were maintained with EBM-2/EGM-2 MV SingleQuot Kit Supplement & Growth Factors (Lonza) containing 15% fetal bovine serum. Cell proliferation in MLEC was measured at 24 and 72 hours after MMI-0100 treatment by direct cell counting after trypsin treatment.
2.2 MMI-0100 reconstitution/dilution
MMI-0100 was synthesized using standard Fmoc chemistry as previously described, with the peptide sequence YARAAARQARAKALARQLGVAA [Ward 2009]. 114mg of MMI-0100 (MW=2283.67g/mol; Moerae Matrix, Inc.) was dissolved in 5ml of phosphate-buffered saline (PBS) to yield a 0.01M stock solution, which was divided into 500 μl aliquots and stored at −20°C. Serial dilutions of stock solution were made to achieve appropriate drug concentrations for each study.
2.3 MTS cell proliferation assay
The CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega) was used to assess drug effects on cell proliferation according to the manufacturer’s instructions. Briefly, HAECs and HASMCs from early passages were grown to 80–90% confluence in 25cm2 tissue culture flasks in a 37°C/5% CO2 incubator prior to harvest. 200 μl of each type of cell suspension (at 20,000 cells/cm2) was seeded onto separate 96-well plates to yield an approximate 60% confluence per well. Cells were allowed to adhere to the plate surface overnight, followed by addition of 20ng/ml of TNF-α to stimulate production of inflammatory agents. After a 4–6 hour incubation period, MMI-0100 peptide drug was added and cells incubated for another 20–24 hours. Each well was then supplemented with 100 μl of fresh medium and 20 μl of CellTiter 96® AQueous One Solution Reagent and incubated for an additional 1.5–2 hours prior to measuring absorbance of each well at 490nm with a SoftMax-equipped plate reader.
2.4 Cell apoptosis analysis
The apoptotic effect of MMI-0100 on MLEC was measured at 24 hours after MMI-0100 treatment. MLEC were removed from the tissue culture plate by trypsin, and re-suspended at 1.0 × 106/ml concentration. Apoptotic cells were detected by AlexaFluor 488 annexin V/Dead Cell Apoptosis Kit (Invitrogen) using flow cytometry sorting analysis.
2.5 ELISA for IL-6, IL-8, and MCP-1 detection
HCAECs were cultured and seeded onto a 96-well plate, using methods described in the MTS proliferation assay, above. Cells were again stimulated with 20ng/ml of TNF-α for 6 hours and then treated with MMI-0100 for approximately 24 hours. Supernatants were then collected and analyzed for drug effect on inflammatory cytokines.
IL-6 (Cat #: 900-K16; Lot #: 0909016) and IL-8 (Cat #: 900-K18; Lot #: 0209018) ELISA kits (Peprotech; Rocky Hill, NJ) were used to measure levels of these cytokines from HCAEC supernatants following treatment with MMI-0100. 9 standards were prepared by following the manufacturer’s protocol. 10 μl of supernatant was diluted with 90 μl of diluents; 3 replicates of each sample were used. Data were collected at 405nm with correction at 650nm on a plate reader. Each plate was monitored for 1 hour with readings taken every 5 minutes. Concentrations of IL-6 and IL-8 in test samples were determined by extrapolating from a standard curve. Data are expressed as means ± SEM.
MCP-1 production from MLEC was analyzed using conditioned culture medium by Quantikine Mouse CCL/JE/MCP-1 Immunoassay (R&D Systems) following the manufacturer’s instructions.
2.6 NO analysis
To measure nitric oxide production, conditioned medium from MLEC was examined at 24 hours after treatment with MMI-0100. The medium was processed for the measurement of nitrite (NO2−) by a NO-specific chemiluminescence analyzer (Sievers) as previously described [Muto 2011].
2.7 Human Saphenous Vein
Following approval by Vanderbilt Medical Center’s Institutional Review Board, de-identified, discarded segments of human saphenous vein (HSV) were collected from consented patients undergoing coronary artery or peripheral vascular bypass surgeries. HSV segments were stored in a saline solution until the end of the surgical procedure, at which time they were placed in cold transplant harvest buffer (100 mM potassium lactobionate, 25 mM KH2PO4, 5 mM MgSO4, 30 mM Raffinose, 5 mM Adenosine, 3 mM Glutathione, 1 mM Allopurinol, 50g/L Hydroxyethyl starch, pH 7.4). The vessels were used within 24 hours of harvest. Using sterile technique, HSV segments were transferred to a 60-mm Petri dish under a sterile hood. The edges (0.5 mm) of each segment were removed with a blade and excess adventitial tissue and fat removed with minimal manipulation. HSV segments were cut into consecutive rings of approximately 1.0mm in width to be utilized for organ culture or muscle bath experiments. Two rings from each segment were immediately fixed in 10% formalin at 37°C for 30 min to obtain pre-culture intimal thickening measurements.
2.8 Effect of MMI-0100 on smooth muscle physiology
In preparation for testing vein segment functional viability, HSV rings were weighed and their lengths recorded. To focus on smooth muscle responses, the endothelium was mechanically denuded by rolling the luminal surface of each ring at the tip of a fine vascular forceps before suspension in a muscle bath containing a bicarbonate buffer (120 mM NaCl, 4.7 mM KCl, 1.0 mM MgSO4, 1.0 mM NaH2PO4, 10 mM glucose, 1.5 mM CaCl2, and 25 mM Na2HCO3, pH 7.4) equilibrated with 95% O2 and 5% CO2 at 37°C. The rings were stretched and the length progressively adjusted until maximal tension was obtained. Normalized reactivity was obtained by determining the passive length-tension relationship for each vessel segment. Rings were maintained at a resting tension of 1g, which produces maximal responses to contractile agonists as previously determined, and equilibrated for 2 hours in buffer. Force measurements were obtained using a Radnoti Glass Technology (Monrovia, CA) force transducer (159901A) interfaced with a Powerlab data acquisition system and Chart software (AD Instruments, Colorado Springs, CO).
HSV rings were first contracted with 110 mM KCl (with equimolar replacement of NaCl in bicarbonate buffer) and force generated was measured. 110 mM KCl causes membrane depolarization, leading to contraction of vessels containing functionally viable smooth muscle. After multiple KCl challenges, rings were washed and allowed to equilibrate in bicarbonate solution for 30 min, and then contracted with phenylephrine (PE, 10−7–10−6M). Rings were relaxed with a cumulative log dose of sodium nitroprusside (SNP), a nitric oxide donor, and force generated was recorded. All rings were again washed and equilibrated in buffer for 15 minutes. Rings were then incubated with either buffer alone or buffer plus 100 μM MMI-0100 for 2 hours, followed by treatment with the same doses of PE and SNP, and the forces generated again recorded. Measured force was normalized for ring weight and length and percent relaxation was calculated; force generated with 10−6M PE was set as 0% relaxation.
2.9 Organ culture
After viability was determined in the muscle bath, additional rings were cut and placed in 8-well chamber slides and maintained in RPMI 1640 medium supplemented with 30% FBS, 1% L-glutamine and 1% penicillin/streptomycin for 14 days at 37° C in an atmosphere of 5% CO2 in air. The rings were either untreated or treated with MMI-0100 peptide (10, 50 or100 μM). The culture medium with treatments was replaced every 2–3 days.
2.10 Vessel morphometry
After 14 days of organ culture, vein segments were fixed in 0.5mL of 10% formalin at 37°C for 30 minutes and embedded in paraffin for sectioning. Beginning at the midportion of each ring, 5 transverse sections, spaced 5 μm apart, were cut for each specimen. Sections were then stained with Verhoeff-van Gieson stain. Each section was examined using light microscopy (Carl Zeiss, Thornwood, NY) and 6 radially parallel measurements of intimal and medial thickness were randomly taken from each section (total of 6–12 measurements per ring). Intima was defined as tissue on the luminal side of the internal elastic lamina or the chaotic organization of the cells contained within it, whereas the medial layer was contained between the intimal layer and the external elastic lamina. Intimal and medial thickening was measured for each section at 5X magnification with the microscope’s computerized image analysis system.
2.11 Mouse vein graft model
All procedures, protocols, and medications were approved by the Institutional Animal Care and Use Committee and were performed and administered within NIH and ethical guidelines. 12-week-old C57Bl/6 wild type mice (Harlan) were used for all experiments, as previously described [Muto 2011]. To obtain veins, an approximately 2.0 mm segment of the intrathoracic inferior vena cava was isolated and excised. Prior to implantation, the vein was treated ex vivo with 100 μM MMI-0100 peptide solution, or control PBS solution, for 20 minutes at room temperature.
To implant the vein graft, a midline incision was made in the abdomen of a recipient mouse and the infrarenal abdominal aorta was exposed. The abdominal aorta was temporarily occluded with atraumatic micro-clamps and a segment corresponding to the length of the vein graft was excised. The vein was sutured into the arterial circulation using 10–0 nylon in continuous fashion.
Vein grafts were followed postoperatively using the Vevo770 High-Resolution Imaging System (VisualSonics, Toronto, Canada), with weekly measurements of graft wall thickness. At 28 days after surgery, mice were sacrificed to allow explantation of the vein graft. Tissue was either frozen with RNA stabilization reagent (Qiagen) or explanted for paraffin embedding after circulatory flushing with ice-cold PBS followed by 4% paraformaldehyde perfusion-fixation. Vein graft wall thickness, lumen diameter, and outer wall diameter (elastic lamina) were measured in elastin-stained sections using computer morphometry (ImageJ).
2.12 Histology and Immunohistochemistry
Vein graft samples were fixed as noted above and harvested for histology. Specimens were embedded in paraffin and cut in cross section (5 μm). Hematoxylin & Eosin, Masson trichrome, and van Gieson elastin staining were performed for all samples. Cells were cultured on gelatin-coated cover slips and fixed with methanol.
All sections analyzed with immunohistochemistry were first treated for antigen retrieval using 10 mmol/L citrate buffer (pH 6.0) prior to boiling or proteinase K (20 μg/ml) treatment, at room temperature, for 10–15 minutes. Immunohistochemical detection was performed using a primary antibody to F4/80 (AbD Serotec) according the manufacturer’s instructions, and then secondary detection was performed using DAB as well as NovaRED substrate (Vector). Sections were counterstained with Mayer’s Hematoxylin. Images were captured with an Axioimager A1 (Carl Zeiss) and density was analyzed by Image J (NIH).
2.13 Statistics
Statistical analysis was performed with one-way ANOVA followed by Tukey test to compare experimental groups. Analyses were done with OriginPro 8 software (Originlab, Northampton, MA) or GraphPad software (La Jolla, CA). Statistical significance was accepted within a 95% confidence limit. Results are presented as arithmetic mean ± SEM graphically.
3.0 Results
3.1 MMI-0100 induces minimal cell proliferation
To determine the effect of MMI-0100 on human endothelial cell (EC) and smooth muscle cell (SMC) proliferation under stress conditions, such as occurs during surgical vein graft harvest and handling, human EC and SMC cultures were treated with three concentrations of MMI-0100 (0.25 mM, 0.5 mM, and 1 mM) following pre-treatment with TNF-α, a cytokine that stimulates cellular inflammation and stress as well as activates MK2. Both 0.25 mM and 0.5 mM concentrations of MMI-0100 slightly increased cell proliferation in both cell types compared to control cells treated with 20 ng/ml TNF-α alone (maximal with 0.5 mM: 30% and 12% increases in EC and SMC cultures, respectively; Figure 1). However, while the 1 mM MMI-0100 treatment also increased both EC (11%) and SMC (7%) proliferation as compared to control, this response was not as robust as that induced by treatment with 0.5 mM MMI-0100 (Figure 1). Phase contrast images of EC and SMC treated with MMI-0100 for 24 hours showed no obvious morphological changes as compared to control cells (Figure 1C). These data suggest that MMI-0100 has no major negative effects on vascular cell proliferation or morphology during stress conditions.
Figure 1.

A, B) Effect of three concentrations of MMI-0100 on HAEC (A) and HASMC (B) proliferation. C) Microscopic images of ECs (A–D) and SMCs (E–H) before and after 24 hours of MMI-0100 treatments. From top to bottom, the concentrations of MMI-0100 are (A, E) no treatment, (B, F) 0.25mM, (C, G) 0.5mM, and (D, H) 1mM.
3.2 MMI-0100 reduces Interleukin-6 expression in endothelial cells
Since MMI-0100 has no effects on TNF-α-stimulated proliferation, we investigated the anti-inflammatory effect of MMI-0100 by assaying expression of Interleukin 6 (IL-6) and Interleukin 8 (IL-8) secreted by human coronary endothelial cells (HCAEC) following TNF-α stimulation. HCAEC were seeded on a multi-well plate at a density of approximately 25,000 cells/cm2. After a 6 hour incubation with TNF-α, which activates MK2 and stimulates IL-6 production, MMI-0100 (0.5 mM) was added to the culture medium. After 24 hours of drug treatment, supernatant from each well was collected and assayed for cytokine expression. MMI-0100 treatment reduced the level of TNF-α-induced IL-6 expression to that of the untreated control (Figure 2A). However, since IL-8 is not under the control of MK2, its expression levels should not be affected by addition of an MK2 inhibitor [Coxon 2003]; consistent with this expectation, MMI-0100 had no effect on the level of TNF-α-induced IL-8 expression (Figure 2B). This data suggests specificity of MMI-0100 on suppressing TNF-α-induced IL-6 production.
Figure 2.

Comparison of IL-6 (A) and IL-8 (B) levels secreted by endothelial cells, without and with MMI-0100. IL6 and IL-8 were measured in pg/ml. Data shown as mean ± SEM from 3 replicates. *, p<0.05 compared to the control group.
3.3 MMI-0100 enhances human saphenous vein relaxation
To examine the direct role of MMI-0100 on smooth muscle relaxation, human saphenous vein (HSV) rings were pre-treated with buffer or MMI-0100 (100 μM) and then rings were contracted with phenylephrine (10−6M) and relaxed with sodium nitroprusside (10−8 and 10−7M; SNP). Pretreatment of HSV rings with MMI-0100 led to a significant increase in relaxation (25.622 ±6.38 and 92.54 ±3.09 for 10−8 and 10−7M SNP, respectively) when compared to untreated control (10.825 ± 5.62 and 72.768 ± 6.99 for 10−8 and 10−7M SNP, respectively) (Figure 3). There was no significant difference in relaxation response when HSV rings were pretreated with the control peptide (transduction domain, PTD peptide, 50 or 100 μM) when compared to the untreated control (data not shown). In addition, MMI-0100 did not induce relaxation in absence of SNP, with no reduction of basal tension and no reduction of phenylephrine-induced force (data not shown).
Figure 3.
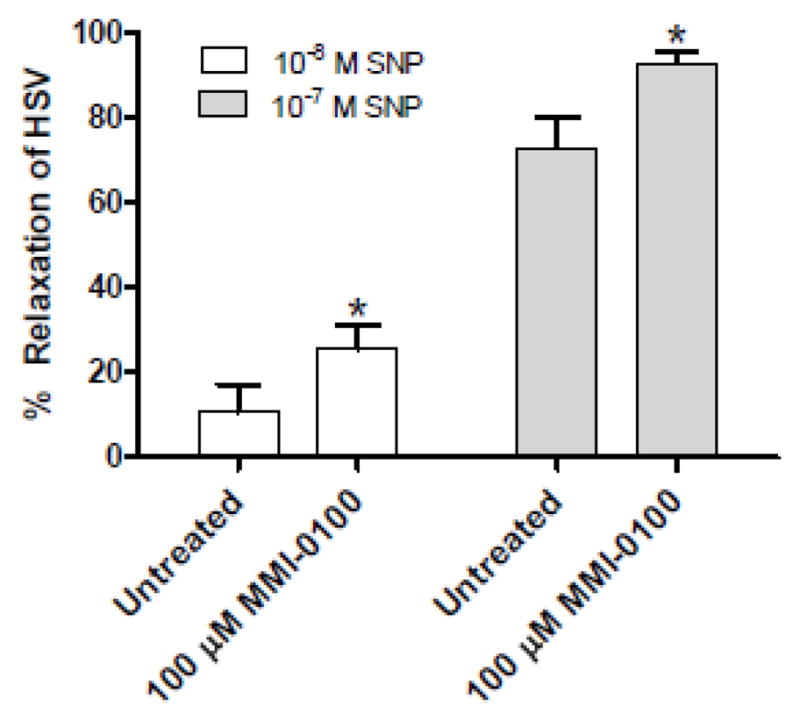
MMI-0100 peptide enhances sodium nitroprusside (SNP)-induced relaxation of human saphenous vein (HSV). *, p<0.05 compared to untreated, n=4.
3.4 MMI-0100 reduces intimal hyperplasia in a human saphenous vein organ culture model
To examine the effect of MMI-0100 on development of intimal hyperplasia, we measured intimal thickness of HSV in a human organ culture model in the presence of high serum and different concentrations (10–100 μM) of MMI-0100. HSV were cultured for 14 days in 30% serum. All veins were deemed viable at the time of culture by adequate contraction with a phenylephrine challenge in a muscle bath. The average intimal thickness of pre-cultured vein segments was 43.7 ± 7.8 μm. After culture, the average intimal thickness of the control was 81.6 ± 17.3 μm. The average intimal thickness in the presence of 50 μM and 100 μM MMI-0100 was 42.7 ± 6.0 μm and 50.4 ± 10.7 μm, respectively, with a significant reduction in intimal thickness (Figures 4A and 5). Measurement of the intima:media (I:M) ratio showed a greater reduction of the I:M ratio at the 100uM concentration of MMI-0100 (Figure 4B).
Figure 4.

Effect of MMI-0100 peptide treatment on (A) intimal layer thickening, and (B) intimal-to-medial ratio, in human saphenous vein (HSV). A) #, p<0.05 compared to pre-culture. *, p<0.05 compared to untreated, n=4–5. A representative graph is shown. B) #, p<0.05 compared to pre-culture. *, p<0.05 compared to untreated. n=5. Graph shows the cumulative data.
Figure 5.

Effect of MMI-0100 peptide treatment on intimal layer thickening in human saphenous vein. Representative sections for each condition are shown at 20X magnification. White bar indicates intimal thickening.
3.5 MMI-0100 inhibits intimal hyperplasia in a mouse vein graft model
To confirm the inhibitory effects of MMI-0100 on intimal hyperplasia development in an ex vivo model, we examined the role of MMI-0100 in an in vivo model of intimal hyperplasia, using a mouse model of vein graft adaptation. Vein grafts were treated with PBS or MMI-0100 (100 μM) for 20 minutes prior to implantation and then followed weekly with ultrasound. Weekly ultrasound examination of the vein graft wall thickness showed diminished wall thickness at all postoperative time points in vein grafts treated with MMI-0100, with a ratio of 2.6-fold thicker at 4 weeks, compared to 4.7-fold thicker at 4 weeks in control grafts (Figure 6A, 6B). Histological staining of the grafts confirmed 72% reduced wall thickness with MMI-0100 treatment compared to control grafts, as seen in vivo with ultrasound (Figures 6C, 6D). Examination of the grafts for F4/80 immunohistochemical reactivity demonstrated fewer F4/80-positive cells infiltrating into vein grafts treated with MMI-0100, consistent with fewer infiltrating macrophages in grafts treated with MMI-0100 (Figures 6E, 6F).
Figure 6.

Effect of MMI-0100 on vein graft adaptation. A) Mouse vein grafts were followed weekly with ultrasound; representative figures are shown. Yellow star shows the lumen of the graft; red arrows show the vein graft wall. B) Graph shows summary of ultrasound measurements of vein graft wall thickness, plotting the ratio of the wall thickness compared to the wall thickness at time of implantation. **, p<.0001 at 4 weeks. C) Photomicrographs show representative portions of vein grafts treated with PBS or MMI-0100. D) Bar graph shows summary of histological measurements of vein graft wall thickness. n=5 or 6 at each time point. **, p=0.016. E) Mouse vein grafts were examined for F4/80 immunohistochemical reactivity; red-brown staining shows immunoreactive cells. F) Bar graph shows summary of F4/80 density showing reduced macrophages in the vein graft wall with MMI-0100 treatment.
Although MMI-0100 induces minimal proliferation of human EC and SMC (Figure 1), we confirmed the effect using physiological doses of MMI-0100 on murine EC. Murine EC were positive for Eph-B4, the marker of venous identity (Figure 7A). MMI-0100 did not induce significant murine EC proliferation at physiological doses (Figure 7B). Similarly, MMI-0100 did not induce EC apoptosis at any dose (Figure 7C). MMI-0100 did not stimulate MCP-1 production, even at high doses (Figure 7D), consistent with reduced number of macrophages in vein grafts treated with MMI-0100 (Figures 6E, 6F). Interestingly, nitric oxide (NO) production was not suppressed, and was even enhanced at physiological doses of MMI-0100 (Figure 7E), suggesting perhaps an additional mechanism of action on endothelial cells.
Figure 7.

Effect of MMI-0100 on murine EC. A) Murine EC analyzed for Eph-B4, by Western blot. Representative of n=4 experiments. B) Graph shows mean of cell counts after treatment with MMI-0100. C) Representative analysis of MMI-0100 effects on EC apoptosis. N=3 experiments. D) Bar graph shows MCP-1 production by murine EC after MMI-0100 treatment. *, p<.0001. E) Bar graph shows NO production by murine EC after MMI-0100 treatment. **, p<.0001.
4.0 Discussion
Recent successes demonstrating that suppression of monocytes prior to vascular injury inhibits intimal hyperplasia [Danenberg 2002, Epstein 2008] led us to test the efficacy of a potent anti-inflammatory compound, MMI-0100, in inhibiting development of intimal hyperplasia. Additional motivation for these studies came from our previous work demonstrating that MMI-0100 suppressed inflammatory cytokine production in human plural mesothelial cells after stimulation with IL-1β or TNFα and also suppressed surgically induced adhesions following bowel anastomosis procedures in rats [Ward 2011]. Together, these data suggest that MMI-0100 inhibits fibrosis as well as inflammation and may also effectively inhibit intimal hyperplasia in conjunction with vascular graft surgeries.
In the current study, consistent with studies in human mesothelial cells, pharmacological MMI-0100 treatment of vascular cells induced minimal effects on cell proliferation or morphology and reduced TNF-α-induced IL-6, but not IL-8, secretion in cultured human vascular cells. Similarly, physiological doses of MMI-0100 did not significantly stimulate proliferation or apoptosis, or suppress NO production, in murine EC. These studies also demonstrate enhanced saphenous vein relaxation and reduced intimal hyperplasia in human saphenous vein rings ex vivo, as well as reduced vein graft intimal hyperplasia in an in vivo mouse model. Taken together, these results show that MMI-0100 prevents vein graft intimal thickening, possibly via reduced inflammatory processes in response to surgical vein graft harvest and during subsequent vein graft adaptation.
A single ex vivo exposure of the vein graft to MMI-0100 at the time of surgery inhibits intimal hyperplasia development in an animal vein graft model for several weeks post-surgery (Figure 6). Since these effects on vein graft adaptation occur over an extended period of time, it is likely that MMI-0100 induces alterations in gene transcription. We have shown previously that MMI-0100 suppressed heterogeneous nuclear ribonucleoprotein A0 (hnRNPA0) phosphorylation [Ward 2011]. Rousseau, et al. showed that hnRNPA0 is phosphorylated by MK2 and its phosphorylated form is released from the AU-rich 3′ untranslated region of IL-6 mRNA to stimulate protein expression [Rousseau 2002]. MK2 is also known to phosphorylate tristetraprolin (TTP), another transcription factor that regulates TNFα and COX2 production [Mahtani 2001, Streicher 2010]. Thus, inhibition of MK2 will down regulate inflammatory cytokine production that can lead to both inflammation and intimal hyperplasia development. In addition to MK2 being required for cytokine production [Kotlyarov 1999, Kotlyarov 2002] as well as cyclooxygenase (COX)-2 protein synthesis [Streicher 2010], MK2 has also been suggested to alter stability of α-actin mRNA [Sousa 2007] and to modulate myofibroblast phenotype [Hagood 2007]. Thus, there are multiple mechanisms by which alteration of MK2 function might impact fibrotic processes such as vein graft intimal hyperplasia.
We have previously shown that inhibition of MK2 with a non-specific cell-permeable peptide inhibits heat shock protein 27 (HSP27) phosphorylation, TGF-β1-induced intracellular HSP27 phosphorylation, as well as TGF-β1-induced expression of connective tissue growth factor and collagen type I [Lopes 2009]. These results show that inhibition of MK2 may affect fibrotic cellular responses and are consistent with our previous study with the more specific MK2 inhibitor peptide, MMI-0100, showing reduced adhesion formation in a rat bowel anastomosis model [Kavalukas 2009; Ward 2011]. These results are also consistent with reduced cellular turnover as well as effects on the TGF-β1 pathway, both of which are associated with vein graft neointimal hyperplasia. Since TGF-β1 can stimulate HSP27 phosphorylation, it is quite possible that the reduced intimal hyperplasia seen in vein grafts treated with MMI-0100 is associated with modulation of the TGF-β1-HSP27 pathway. Inhibition of MK2 may also alter other downstream pathways that affect vein graft neointimal hyperplasia. For example, Nogo-B is phosphorylated at Serine-107 by MK2 or MK3, but not by other kinases that are activated by p38 [Rousseau 2005]. Although the function of Nogo-B is not currently understood, Nogo-B has a positive effect on vascular injury-induced remodeling and reduced neointimal development in both arterial and venous models of vascular injury [Kritz 2008]. Therefore MMI-0100 may alter Nogo-B function indirectly through downstream effects; however, exactly how phosphorylation of Nogo-B affects its function, or development of intimal hyperplasia, is not clear.
Although basic cell penetrating peptides may lead to nonspecific kinase inhibition or increased toxicity, we have previously shown that several novel domains lead to increased specificity; in particular, domains based on the antithrombin III-heparin binding domain lead to increased specificity of MK2 inhibition [Ward 2009] compared to another, less-specific MK2 peptide inhibitor [Lopes 2009]. MMI-0100 is a relatively specific inhibitor of MK2, with preserved mitogen-activated protein kinase-activated protein kinase 5, p38, Protein kinase B beta, Protein kinase C delta, and Rho-associated coiled-coil containing protein kinase 1 activity at concentrations of MMI-0100 that completely inhibit MK2 activity [Ward 2009]. However, MMI-0100 can also inhibit calcium/calmodulin-dependent protein kinase I (CaMKI) as well as Trk-B [Ward 2009], both of which can alter smooth muscle function [Passier 2000, Donovan 1995], suggesting the possibility of selective gene expression mediating potential other effects of MMI-0100 [Wamhoff 2006]. However, it is likely that these other effects on smooth muscle cells would induce smooth muscle cell relaxation, augmenting MMI-0100 function. Furthermore, other peptide inhibitors of MK2 have similar inhibition of CaMKI, MK3, as well as other kinases, suggesting that MMI-0100 would have the fewest other effects of any tested MK2 peptide inhibitor [Ward 2009]. Therefore we believe that the inhibitory effects of MMI-0100 may be specific for fibrotic responses secondary to inflammation, such as vein graft intimal hyperplasia, and are likely to have few side effects if given clinically, particularly when locally vs. systemically-delivered.
We show, using the novel cell-permeant peptide inhibitor MMI-0100, that inhibition of MK2 inhibits intimal thickening in both ex vivo and in vivo models of intimal hyperplasia. Although there are several mechanisms by which MMI-0100 may inhibit intimal thickening, the sustained in vivo effects from a single ex vivo graft treatment at the time of graft surgery suggest clinical utility, especially in vein graft disease that is amenable to ex vivo treatment. As such, MMI-0100 may represent a novel strategy to inhibit fibrotic processes such as vein graft disease.
Acknowledgments
This work was supported in part by the National Institute of Health grant R01-HL095498-01 (A.D.) and 2R01HL070715 (C.M.B.), the American Vascular Association William J. von Liebig Award (A.D.), as well as with the resources and the use of facilities at the VA Connecticut Healthcare System, West Haven, Conn (A.D.). MMI-0100 peptide was provided by Moerae Matrix, Inc. A.P. and C.M.B. have significant ownership interests in Moerae Matrix, Inc.; A.P. and C.M.B are consultants to Moerae Matrix, Inc.
Abbreviations
- CaMKI
calcium/calmodulin-dependent protein kinase I
- EC
endothelial cell
- GA
gentamycin/amphotericin
- HAEC
human aortic endothelial cells
- HASMC
human aortic smooth muscle cells
- HCAEC
human coronary artery endothelial cells
- hnRNPA0
heterogeneous nuclear ribonucleoprotein A0
- HSP27
heat shock protein 27
- HSV
human saphenous vein
- IL
interleukin
- I:M
intima:media
- MAPK
mitogen activated protein kinase
- MK
mitogen activated protein kinase activated protein kinase
- MLEC
mouse lung endothelial cells, PBS, phosphate-buffered saline
- PE
phenylephrine
- SMC
smooth muscle cell
- SNP
sodium nitroprusside
- TTP
tristetraprolin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. J Clin Invest. 2005 Aug;115(8):2119–27. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JH, Hafley G, Harrington RA, Peterson ED, Ferguson TB, Jr, Lorenz TJ, Goyal A, Gibson M, Mack MJ, Gennevois D, Califf RM, Kouchoukos NT PREVENT IV Investigators. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: a randomized controlled trial. JAMA. 2005;294(19):2446–54. doi: 10.1001/jama.294.19.2446. [DOI] [PubMed] [Google Scholar]
- Allaire E, Clowes AW. Endothelial cell injury in cardiovascular surgery: the intimal hyperplastic response. Ann Thorac Surg. 1997 Feb;63(2):582–91. doi: 10.1016/s0003-4975(96)01045-4. [DOI] [PubMed] [Google Scholar]
- Clowes AW, Reidy MA. Prevention of stenosis after vascular reconstruction: pharmacologic control of intimal hyperplasia--a review. J Vasc Surg. 1991 Jun;13(6):885–91. doi: 10.1067/mva.1991.27929. [DOI] [PubMed] [Google Scholar]
- Conte MS, Bandyk DF, Clowes AW, Moneta GL, Seely L, Lorenz TJ, Namini H, Hamdan AD, Roddy SP, Belkin M, Berceli SA, DeMasi RJ, Samson RH, Berman SS PREVENT III Investigators. Results of PREVENT III: A multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg. 2006;43(4):742–751. doi: 10.1016/j.jvs.2005.12.058. [DOI] [PubMed] [Google Scholar]
- Coxon PY, Rane MJ, Uriarte S, Powell DW, Singh S, Butt W, Chen Q, McLeish KR. MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils. Cell Signal. 2003 Nov;15(11):993–1001. doi: 10.1016/s0898-6568(03)00074-3. [DOI] [PubMed] [Google Scholar]
- Danenberg HD, Fishbein I, Gao JC, Monkkonen J, Reich R, Gati I, Moerman E, Golomb G. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106(5):599–605. doi: 10.1161/01.cir.0000023532.98469.48. [DOI] [PubMed] [Google Scholar]
- Dashwood M, Anand R, Loesch A, Souza D. Surgical trauma and vein graft failure: further evidence for a role of ET-1 in graft occlusion. J Cardiovasc Pharmacol. 2004 Nov;44( Suppl 1):S16–9. doi: 10.1097/01.fjc.0000166222.50346.c6. [DOI] [PubMed] [Google Scholar]
- Dashwood MR, Loesch A. Surgical damage of the saphenous vein and graft patency. J Thorac Cardiovasc Surg. 2007 Jan;133(1):274–5. doi: 10.1016/j.jtcvs.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Miranda RC, Kraemer R, McCaffrey TA, Tessarollo L, Mahadeo D, Sharif S, Kaplan DR, Tsoulfas P, Parada L, Toran-Allerand CD, Hajjar DP, Hempstead BL. Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am J Pathol. 1995 Aug;147(2):309–24. [PMC free article] [PubMed] [Google Scholar]
- Epstein H, Grad E, Golomb M, Koroukhov N, Edelman ER, Golomb G, Danenberg HD. Innate immunity has a dual effect on vascular healing: suppression and aggravation of neointimal formation and remodeling post-endotoxin challenge. Atherosclerosis. 2008 Jul;199(1):41–6. doi: 10.1016/j.atherosclerosis.2007.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funding AT, Johansen C, Gaestel M, Bibby BM, Lilleholt LL, Kragballe K, Iversen L. Reduced oxazolone-induced skin inflammation in MAPKAP kinase 2 knockout mice. J Invest Dermatol. 2009 Apr;129(4):891–8. doi: 10.1038/jid.2008.322. [DOI] [PubMed] [Google Scholar]
- Hagood JS, Olman MA. Muscle fatigue: MK2 signaling and myofibroblast differentiation. Am J Respir Cell Mol Biol. 2007 Nov;37(5):503–6. doi: 10.1165/rcmb.2007-0005ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayess K, Benndorf R. Effect of protein kinase inhibitors on activity of mammalian small heat-shock protein (HSP25) kinase. Biochem Pharmacol. 1997 May 9;53(9):1239–47. doi: 10.1016/s0006-2952(96)00877-5. [DOI] [PubMed] [Google Scholar]
- Kavalukas SL, et al. MK2 inhibitor peptide reduces adhesion formation without affecting colonic anastomotic healing. Journal of the American College of Surgeons. 2009;209(3):S17–S17. [Google Scholar]
- Kent KC, Liu B. Intimal hyperplasia--still here after all these years! Ann Vasc Surg. 2004 Mar;18(2):135–7. doi: 10.1007/s10016-004-0019-4. [DOI] [PubMed] [Google Scholar]
- Klyachkin ML, Davies MG, Svendsen E, Kim JH, Massey MF, Barber L, McCann RL, Hagen PO. Hypercholesterolemia and experimental vein grafts: accelerated development of intimal hyperplasia and an increase in abnormal vasomotor function. J Surg Res. 1993 May;54(5):451–68. doi: 10.1006/jsre.1993.1071. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999 Jun;1(2):94–7. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, Lin LL, Gaestel M. Distinct cellular functions of MK2. Mol Cell Biol. 2002 Jul;22(13):4827–35. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritz AB, Yu J, Wright PL, Wan S, George SJ, Halliday C, Kang N, Sessa WC, Baker AH. In vivo modulation of Nogo-B attenuates neointima formation. Mol Ther. 2008 Nov;16(11):1798–804. doi: 10.1038/mt.2008.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner MD, Schwoebel F, Kotlyarov A, Leist M, Gaestel M, Hartung T. Mitogen-activated protein kinase-activated protein kinase 2-deficient mice show increased susceptibility to Listeria monocytogenes infection. J Immunol. 2002 May 1;168(9):4667–73. doi: 10.4049/jimmunol.168.9.4667. [DOI] [PubMed] [Google Scholar]
- LoGerfo FW, Quist WC, Cantelmo NL, Haudenschild CC. Integrity of vein grafts as a function of initial intimal and medial preservation. Circulation. 1983 Sep;68(3 Pt 2):II117–24. [PubMed] [Google Scholar]
- Lopes LB, Flynn C, Komalavilas P, Panitch A, Brophy CM, Seal BL. Inhibition of HSP27 phosphorylation by a cell-permeant MAPKAP Kinase 2 inhibitor. Biochem Biophys Res Commun. 2009 May 8;382(3):535–9. doi: 10.1016/j.bbrc.2009.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LB, Brophy CM, Flynn CR, Yi Z, Bowen BP, Smoke C, Seal B, Panitch A, Komalavilas P. A novel cell permeant peptide inhibitor of MAPKAP kinase II inhibits intimal hyperplasia in a human saphenous vein organ culture model. J Vasc Surg. 2010 Dec;52(6):1596–607. doi: 10.1016/j.jvs.2010.06.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso R, De Cicco G, Beghi C, Gherli T, Poli E, Corradi D, Maestri R, Bonadonna S, Mancini T, Giustina A. Functional effects of nitric oxide-releasing aspirin on vein conduits of diabetic patients undergoing CABG. Int J Cardiol. 2007 May 31;118(2):164–9. doi: 10.1016/j.ijcard.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Mahtani KR, Brook M, Dean JLE, Sully G, Saklatvala J, Clark A. Mitogen-Activated Protein Kinase p38 controls the expression and posttranslational modification of Tristetraprolin, a regulator of Tumor Necrosis Factor Alpha mRNA Stability. Mol Cell Biol. 2001 October;21(19):6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosse PR, Campbell GR, Wang ZL, Campbell JH. Smooth muscle phenotypic expression in human carotid arteries. I. Comparison of cells from diffuse intimal thickenings adjacent to atheromatous plaques with those of the media. Lab Invest. 1985 Nov;53(5):556–62. [PubMed] [Google Scholar]
- Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: pathogenesis, predisposition, and prevention. Circulation. 1998 Mar 10;97(9):916–31. doi: 10.1161/01.cir.97.9.916. [DOI] [PubMed] [Google Scholar]
- Muto A, Yi T, Harrison KD, Dávalos A, Fancher TT, Ziegler KR, Feigel A, Kondo Y, Nishibe T, Sessa WC, Dardik A. Eph-B4 prevents venous adaptive remodeling in the adult arterial environment. J Exp Med. 2011;208(3):561–575. doi: 10.1084/jem.20101854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002 Feb 1;277(5):3065–8. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000 May;105(10):1395–406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994 Sep 23;78(6):1027–37. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 2002 Dec 2;21(23):6505–14. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau S, Peggie M, Campbell DG, Nebreda AR, Cohen P. Nogo-B is a new physiological substrate for MAPKAP-K2. Biochem J. 2005 Oct 15;391(Pt 2):433–40. doi: 10.1042/BJ20050935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler H, Stoecklin G. Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans. 2008 Jun;36(Pt 3):491–6. doi: 10.1042/BST0360491. [DOI] [PubMed] [Google Scholar]
- Sousa AM, Liu T, Guevara O, Stevens J, Fanburg BL, Gaestel M, Toksoz D, Kayyali US. Smooth muscle alpha-actin expression and myofibroblast differentiation by TGF-β are dependent upon MK2. J Cell Biochem. 2007;100:1581–1592. doi: 10.1002/jcb.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JM, Ren S, Herschman H, Wang Y. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010 Apr 30;106(8):1434–43. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Timmer M, Cesnulevicius K, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation--relevance in a mouse model of Parkinson’s disease. J Neurochem. 2008 Jun;105(5):2039–52. doi: 10.1111/j.1471-4159.2008.05310.x. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006 Apr 14;98(7):868–78. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- Wang X, Xu L, Wang H, Young PR, Gaestel M, Feuerstein GZ. Mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 deficiency protects brain from ischemic injury in mice. J Biol Chem. 2002 Nov 15;277(46):43968–72. doi: 10.1074/jbc.M206837200. [DOI] [PubMed] [Google Scholar]
- Ward B, Seal BL, Brophy CM, Panitch A. Design of a bioactive cell-penetrating peptide: when a transduction domain does more than transduce. J Pept Sci. 2009 Oct;15(10):668–74. doi: 10.1002/psc.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BC, Kavalukas S, Brugnano J, Barbul A, Panitch A. Peptide inhibitors of MK2 show promise for inhibition of abdominal adhesions. J Surg Res. 2011 doi: 10.1016/j.jss.2011.01.043. epub Feb 23, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Müller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999 Sep 15;18(18):4969–80. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]