

Abstract
It is widely believed that microglia and monocyte-derived macrophages (collectively referred to as central nervous system (CNS) macrophages) cause excitotoxicity in the diseased or injured CNS. This view has evolved mostly from in vitro studies showing that neurotoxic concentrations of glutamate are released from CNS macrophages stimulated with lipopolysaccharide (LPS), a potent inflammogen. We hypothesized that excitotoxic killing by CNS macrophages is more rigorously controlled in vivo, requiring both the activation of the glutamate/cystine antiporter (system xc-) and an increase in extracellular cystine, the substrate that drives glutamate release. Here, we show that non-traumatic microinjection of low-dose LPS into spinal cord gray matter activates CNS macrophages but without causing overt neuropathology. In contrast, neurotoxic inflammation occurs when LPS and cystine are co-injected. Simultaneous injection of NBQX, an antagonist of AMPA glutamate receptors, reduces the neurotoxic effects of LPS+cystine, implicating glutamate as a mediator of neuronal cell death in this model. Surprisingly, neither LPS nor LPS+cystine adversely affects survival of oligodendrocytes or oligodendrocyte progenitor cells. Ex vivo analyses show that redox balance in microglia and macrophages is controlled by induction of system xc- and that high GSH:GSSG ratios predict the neurotoxic potential of these cells. Together, these data indicate that modulation of redox balance in CNS macrophages, perhaps through regulating system xc-, could be a novel approach for attenuating injurious neuroinflammatory cascades.
Keywords: system xc-, redox, neuroinflammation, spinal cord injury, glutamate
INTRODUCTION
Glutamate “excitotoxicity” is a mechanism of pathology in most neurological diseases (Mattson, 2003, Olney, 1994). In the normal CNS, physiological concentrations of extracellular glutamate are maintained by energy-dependent synaptic transmission and glial metabolism. However, these mechanisms become dysfunctional or are insufficient to buffer the high levels of glutamate that accumulate in the diseased or injured CNS. As extracellular concentrations of glutamate rise, cells undergo necrosis and apoptosis or prolonged depolarization (Liu, et al., 1991, Xu, et al., 2004). It is widely believed that microglia and macrophages (i.e., CNS macrophages) contribute to excitotoxicity. This view has evolved mostly from in vitro data showing that lipopolysaccharide (LPS), a component of gram-negative bacteria and a toll-like receptor 4 (TLR4) agonist, causes CNS macrophages to release neurotoxic concentrations of glutamate (Domercq, et al., 2007, Piani and Fontana, 1994). In macrophages and microglia, this requires activation of the cystine-glutamate antiporter, i.e., system xc-, (Piani and Fontana, 1994).
System xC− is a membrane bound heterodimer comprised of an xCT catalytic subunit and a 4F2hc/CD98 heavy chain. In the intact CNS, basal activity of system xc- is low, and expression of xCT is limited to cells, mostly macrophages, in the choroid plexus, meninges and ependyma (Sato, et al., 2002). In response to inflammation or oxidative stress, induction of xCT corresponds with transporter activation, resulting in a 1:1 exchange of extracellular cystine for intracellular glutamate (Bannai, 1986). In microglia and macrophages, this antiporter is needed to maintain intracellular redox balance (Sasaki, et al., 2002, Sato, et al., 2002). Specifically, extracellular cystine, the oxidized form of the amino acid cysteine, is exchanged for glutamate. Within the cell, cystine is then reduced to cysteine, the limiting reagent for glutathione (GSH) biosynthesis.
In vitro, it is relatively simple to establish a relationship between system xc- activation in macrophages and the ability of these cells to release neurotoxic concentrations of glutamate. However, it is difficult to prove unequivocally that macrophages release glutamate and cause excitotoxicity via this mechanism in vivo. Indeed, there are no truly “selective” system xc- antagonists and even newer in vivo voltammetry techniques cannot resolve glutamate production at the cellular level. Thus, a cause/effect relationship between macrophage activation, glutamate release and excitotoxicity must be assessed through indirect means.
Here, using an intraspinal microinjection model, we examined the relationship between activated macrophages, system xc- and excitotoxicity. Specifically, selective activation of CNS macrophages is achieved in naïve spinal cord via focal microinjection of low-dose LPS. In brain and spinal cord, microglia and macrophages are the predominant cell types that express toll-like receptor-4 (TLR4), the receptor for LPS (Kigerl, et al., 2007, Lehnardt, et al., 2002, Lehnardt, et al., 2003). When LPS is injected together with cystine, a focal inflammatory reaction occurs marked by induction of system xc- in the presence of a large transmembrane gradient for intracellular transport of cystine.
This model mimics aspects of the lesion microenvironment found at sites of acute ischemia or trauma where numerous TLR4 agonists exist and micromolar concentrations of cystine accumulate from cysteine oxidation and blood extravasation (Droge, et al., 1991, Hosoya, et al., 2001, Wade and Brady, 1981, Wang and Cynader, 2000). This model also facilitates analysis of the effects of increased cystine/glutamate exchange at sites of neuroinflammation without the confounding variables that accompany trauma, i.e., hemorrhage, axonal shearing, necrosis and hypoxia.
Using this approach, we show that in the presence of high extracellular cystine, CNS macrophages become neurotoxic effector cells. The enhanced toxicity was specific for neurons and was dependent on activation of ionotropic glutamate receptors (e.g., AMPA). (S)-4-Carboxyphenylglycine (S-4-CPG) is a competitive inhibitor of group I metabotropic glutamate receptors (mGluR) that also has been reported to be a non-specific antagonist of system xc- (Chung, et al., 2005, Ye, et al., 1999). However, unlike NBQX, co-injecting S-4-CPG did not reverse the neurotoxicity caused by LPS+cystine. In fact, a non-significant increase in neuron loss was observed, suggesting that type I mGluRs may modulate microglia/macrophage functions in this model. To provide additional insight to how LPS+cystine microinjections could elicit neurotoxic functions in CNS macrophages, redox status was measured in both microglia and macrophages ex vivo. Those data show that excess extracellular cystine causes intracellular GSH levels to rise, i.e., microglia and macrophages become “reduced”. This redox signature predicts the ability of CNS macrophages to release neurotoxic levels of glutamate and cause neuron killing. Collectively, these data indicate that modulation of redox status, perhaps through inhibition of system xc-, could limit the acute destructive potential of macrophages in the ischemic or traumatized CNS.
MATERIALS AND METHODS
Intraspinal Microinjection
Mice were anesthetized with a cocktail of ketamine/xylazine (80 and 10 mg/kg, respectively). Using aseptic technique, a laminectomy was performed at the T12–13 vertebral level after which the spinal column was secured via the spinous processes adjacent to the laminectomy site using Adson forceps fixed in a spinal frame. Sterile glass micropipettes (pulled to an external diameter of ~25 µm and pre-filled with sterile 0.1M PBS, LPS [500ng], L-cystine [0.6 mM], LPS+cystine, LPS/cystine + NBQX [15nmol], or LPS/cystine + (S)-4-CPG (0.5 or 1.0μM) were positioned at 0.4mm lateral from midline. From the meningeal surface, pipettes were lowered 0.8 mm (dorso-ventral) using a hydraulic micropositioner (David Kopf Instruments, Tujunga, CA). Using a PicoPump (World Precision Instruments, Sarasota, FL), 1 µl of solution was injected over a period of 15 min. To minimize fluid reflux, pipettes remained in place for 2 additional minutes to allow the injectate to dissipate into the parenchyma. To facilitate localization of the injection sites for anatomical analysis, a small amount of sterile charcoal was placed on the adjacent dura before closing the overlying tissues. LPS and cystine were obtained from Sigma Chemicals (St. Louis, MO), and NBQX and (S)-4-CPG from Tocris Bioscience (Ellisville, MO). All chemicals were diluted in sterile, endotoxin-free 0.1 M PBS.
Tissue processing
At designated times post-microinjection, mice were anesthetized then perfused intracardially with 100 ml of 0.1M PBS (pH 7.4) followed by 100 ml of 4% paraformaldehyde (PF). Perfused spinal cords were post-fixed via immersion in 4% PF for 2 hours. Fixed tissues were rinsed and stored overnight at 4°C in 0.2M PB, then cryoprotected in 20% sucrose for 48 hours. Spinal cords were blocked into 1 cm segments centered on the injection site then were embedded in OCT (VWR International). Serial cross-sections (10μm) were cut through each block using a Microm cryostat (HM 505 E) then were collected on SuperFrost Plus slides (Fisher Scientific, Fair Lawn, NJ) and stored at −20°C.
Immunohistochemistry
Immunohistochemistry was done as previously described (Kigerl et al., 2006). Sections were rinsed (0.1M PBS) then overlaid with blocking serum for 1 hour at room temperature. Primary antibody (Table 1) was incubated overnight at 4°C in humidified chambers. Sections were rinsed with 0.1M PBS then overlaid with biotinylated- or AlexaFluor- (Invitrogen) conjugated secondary antibodies for 2 hours at room temperature. Biotinylated antibody was visualized using Elite-ABC reagent (Vector Laboratories, Burlingame CA) with DAB as a substrate (Vector Laboratories). Immunoperoxidase-labeled sections were dehydrated through ascending alcohols, cleared in xylene then coverslipped with Permount (Fisher Scientific, Fair Lawn, NJ). Immunofluorescent slides were counterstained with DAPI and coverslipped with ImmuMount (Thermo Fisher Scientific). Antibody specificity was confirmed by incubating slides without primary antibody. Images were collected on an Olympus Fluoview 1000 Laser Scanning Confocal microscope.
Table 1.
Antibodies for Immunohistochemistry
| Antibody | Specificity | Vendor | Host | Dilution |
|---|---|---|---|---|
| F4/80 | F4/80 antigen | Serotec (MCA497) | Rat | 1:50 |
| Mac-1 | CD11b(M1/70.15) | Serotec (MCA-74G) | Rat | 1:800 |
| HuC/HuD | HuC, HuD, Hel-N1 | Invitrogen (16A11) | Mouse | 1:2000 |
| NG2 | Rat NG2 CSPG (~270–300kD) | US Biological (C5067-70D) | Rabbit | 1:1000 |
| GSTπ | Glutathione-S-Transferase-π | Chemicon (AB8902) | Rabbit | 1:4000 |
| xCT | xCT (extracellular domain) | Santa Cruz (Q-18) | Goat | 1:100 |
| GFAP | GFAP from bovine spinal cord | DAKO (Z0334) | Rabbit | 1:10000 |
| Tomato Lectin | Lycopersicon esculentum | Sigma (L0651) | 1:500 |
Neuron and glial cell quantitation
Equidistant sections spanning the injection site (100-μm section spacing) were stained with HuC/HuD, GSTπ, or NG2 to label neurons, oligodendrocytes, or OPCs respectively. Labeled cells in the ventral horn were counted under high power on a Zeiss Axioplan 2 Imaging light microscope equipped with an MCID ELITE image analysis system (InterFocus Imaging Ltd., Cambridge, England). Data were then expressed as a percentage of contralateral (HuC/HuD; GSTπ) or total number of cells per ventral horn (NG2). Neurons were only counted when a clear nucleolus was visible. Activated microglia (F4/80+ cells) were quantified using proportional area measurements as described previously by our group (Donnelly, et al., 2009).
RNA Extraction
RNA was isolated from a 5mm segment of homogenized spinal cord or cell cultures via Trizol extraction followed by treatment with DNase I (1 μg/μl) to eliminate genomic DNA (Invitrogen). RNA quantity/quality was assessed (BioPhotometer, Eppendorf) and 1.75 μg of DNase-treated RNA was primed with random hexamers (1 μM, Applied Biosystems) and reverse transcribed using SuperScript II reverse transcriptase (Applied Biosystems) in a 50 μl reaction. cDNA was stored at −20°C until analyzed by PCR.
Laser capture microdissection
Laser capture microdissection of injection sites was done as described previously (Kigerl, et al., 2007). Briefly, using an Axiovert 200 microscope (Zeiss) fitted with a P.A.L.M.® MicroBeam (P.A.L.M. Microlaser Technologies AG, Bernried,Germany), PALM.® RoboSoftware was used to identify then remove phenotypically-distinct cells from microinjection sites at 3dpi. Tissue was collected into lysis buffer (RNAqueous® Micro-kit; Ambion, Austin, TX, USA) then stored at −20°C until RNA isolation. RNA was isolated using the RNAqueous® Microkit.
Quantitative Real-Time PCR
Reactions were performed in triplicate using 1 μl of cDNA per reaction. RNA was analyzed using primers specific to the gene of interest [designed using PrimerExpress (Applied Biosystems)] and SYBR Green master mix (Applied Biosystems) in 20 μl reactions. All PCR reactions were performed using an Applied Biosystems 7300 system, and melting point analyses were performed for each reaction to confirm single amplified products. Gene expression was extrapolated from standard curves generated for each gene using a control cDNA dilution series then was normalized to 18s.
Cultures of bone marrow derived macrophages and BV2 microglia
BMDM cultures were derived as previously described (Longbrake, et al., 2007). Briefly, bone marrow was harvested from the femur and tibia of adult C57BL/6 mice and cultured in media containing 20% sL929 fibroblast-conditioned media (which contains M-CSF) for 7days to drive macrophage differentiation. BMDMs and BV-2 cells were then plated at a concentration of 5 × 105 cells/500μl and stimulated with media (RPMI+10%FBS or DMEM+10%FBS, respectively), LPS (100ng/ml), or LPS (100ng/ml) +cystine (0.6mM) for 24hrs. Culture supernatants were harvested and cells were washed with PBS and immediately frozen on dry ice for GSH and GSSG measurements.
Metabolite analyses
Glutamate in the culture medium was analyzed by HPLC as described previously (Garg, et al., 2008). In brief, cell culture supernatant was mixed with an equal volume of metaphosphoric acid solution (16.8 mg/ml HPO3, 2 mg/ml EDTA, and 9 mg/ml NaCl), vortexed, and proteins were sedimented by centrifugation at ~14,000 × g for 10 min at 4°C. Metabolites in protein-free extracts were derivatized with monoiodoacetic acid followed by 2,4-dinitrofluorobenzene and analyzed by HPLC on a μ-Bondapak NH2 column (Waters, 300 mm × 3.9 mm, 10 μm) with a methanol-acetate gradient as previously described (Garg, et al., 2008). For analysis of intracellular GSH, cells were washed three times with ice cold PBS and detached by gentle scraping on ice. An aliquot of the cell suspension was mixed with an equal volume of metaphosphoric acid solution and protein free cell lysate were either stored at −80°C until further use or derivatized immediately with monoiodoacetic acid, 2, 4-dinitrofluorobenzene solution (1.5% v/v in absolute ethanol) and analyzed by HPLC as described previously (Garg, et al., 2006). To measure protein concentration, an aliquot of the cell suspension was mixed with an equal volume of lysis buffer (0.1 M sodium phosphate, pH 7.4, containing 0.1% Triton-X100, 10 μl/ml protease inhibitor cocktail (Sigma), 25 μg/ml tosyllysine chloromethylketone and 5 μg/ml phenylmethylsulfonyl fluoride (Sigma) and centrifuged at 12,000×g for 10 min at 4°C. The protein concentration in the cell lysate was measured by the Bradford method (Bio-Rad) using bovine serum albumin as a standard. The concentration of each metabolite was determined using a calibration curve generated for each compound. Results were normalized to protein concentration in each sample.
RESULTS
Intraspinal injection of LPS and cystine causes focal inflammation and induction of system xc- (xCT)
Macrophages (and microglia) stimulated in vitro with LPS release neurotoxic concentrations of glutamate, a process that is dependent on xCT induction and extracellular cystine (Piani and Fontana, 1994, Sato, et al., 1995). In an effort to elicit glutamate release from CNS macrophages in vivo, we selectively activated microglia/macrophages with focal microinjections into the ventral horn of the intact thoracic spinal cord (T12–13) of low-dose LPS in the presence or absence of high extracellular cystine.
An initial goal was to confirm induction of the system xc- transporter in activated microglia/macrophages at sites of injection, especially under conditions where the transmembrane gradient for cystine uptake was increased. Using laser capture microdissection to isolate mRNA from CNS macrophages, a robust increase in xCT expression was found at sites of LPS+cystine injection (Fig. 1A). Thus, the transporter and substrate necessary for glutamate exchange are induced with this model. LPS alone also elicits xCT in microglia and macrophages (Piani and Fontana, 1994; data not shown); however, without cystine as a substrate, glutamate cannot be released. Notably, co-injecting LPS+cystine consistently elicits a florid CNS macrophage response, one that is qualitatively enhanced relative to injections of LPS or cystine alone (Fig. 1B–D).
Figure 1.

Co-injections of LPS and cystine cause focal intraspinal inflammation with concurrent activation of system Xc-. (A) Real-time PCR analysis of mRNA obtained via laser capture microdissection from control (PBS) or LPS+cystine microinjection sites reveals focal increases in xCT mRNA expression at sites of CNS macrophage activation. (B–D) A qualitative comparison of F4/80 labeling (for CNS macrophages) at sites injected with cystine (B), LPS (C) or LPS+cystine reveals the enhanced pro-inflammatory effects of LPS+cystine injections. (*indicates central canal;*** p<0.001; scale bar = 100 μm; n=5–8/group)
Immunofluorescent labeling and confocal microscopy was used to reveal the phenotype of cells that upregulate xCT. Neurons and astrocytes have been shown to express system xc- (Burdo, et al., 2006, Piani and Fontana, 1994, Qin, et al., 2006, Sato, et al., 1999); however, after intraspinal injection of LPS or LPS+cystine xCT is upregulated only on microglia/macrophages (4.7- and 5.0-fold increase, respectively, compared to PBS; data not shown) (Fig. 2A–C, G). Expression of xCT mRNA or protein, was minimal in spinal cords injected with PBS (Figs. 1A&2A). This is consistent with previous data showing that xCT expression is low in naïve CNS, except adjacent to the meninges and cerebral ventricles (Sato, et al., 2002). Collectively, data in Figs. 1&2 indicate that co-injecting LPS+cystine creates inflammatory foci in which system xc- activity is enhanced predominantly in CNS macrophages.
Figure 2.

Immunohistochemical labeling reveals xCT expression in activated microglia/macrophages. Three days after injecting LPS (B) or LPS+cystine (C), xCT expression is increased in microglia/macrophages (A–C) but not astrocytes (D), neurons (E) or oligodendrocyte progenitor cells (F). (arrows delineate double-labeled cells in B&C, A–Fscale bar = 50 μm; G–Jscale bar= 20 μm)
Intraspinal injection of LPS and cystine cause focal inflammation and glutamate-mediated killing of neurons but not oligodendrocytes
Increased activity of system xc- in microglia/macrophages should predict focal increases in extracellular glutamate, especially when the gradient to promote cystine entry into cells is optimal. To test whether increasing system xc- activity in CNS macrophages corresponded with changes in neuron or glial survival, microinjections of LPS, with or without cystine, were completed as before, then 3 days later numbers of neurons (HuC/HuD+), oligodendrocytes (GSTπ+) and oligodendrocyte progenitor cells (OPCs; NG2+) were quantified at the injection site (Figs. 3&4). Injection of LPS or cystine alone caused minimal neuron loss; total numbers of neurons was reduced ~10% as compared to the contralateral uninjected ventral horn (Fig. 3A&B). Conversely, neuronal loss was increased ~2-fold after co-injection of LPS+cystine (p<0.05 vs. LPS alone; Fig. 3A&C).
Figure 3.

Intraspinal activation of system xc- is neurotoxic. Neurons were quantified in the spinal cord ventral horn 3 days after injecting cystine, LPS or LPS+cystine. (A–C) Significant neuron loss was evident nearby the site of LPS+cystine injection (ANOVA; *p<0.05 vs. LPS alone). Immunohistochemical labeling of Huc/HuD (AF546 with DAPI counterstain) reveals differences in numbers of spinal cord ventral horn neurons after injecting LPS (B) or LPS+cystine (C). (* = central canal; scale bar = 100μm; n=5–8per group (n=150–200neurons counted on contralateral (intact) side per animal).
Figure 4.

Enhancing cystine/glutamate exchange in CNS macrophages is not toxic to oligodendrocytes or oligodendrocyte progenitor cells (OPCs). Neither LPS or LPS+cystine reduced the number of GSTπ+ oligodendrocytes (A) or NG2+ OPCs (B) (3 days post intraspinal injection). In fact, microinjections of either LPS (not shown) or LPS+cystine [D,F] increased NG2+ OPCs (red) in the spinal cord ventral horn compared to cystine alone (C,E). (All sections counterstained with DAPI to label cell nuclei (blue); ANOVA; **p<0.01 vs. cystine alone; n=5–8per group (n=300–400 OLs counted on contralateral side per animal).
Despite the reported sensitivity of oligodendrocytes to glutamate excitotoxicity, focal activation of CNS macrophage system xc- (with or without changes in extracellular cystine) did not enhance killing of oligodendrocytes or OPCs (Fig. 4A&B). In fact, consistent with our previous data (Schonberg, et al., 2007), NG2+ OPCs appeared to thrive in the inflammatory environment (Fig. 4B,D,F). Injections of cystine alone had no effect on total numbers of oligodendrocytes or NG2+ cells (Fig. 4B,C,E).
Since xCT induction predicts glutamate efflux from macrophages/microglia, glutamate excitotoxicity was suspected as a possible mechanism for the loss of ventral horn motor neurons at sites of intraspinal inflammation. To test this hypothesis, NBQX, an antagonist of AMPA-glutamate receptors, was co-injected with LPS+cystine (Fig. 5). The addition of NBQX reversed the neurotoxic effects of LPS+cystine with neuron numbers approaching those found in PBS-injected spinal cords (Fig. 5A).
Figure 5.
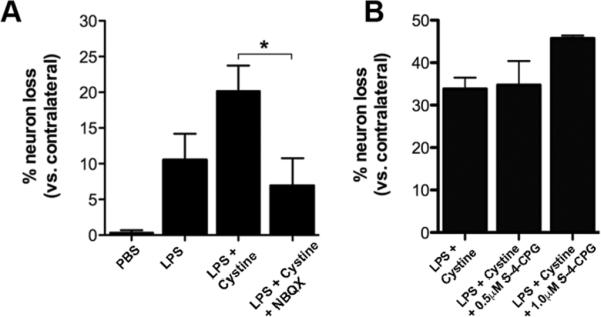
NBQX, an AMPA/kainate glutamate receptor antagonist, blocks the neurotoxic effects of macrophage system Xc- activation. Ventral horn motor neurons were quantified 3 days after intraspinal injection with PBS, LPS, LPS+cystine or LPS+cystine+ NBQX. (A) Co-injecting NBQX with LPS+cystine attenuates neuron loss indicating that microglia/macrophage-derived glutamate is a principal mediator of toxicity in this model; ANOVA; *p<0.05 vs. LPS+cystine; n=5–6per group (n=300–350neurons counted on contralateral side per animal). (B) Co-injecting S-4-CPG, an mGluR1 and presumed system Xc- antagonist, with LPS+cystine did not reduce neurotoxicity at 3 days post injection. n=4per group (n≃100 neurons counted on contralateral side per animal).
Since blocking glutamate activation of AMPA-receptors was effective in protecting neurons after LPS+cystine injection (Fig. 5A), we tested whether an antagonist of system xc- would also improve neuron survival. We co-injected (S)-4-Carboxyphenylglycine (CPG), a system xc- and mGluR1 antagonist, with LPS+cystine into the ventral horn. The addition of S-4-CPG did not improve neuronal survival following LPS+cystine injection (Fig. 5B).
The intracellular redox status of CNS macrophages predicts their ability to cause excitotoxicity
Macrophages maintain intracellular redox balance in part through activation of system xc-. When activated, system xc- moves extracellular cystine down a concentration gradient into cells resulting in an increase in intracellular GSH. As GSH rises, the intracellular milieu is “reduced”. In addition to the glutamate that is released during cystine exchange, reduced macrophages also release large quantities of potentially neurotoxic oxidants (e.g., nitric oxide) and inflammatory cytokines (e.g., IL-12) (Bethea, et al., 1999, Dobashi, et al., 2001, Murata, et al., 2002, Penkowa, et al., 2003). Thus, the redox status of CNS macrophages could predict their excitotoxic potential. We hypothesized that microglia and macrophages activated in the presence of cystine would become “reduced” and would produce more glutamate relative to cells activated by LPS alone. To test this hypothesis, we examined redox regulation ex vivo in a microglial cell line (BV-2) and primary bone-marrow derived macrophages (BMDMs).
Both BV-2 cells and BMDMs stimulated with LPS+cystine released significantly more glutamate into the extracellular milieu than when either cell type was stimulated with media or LPS alone (Fig. 6A). Corresponding with these changes, intracellular cysteine levels also were higher in microglia and BMDMs treated with LPS+cystine (Fig. 6B).
Figure 6.

Redox balance in microglia and macrophages is differentially affected by LPS and LPS+cystine. Redox status was measured in primary bone marrow-derived macrophages (BMDMs) and a microglial cell line (BV-2) after stimulating cells ex vivo with LPS or LPS+cystine. (A&B) When compared with cells stimulated with media or LPS (100ng/ml) alone, microglia and macrophages activated with LPS (100ng/ml) in the presence of high extracellular cystine (0.6mM) released significantly more glutamate (A) with a corresponding increase in intracellular cysteine (B). (C) Significantly higher intracellular glutathione (GSH) levels and high ratios of reduced:oxidized glutathione (GSH:GSSG) confirm the “reduced” intracellular redox status of microglia and macrophages activated with LPS+cystine. (ANOVA, *p<0.05, **p<0.001, ***p<0.0001 (vs. BMDM or BV-2 cells stimulated with LPS or media alone; data are representative of two independent experiments).
To assess the intracellular redox status of activated microglia and macrophages, intracellular concentrations of GSH (and the ratio of GSH:GSSG) was measured. In both microglia and BMDMs, LPS+cystine significantly increased intracellular GSH with a corresponding increase in GSH:GSSG (BMDM data shown in Fig. 6C; BV2 data not shown). These data confirm the in vivo observations and show that high levels of extracellular cystine enhance glutamate release from activated microglia and macrophages with a corresponding change in their redox potential.
DISCUSSION
The presence of activated microglia and macrophages in the diseased or injured CNS is perceived as a sign of imminent tissue destruction (Banati, et al., 1993, Boje and Arora, 1992, McMillian, et al., 1997, Wang, et al., 2003). This perception has emerged from a collection of in vitro data showing that macrophages release neurotoxic concentrations of glutamate when activated by LPS, a potent inflammatory stimulus (Domercq, et al., 2007, Piani, et al., 1991, Piani, et al., 1992). It is assumed that activated macrophages produce similar levels of glutamate in vivo and that this underlies inflammatory excitotoxicity. However, there is little in vivo data to support this hypothesis. The current model was developed to determine whether neurotoxic concentrations of glutamate could be produced in vivo by selectively activating microglia/macrophages in the presence of increased levels of extracellular cystine, the requisite substrate for the cystine/glutamate exchanger, i.e., system xc-.
Non-traumatic microinjections of low-dose LPS create focal zones of neuroinflammation. While some may consider LPS to be a “sledge hammer” inflammatory stimulus and irrelevant for studying mechanisms of acute innate immune cell activation, we and others have proven that low doses of LPS and other canonical pathogen associated molecular patterns (PAMPs) are useful tools for eliciting signaling pathways in microglia and macrophages that also are activated by ischemia, trauma or neurodegenerative disease (Gensel, et al., 2009, Glezer, et al., 2007, Hauss-Wegrzyniak, et al., 1998, Kitazawa, et al., 2005, Schonberg, et al., 2007). This is likely because the network of pattern recognition receptors (PRRs) that recognize LPS (and other PAMPs) also recognize endogenous proteins that are released in the injured or degenerating CNS (e.g., heat shock proteins, extracellular matrix molecules). This dual recognition capacity of PRRs has evolved to ensure that innate immune responses are amplified quickly, regardless of the origin of the stimulus (Nathan and Ding, 2010). Although simplistic in its approach, our current model is relevant to more complex forms of traumatic or ischemic CNS injury where extracellular cystine accumulates due to vascular damage and inflammation (Droge, et al., 1991, Hosoya, et al., 2001, Wade and Brady, 1981, Wang and Cynader, 2000). In mammalian plasma, cystine concentrations are ~30–70 μM or ~30× higher than those measured in CSF (Anderson, et al., 1989, Johnson, et al., 2008, Shaw, et al., 1995, Wang and Cynader, 2000). Thus, hemorrhage and inflammation create large extracellular cystine gradients that enhance the excitotoxic potential of microglia and macrophages. This potential is expected to vary as a function of distance from the site of vascular injury (Popovich and Longbrake, 2008).
Data presented here show that when the catalytic subunit of system xc- is induced in CNS macrophages and extracellular cystine levels are high, excitotoxicity occurs. Toxicity was restricted to neurons and involved activation of AMPA glutamate receptors; oligodendrocytes and OPCs were not adversely affected. On the contrary, as we have reported previously, cells of the oligodendroglial lineage seem to flourish in this inflammatory milieu (Schonberg, et al., 2007). Interestingly, cystine has been shown to protect oligodendrocytes from glutamate excitotoxicity in vitro (Oka, et al., 1993). This may be because oligodendrocytes use this amino acid to counteract the neurotoxic effects of activated CNS macrophages (Domercq, et al., 2007). These data indicate that demyelination, neuron loss and axon pathology cannot be predicted by the mere presence or absence of activated macrophages. Instead, unique molecular mechanisms regulate the divergent effects that these innate immune cells have on neurons and glia.
There is little doubt that microglia and macrophages secrete factors, including glutamate, that can kill neurons and glia. However, it is unlikely that system xc- induction and release of glutamate from microglia or macrophages has evolved as a mechanism to kill CNS cells. Instead, since activated CNS macrophages create focal “hot-spots” of oxidative stress through the release of superoxide, nitric oxide and inflammatory cytokines, these cells utilize the cystine/glutamate exchanger for ensuring redox maintenance and cell survival within the inhospitable environments that they create. The present data reveal a relationship between the induction of excitotoxic microglia/macrophages in vivo and high GSH:GSSG ratios ex vivo; LPS+cystine activated macrophages are reduced relative to those activated with LPS alone. Previously, it was shown that reduced macrophages release large quantities of oxidative metabolites (e.g., nitric oxide) and proinflammatory cytokines but low levels of neuroprotective cytokines like IL-6 and IL-10 (Dobashi, et al., 2001, Murata, et al., 2002). Since proinflammatory cytokines and glutamate synergize to induce cell death in the CNS (Hermann, et al., 2001, Miller, et al., 2005), manipulating the redox status of macrophages may prove useful in modifying the effects of neuroinflammation on neurodegenerative and/or repair cascades.
Although other cells in the CNS express functional system xc-, the current model seems to selectively induce this exchanger in microglia/macrophages with subsequent release of glutamate. Antibodies specific for xCT, the catalytic subunit of system xc-, failed to label astrocytes and only marginally labeled a subset of neurons. This is likely because only microglia (and macrophages) express high levels of the LPS receptor (TLR4) in the CNS (Lehnardt, et al., 2002, Lehnardt, et al., 2003). We cannot dismiss the possibility that astrocytes could contribute to focal accumulation of glutamate and toxicity in our model but this likely occurs subsequent to microglial activation. In response to microglial cytokines, astrocytes release glutamate via vesicular exocytosis (Bezzi, et al., 2001, Bezzi, et al., 2004), a phenomenon that can explain why presumed system xc- antagonists (e.g., sulfasalazine, α-aminoadipate) do not directly block glutamate release from astrocytes (Chung, et al., 2005).
Currently, there are no drugs that specifically target system xc-. Sulfasalazine is widely used as an antagonist of system xc-; however, it also blocks parallel mechanisms of inflammation and neuron pathology including NMDA receptor activation and NFκB signaling (Gan, et al., 2005, Noh, et al., 2006). Similarly, other drugs, including the phenylcycline derivatives 4-CPG and LY367385, act as system xc- antagonists but they also are competitive antagonists of metabotropic glutamate receptors (mGluRs) (Baker, et al., 2003, Fogal, et al., 2007).
We did try blocking cystine/glutamate exchange by co-injecting S-4-CPG with LPS and cystine; however, unlike NBQX, S-4-CPG failed to protect neurons nearby the injection site. There are a number of possible explanations for this result. Although S-4CPG is effective in reducing cystine uptake and glutamate release from astrocytes and glioma cells (Chung, et al., 2005, Ye, et al., 1999) in vitro, at/nearby the injection sites in our model S-4-CPG also will affect mGluRs on neurons and microglia. Emerging data show that glutamate signaling via mGluR1 (and mGluR5) suppresses microglial activation and neurotoxicity caused by these cells, especially under conditions of oxidative stress (Byrnes, et al., 2009, Chong, et al., 2005, Farso, et al., 2009). Thus, to fully understand the functional significance of system xc- or temporarily block its activity for therapeutic gain, selective antagonists must be developed.
Research Highlights
We show that CNS macrophages can cause neuronal excitotoxicity in vivo.
Activated CNS microglia/macrophages exert divergent effects on neurons and glia.
Demyelination, neuron loss and axon pathology cannot be predicted by the mere presence or absence of activated macrophages.
The redox signature CNS macrophages may regulate acute neuroinflammatory-mediated injury.
Acknowledgments
This work was supported in part by grants NIH -NS37846 (PGP), NS059776 (DMM) and DK64959 (RB), P30-NS045758, and Craig H. Neilsen Foundation (PGP).
Abbreviations
- CNS
central nervous system
- LPS
lipopolysaccharide
- OPC
oligodendrocyte progenitor cell
- GSH
glutathione
- TLR
toll-like receptor
- NBQX
2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
- DAB
diaminobenzidine
- DAPI
4',6-diamidino-2-phenylindole
- S-4-CPG
(S)-4-Carboxyphenylglycine
- BMDM
bone marrow derived macrophage
- M-CSF
macrophage colony stimulating factor
- HMGB1
high mobility group box 1
- (mGluRs)
metabotropic glutamate receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Anderson ME, Underwood M, Bridges RJ, Meister A. Glutathione metabolism at the blood-cerebrospinal fluid barrier. FASEB J. 1989;3:2527–2531. doi: 10.1096/fasebj.3.13.2572501. [DOI] [PubMed] [Google Scholar]
- 2.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 3.Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. GLIA. 1993;7:111–118. doi: 10.1002/glia.440070117. [DOI] [PubMed] [Google Scholar]
- 4.Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. Journal of Biological Chemistry. 1986;261:2256–2263. [PubMed] [Google Scholar]
- 5.Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich WD. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J.Neurotrauma. 1999;16:851–863. doi: 10.1089/neu.1999.16.851. [DOI] [PubMed] [Google Scholar]
- 6.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat.Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 7.Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- 8.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 9.Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- 10.Byrnes KR, Stoica B, Loane DJ, Riccio A, Davis MI, Faden AI. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. 2009;57:550–560. doi: 10.1002/glia.20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong ZZ, Kang J, Li F, Maiese K. mGluRI targets microglial activation and selectively prevents neuronal cell engulfment through Akt and caspase dependent pathways. Curr.Neurovasc.Res. 2005;2:197–211. doi: 10.2174/1567202054368317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J.Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobashi K, Aihara M, Araki T, Shimizu Y, Utsugi M, Iizuka K, Murata Y, Hamuro J, Nakazawa T, Mori M. Regulation of LPS induced IL-12 production by IFN-gamma and IL-4 through intracellular glutathione status in human alveolar macrophages. Clin.Exp.Immunol. 2001;124:290–296. doi: 10.1046/j.1365-2249.2001.01535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J.Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- 15.Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Droge W, Eck HP, Gmunder H, Mihm S. Modulation of lymphocyte functions and immune responses by cysteine and cysteine derivatives. Am.J.Med. 1991;91:140S–144S. doi: 10.1016/0002-9343(91)90297-b. [DOI] [PubMed] [Google Scholar]
- 17.Farso MC, O'Shea RD, Beart PM. Evidence group I mGluR drugs modulate the activation profile of lipopolysaccharide-exposed microglia in culture. Neurochem Res. 2009;34:1721–1728. doi: 10.1007/s11064-009-9999-3. [DOI] [PubMed] [Google Scholar]
- 18.Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gan HT, Chen YQ, Ouyang Q. Sulfasalazine inhibits activation of nuclear factor-kappaB in patients with ulcerative colitis. J.Gastroenterol.Hepatol. 2005;20:1016–1024. doi: 10.1111/j.1440-1746.2005.03862.x. [DOI] [PubMed] [Google Scholar]
- 20.Garg S, Vitvitsky V, Gendelman HE, Banerjee R. Monocyte differentiation, activation, and mycobacterial killing are linked to transsulfuration-dependent redox metabolism. Journal of Biological Chemistry. 2006;281:38712–38720. doi: 10.1074/jbc.M606235200. [DOI] [PubMed] [Google Scholar]
- 21.Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J.Immunol. 2008;180:3866–3873. doi: 10.4049/jimmunol.180.6.3866. [DOI] [PubMed] [Google Scholar]
- 22.Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG. Macrophages promote axon regeneration with concurrent neurotoxicity. J.Neurosci. 2009;29:3956–3968. doi: 10.1523/JNEUROSCI.3992-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glezer I, Chernomoretz A, David S, Plante MM, Rivest S. Genes involved in the balance between neuronal survival and death during inflammation. PLoS One. 2007;2:e310. doi: 10.1371/journal.pone.0000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer's disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- 25.Hermann GE, Rogers RC, Bresnahan JC, Beattie MS. Tumor necrosis factor-alpha induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol.Dis. 2001;8:590–599. doi: 10.1006/nbdi.2001.0414. [DOI] [PubMed] [Google Scholar]
- 26.Hosoya K, Saeki S, Terasaki T. Activation of carrier-mediated transport of L-cystine at the blood-brain and blood-retinal barriers in vivo. Microvasc.Res. 2001;62:136–142. doi: 10.1006/mvre.2001.2328. [DOI] [PubMed] [Google Scholar]
- 27.Johnson JM, Strobel FH, Reed M, Pohl J, Jones DP. A rapid LC-FTMS method for the analysis of cysteine, cystine and cysteine/cystine steady-state redox potential in human plasma. Clin.Chim.Acta. 2008;396:43–48. doi: 10.1016/j.cca.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J.Neurochemistry. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 29.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J.Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc.Natl.Acad.Sci.U.S.A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu D, Thangnipon W, McAdoo DJ. Excitatory amino acids rise to toxic levels upon impact injury to the rat spinal cord. Brain Res. 1991;547:344–344. doi: 10.1016/0006-8993(91)90984-4. [DOI] [PubMed] [Google Scholar]
- 33.Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J.Neurochemistry. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- 34.Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular.Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- 35.McMillian MK, Vainio PJ, Tuominen RK. Role of protein kinase C in microglia-induced neurotoxicity in mesencephalic cultures. Journal of Neuropathology and Experimental Neurology. 1997;56:301–307. doi: 10.1097/00005072-199703000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Miller BA, Sun F, Christensen RN, Ferguson AR, Bresnahan JC, Beattie MS. A sublethal dose of TNFalpha potentiates kainate-induced excitotoxicity in optic nerve oligodendrocytes. Neurochem.Res. 2005;30:867–875. doi: 10.1007/s11064-005-6880-x. [DOI] [PubMed] [Google Scholar]
- 37.Murata Y, Ohteki T, Koyasu S, Hamuro J. IFN-gamma and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur.J.Immunol. 2002;32:2866–2873. doi: 10.1002/1521-4141(2002010)32:10<2866::AID-IMMU2866>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 38.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 39.Noh JH, Gwag BJ, Chung JM. Underlying mechanism for NMDA receptor antagonism by the anti-inflammatory drug, sulfasalazine, in mouse cortical neurons. Neuropharmacology. 2006;50:1–15. doi: 10.1016/j.neuropharm.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 40.Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J.Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olney JW. New mechanisms of excitatory transmitter neurotoxicity. J.Neural Transm.Suppl. 1994;43:47–51. [PubMed] [Google Scholar]
- 42.Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernandez J, Molinero A, Campbell IL, Hidalgo J. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp.Neurol. 2003;181:130–148. doi: 10.1016/s0014-4886(02)00051-1. [DOI] [PubMed] [Google Scholar]
- 43.Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J.Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- 44.Piani D, Frei K, Do KQ, Cuenod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci.Lett. 1991;133:159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- 45.Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur.J.Immunol. 1992;22:2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- 46.Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat.Rev.Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- 47.Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J.Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. Journal of Biological Chemistry. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 49.Sato H, Fujiwara K, Sagara J, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochemical Journal. 1995;310(Pt 2):547–551. doi: 10.1042/bj3100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 51.Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J.Neurosci. 2002;22:8028–8033. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schonberg DL, Popovich PG, McTigue DM. Oligodendrocyte Generation is Differentially Influenced by Toll-Like Receptor (TLR) 2 and TLR4-Mediated Intraspinal Macrophage Activation. Journal of Neuropathology and Experimental Neurology. 2007;66 doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- 53.Shaw PJ, Forrest V, Ince PG, Richardson JP, Wastell HJ. CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration. 1995;4:209–216. doi: 10.1006/neur.1995.0026. [DOI] [PubMed] [Google Scholar]
- 54.Wade LA, Brady HM. Cysteine and cystine transport at the blood-brain barrier. J.Neurochemistry. 1981;37:730–734. doi: 10.1111/j.1471-4159.1982.tb12548.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J.Neurosci.Res. 2003;72:508–519. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- 56.Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J.Neurochemistry. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- 57.Xu GY, Hughes MG, Ye Z, Hulsebosch CE, McAdoo DJ. Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Exp.Neurol. 2004;187:329–336. doi: 10.1016/j.expneurol.2004.01.029. [DOI] [PubMed] [Google Scholar]
- 58.Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J.Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]