

Abstract
There is a clear link between dysregulation of glutamatergic signaling and mood disorders. Genetic variants in the glutamate receptor gene GRIK4, which encodes the kainate receptor subunit GluK4, alter the susceptibility for depression, bipolar disorder and schizophrenia. Here we demonstrate that Grik4−/− mice have reduced anxiety and an antidepressant-like phenotype. In the elevated zero-maze, a test for anxiety and risk taking behavior, Grik4−/− mice spent significantly more time exploring the open areas of the maze. In anxiogenic tests of marble-burying and novelty-induced suppression of feeding, anxiety-like behavior was consistently reduced in knockout animals. In the forced swim test, a test of learned helplessness that is used to determine depression-like behavior, knockout mice demonstrated significantly less immobility suggesting that Grik4 ablation has an antidepressant-like effect. Finally, in the sucrose preference test, a test for anhedonia in rodents, Grik4−/− mice demonstrated increased sucrose preference. Expression of the GluK4 receptor subunit in the forebrain is restricted to the CA3 region of the hippocampus and dentate gyrus regions where KARs are known to modulate synaptic plasticity. We tested whether Grik4 ablation had effects on mossy fiber (MF) plasticity and found there to be a significant impairment in LTP likely through a loss of KAR modulation of excitability of the presynaptic MF axons. These studies demonstrate a clear anxiolytic and antidepressant phenotype associated with ablation of Grik4 and a parallel disruption in hippocampal plasticity, providing support for the importance of this receptor subunit in mood disorders.
Keywords: kainate receptor, GluK4, depression, anxiety, mossy fiber, hippocampus
1. Introduction
Disruption of neuroplasticity caused by dysfunction of the glutamatergic system has been proposed as a major factor in the pathology of anxiety and depression [1, 2]. Levels of glutamate in the blood and cerebrospinal fluid have been reported to be altered in patients with major depressive disorder [3–6] and imaging studies have confirmed that glutamate levels are either up or down regulated in a brain region-specific manner in depressed patients [7–11]. Moreover, there has been a resurgence of interest in the link between glutamatergic neurotransmission and depression because glutamate receptor ligands, particularly non-competitive antagonists of NMDA receptors, have demonstrated efficacy as anti-depressants both in animal models [12–16] and human studies [17, 18].
Recent studies have directly implicated members of the ionotropic glutamate kainate receptor (KAR) family in several neuropsychiatric disorders including mood disorders and schizophrenia [19, 20]. The GRIK1–5 genes encode the GluK1–5 receptor subunits, which play roles in modulating synaptic transmission and cellular excitability in the brain [21]. The principal subunits GluK1, GluK2 and GluK3 can combine to form functional homomeric channels; however in the brain, these subunits are always assembled into heterotetrameric complexes incorporating the high-affinity subunits, GluK4 and GluK5 [22]. GluK4 expression is limited to only a few regions of the brain, and expression is highest in the CA3 region of the hippocampus and dentate gyrus [23–25] where KAR modulation of synaptic and circuit activity has been well documented [22, 26–29].
The GluK4 receptor subunit gene, GRIK4, is located near the tip of the long arm of chromosome 11. Genetic variants in the GRIK4 gene have been demonstrated to associate with several psychiatric disorders. In depressed patient cohorts, tested in the large-scale “sequenced treatment alternatives to relieve depression” (STAR*D) clinical trial, a polymorphism in GRIK4 was identified that is predictive of response to treatment with the antidepressant citalopram [30]. GRIK4 has also been identified as a susceptibility gene in schizophrenia and bipolar disorder [31], two neuropsychiatric disorders that often show comorbidity and shared genetic factors [32]. These studies identified a deletion variant in the 3′ untranslated region of GRIK4 in carriers of a haplotype that is protective against bipolar disorder [31]. In post-mortem hippocampal tissue from subjects carrying the deletion, mRNA transcripts and GluK4 protein levels are increased [33, 34]. In addition, imaging studies on healthy carriers of the protective haplotype demonstrated increased neural activity in the hippocampus during a face recognition task, demonstrating a functional effect of this genetic variation [35].
In order to investigate the role of GluK4-containing KARs in anxiety and depression, we have generated a Grik4 knockout mouse (Grik4−/−; [22]). We tested these mice in several well-characterized paradigms used to evaluate depressive-like and anxiety-like behaviors in rodent models. We found that Grik4−/− mice demonstrated a robust and consistent reduction in anxiety and an antidepressant-like phenotype consistently across several distinct behavioral tests. We also demonstrated that plasticity in the CA3 region of the hippocampus, where this receptor subunit is principally expressed, was impaired after Grik4 ablation. These studies further highlight the importance of GRIK4 as a susceptibility gene for mood disorders and directly demonstrate the functional importance of this receptor for plasticity in the hippocampus.
2. Materials and methods
2.1 Animals
Generation of Grik4 knockout (KO) and Grik4/Grik5 double-KO mice has been previously described [22]. Mice were backcrossed into an isogenic hybrid 129Sv/C57 strain in order to avoid strain dependent effects of congenic strains in some of the behaviors to be tested, and housed with free access to food and water in a barrier facility with a 14:10 hour light/dark cycle. For in vitro electrophysiological experiments, P14–P28 knockout mice and wildtype littermates were used for all recordings. Experiments were performed blind to the genotype of the animal and post-hoc genotyping was performed from tail biopsies using PCR. For behavioral experiments, adult male mice (age > 8 weeks) were used in all tests and comparisons made to age matched littermate control cohorts. All testing was carried out in a behavioral suite adjacent to the holding room in the mouse facility at Northwestern University. Mice were acclimated to the experimental room for at least 30 minutes prior to the start of testing. Experiments took place between 1000 h and 1900 h. Each apparatus was cleaned with 70% ethanol before use. For behavioral tests requiring video-tracking, video was acquired at 4 fps and 240 × 240 pixel spatial resolution using Limelight tracking software (Actimetrics, Wilmette, IL). Mice were handled in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. All procedures for testing and handling were approved by The Institutional Animal Care and Use Committee of Northwestern University.
2.2 Elevated zero-maze
The zero-maze consisted of a gray annular runway (5.5 cm width, 56 cm outer diameter, and 33 cm above ground level) inside a soundproof testing chamber. Two opposing 90° quadrants of the track were surrounded by inner and outer walls of gray polyvinyl-chloride (14 cm high). Animals were placed in an open quadrant at the start of the test and tracked for 5 min using LimeLight software.
2.3 Marble-burying test
Twenty-four dark marbles were placed on top of wood chip bedding (3 cm deep) in six rows of four in a cage measuring 36 × 31 × 19 cm placed in a brightly lit, open area. Animals were introduced to this new cage and remained in the testing cage for 15 min. At the end of the test period, quantification was made of the number of marbles that were buried by more than two-thirds of their diameter.
2.4 Locomotor activity
The open field chamber consisted of a novel arena (56 × 56 × 30 cm) placed in a dimly-lit and soundproof testing chamber. Mice were released in the center of the arena at the start of the experiment. Activity was recorded for 30 min. During offline analysis, the arena was divided into a 7 × 7 grid consisting of two zones of approximate equal area: the outer zone (outer 24 squares along the wall) and the open zone (inner 25 squares).
2.5 Y-maze
The Y-maze consisted of three arms of identical dimensions radiating from a central area at 120° angles. At the start of the trial, a single animal was placed in the base of the start arm with its nose towards the center of the maze. After all four paws crossed into the second arm, the exit to the arm was blocked and the animal remained in the arm for 30 s. The animal was removed to a clean cage for 30 s then returned to the start arm. The animal was allowed to move freely into a second arm. The trial was scored as an alternation if the arm chosen on the second entry was different than the first entry. Mice were tested in a single trial each day for 10 days, and the position of the Y-maze was changed for each trial to provide a novel test environment.
2.6 Novelty-induced suppression of feeding
The testing apparatus consisted of a plastic box (38 × 46 × 14 cm) covered with wood-chip bedding with a food pellet placed in the center. Animals were food deprived for 24 hours prior to testing. On the test day mice were individually placed in the corner of the test box, and the latency to bite the food pellet was measured. Mice were immediately returned to their home cage with access to food and the total consumption of food pellets measured for a further 5 min period.
2.7 Forced swim test
Glass beakers (13 cm diameter × 24 cm high) were filled with fresh water (23–25 °C) to a depth of 10 cm. Mice were placed into the test beaker and were unable to escape or rest by touching the bottom of the beaker. Sessions were video recorded for 10 minutes and analyzed offline. Mice were scored by visual inspection for immobility defined as motionless floating in the water.
2.8 Sucrose preference test
Mice were individually housed with water bottles for 3 days prior to testing. On day 1 of the test, water bottles were replaced with two 50-ml tubes (bottle “A” and bottle “B”) fitted with bottle stoppers containing one-balled sipper tubes. During days 1 and 2, both bottles A and B were filled with drinking water (w/w). During days 3 and 4, both A and B were filled with a solution of 1 % sucrose (s/s). On days 5–8, bottle A contained 1 % sucrose, and bottle B contained drinking water (s/w). The position of bottles A and B was switched daily to avoid a side bias. Total fluid consumed was calculated as [Sucrose + Water] and averaged across each day for a given condition (w/w, s/s, or s/w). Sucrose preference was calculated as 100*(Sucrose/[Sucrose + Water]). In experiments designed to test the dose response of sucrose preference, animals were habituated for one day to w/w and one day s/s and tested on subsequent days with increasing sucrose concentration (0.5 % to 2 %).
2.9 Slice preparation
For electrophysiological recordings, transverse hippocampal slices (350 μm) were prepared from P14–P28 mice. Mice were anesthetized with isoflurane, decapitated and the brain rapidly removed in ice-cold oxygenated sucrose-slicing ACSF containing 85 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 75 mM sucrose, 0.5 mM CaCl2, and 4 mM MgCl2, equilibrated with 95 % O2/5 % CO2. Slices were incubated at 28 °C for 30 min, and a slow exchange was made of the oxygenated sucrose-ACSF for oxygenated sodium-ACSF solution containing 125 mM NaCl, 2.4 mM KCl, 1.2 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 1 mM CaCl2, and 2 mM MgCl2. Individual slices were transferred to a recording chamber and continuously perfused with oxygenated sodium ACSF containing 2 mM CaCl2 and 1 mM MgCl2.
2.10 Mossy fiber long-term potentiation
Whole-cell voltage-clamp recordings were made from visually identified CA3 neurons in slices maintained at 30 °C. The internal solution for whole-cell recording contained 95 mM CsF, 25 mM CsCl, 10 mM Cs-HEPES, 10 mM Cs-EGTA, 2 mM NaCl, 2 mM Mg-ATP, 10 mM QX-314, 5 mM TEA-Cl, 5 mM 4-AP (pH adjusted to 7.3 with CsOH). Series resistance was continuously monitored using hyperpolarizing voltage steps, and recordings were discarded if there was a > 20 % change during the course of the experiment. AMPA/kainate receptor mediated mossy fiber (MF) excitatory postsynaptic currents (EPSCs) were isolated using the GABAA antagonists bicuculline (10 μM) and picrotoxin (100 μM), and the NMDA receptor antagonist, D-APV (50 μM). EPSCs were evoked with a monopolar glass electrode filled with oxygenated ACSF positioned in the stratum lucidum [22]. MF EPSCs were identified by large paired pulse facilitation (>2), brief onset latencies, and rapid EPSC rise times and by the addition of the group II mGluR agonist DCG-IV at the end of each experiment which selectively inhibited MF EPSCs greater that 70 % [22]. During baseline recording, MF EPSCs were evoked at a frequency of 0.05 Hz, and LTP was induced using three trains of tetanic simulation of 100 Hz for 1 s.
Presynaptic fiber volley recordings were performed with low-resistance patch pipettes filled with external ACSF placed in stratum lucidum in CA3. Bipolar tungsten electrodes were placed in the hilus region to stimulate MF axons. The presynaptic fiber volley was isolated using GABAA receptor antagonists, bicuculline (10 μM) and picrotoxin (50 μM), NMDA receptor antagonist, D-2-amino-5-phosphovalerate (APV; 50 μM), and AMPA receptor antagonist, GYKI-53655 (30 μM).
2.11 Statistical analysis
All experiments were subjected to a between-subjects design with genotype as the independent variable. For the locomotor activity test, repeated measures were collected from a single animal at different time points and data were analyzed using a one-way repeated measure ANOVA. For all other behavioral tests, data were analyzed with a Student’s t-test. Data are expressed as mean ± S.E.M. For all tests, significance was defined as p < 0.05. For electrophysiological experiments, n represents the number of individual recordings. Only one recording was made from each slice and at least 3 animals were used for each experiment.
3. Results
3.1 Elevated zero-maze
GRIK4 has been associated with mood disorders, and anxiety is often comorbid with depression and other mood disorders; therefore, we tested Grik4−/− mice in anxiety-related behaviors. In the elevated zero-maze (EZM), the natural inclination of mice to explore novel environments is offset by an aversive response to potentially dangerous open and exposed areas. Mice were released into the open area of the maze and exploratory behavior tracked for five minutes. Grik4−/− mice spent significantly more time than Grik4+/+ littermates exploring the open areas of the EZM (% time in open quadrants Grik4+/+: 23.1 ± 9.6 %, n = 11; % time in open quadrants Grik4−/−: 53.6 ± 9.8 %, n = 13, p < 0.05; Fig 1B). Consistent with this increased exploratory behavior in the open quadrants, Grik4−/− mice traveled a greater distance in the open areas (open quadrant distance Grik4+/+: 79.3 ± 23.9 cm, n = 11; open quadrant distance Grik4−/−: 321.1 ± 77.3 cm, n = 13; p < 0.01; Fig 1C). However the total distance traveled by Grik4−/− and Grik4+/+ mice in all areas of the maze was equivalent, suggesting there were no genotype dependent effects on activity or major motor deficits (total distance Grik4+/+: 819.0 ± 106.0 cm, n = 11; total distance Grik4−/−: 958.0 ± 154.6 cm, n = 13; p > 0.05). A cohort of heterozygous Grik4 mice (Grik4+/−) was also tested and their exploratory behavior was indistinguishable from Grik4+/+ mice. Grik4+/− mice spent equivalent time in the open areas (% time in open quadrants Grik4+/−: 21.5 ± 8.7 %; n = 11, p > 0.05; Fig 1B), traveled an equivalent distance in the open areas (open quadrant distance Grik4+/−: 100.0 ± 22.6 cm; n = 11, p > 0.05; Fig 1C), and traveled an equivalent total distance (total distance Grik4+/−: 884.4 ± 98.7 cm; n = 11, p > 0.05) when compared to wildtype mice. Increased exploration of the open areas of the EZM suggests that genetic ablation of Grik4 decreases anxiety-like behaviors in mice.
Figure 1. Anxiety-like behavior is reduced in Grik4−/− mice in the elevated zero-maze and marble-burying tests.

(A) Representative path vectors of Grik4+/+ (top) and Grik4−/− (bottom) mice during exploration of elevated zero-maze (EZM). Shaded regions represent closed quadrants. (B) Grouped data on percentage of time spent exploring open quadrants of EZM during the 300 s test. (C) Grouped data of total distance traveled while exploring the open quadrants of EZM. Grik4−/− spent significantly more time exploring open quadrants, and traveled a greater distance in the open areas although there was no difference in the total distance travelled in all quadrants between cohorts. (D) Grouped data of marbles buried by ≥ 2/3 diameter during marble-burying test. Grik4−/− mice buried significantly fewer marbles. (B – D) Data for Grik4+/+ (black bar), Grik4+/− (gray bar), and Grik4−/− (white bar). Data are expressed as mean ± S.E.M. * p < 0.05, ** p < 0.01.
3.2 Marble-burying test
To further examine whether Grik4−/− mice exhibit less anxiety-like behavior, mice were tested in the marble-burying test. The brightly lit novel environment in this test is anxiogenic, and after initial cage exploration, mice will dig into the bedding material resulting in partial or complete burial of marbles [36]. We quantified the number of marbles buried by ≥ 2/3 of their diameter and found that this was significantly decreased for the Grik4−/− group compared to Grik4+/+ mice (marbles buried Grik4+/+: 9.1 ± 0.77; marbles buried Grik4−/−: 5.3 ± 1.2; n = 10, p < 0.05; Fig 1D), but there was no difference in Grik4+/− mice (marbles buried Grik4+/−: 7.5 ± 1.5; n = 10, p > 0.05; Fig 1D). Decreased marble burying is an index of anxiety-induced digging behavior, and the results suggest that the absence of Grik4 is anxiolytic, consistent with results in the EZM test.
3.3 Locomotor activity
To test for dysfunction in locomotor activity or exploratory behavior that might account for deficits in anxiety tests, we monitored mice in a novel, dimly-lit open field apparatus. After introduction to the open field chamber, mice in all three genotype cohorts displayed normal habituation to the arena as indicated by a gradual reduction in movement over 30 minutes. Analysis of mouse trajectories determined that there was no effect of genotype on the total amount of time spent exploring the open zone during the test (30 min) (% time in open Grik4+/+: 12.1 ± 3.7 %; % time in open Grik4+/−: 7.1 ± 2.2 %; % time in open Grik4−/−: 12.6 ± 2.3 %; n = 16, p > 0.05; Fig 2A) or in the total distance traveled while exploring the arena (total distance Grik4+/+: 4.7 ± 1.1 m; total distance Grik4+/−: 4.1 ± 3.1 m; total distance Grik4−/−: 5.4 ± 0.8 m; n = 16, p > 0.05; Fig 2B). When we analyzed the data during the initial 15 min of the test when habituation plays a role in activity (Fig 2A), and during the final 15 min of the test once animals are habituated to the environment (Fig 2B), again there was no observable difference in time in open areas or total distance travelled between the genotypes. In addition, there was no difference in the number of times mice crossed between the outer zone and open zone (crossings Grik4+/+: 102.3 ± 36.4; crossings Grik4+/−: 59.6 ± 15.8; crossings Grik4−/−: 114.5 ± 24.2; n = 16, p > 0.05; Fig 2C). These results suggest normal locomotor activity and exploratory activity of Grik4−/− mice.
Figure 2. Activity of Grik4−/− mice in a non-anxiogenic environment is normal and there are no observed deficits in working memory.

(A) Grouped data on percentage of time spent in open area of novel open field arena. Data are segmented into 15 min sessions representing the first (0 – 15 min) and second (16 – 30 min) half of 30 min test. (B) Grouped data of total distance traveled in arena during the first 15 min and minutes 16 – 30 of the 30 min test. (C) Number of crossings between open zone and edge zone during the test. No genotype dependent differences in activity or exploration were observed (D) Grouped data from the Y-maze test of the percentage of alternations made by mice. No genotype dependent differences were observed in this test for working memory. (A – D) Data for Grik4+/+ (black bar/closed squares), Grik4+/− (gray bar), and Grik4−/− (white bar/open squares). Data are expressed as mean ± S.E.M.
3.4 Y-maze
To examine working memory, mice were tested in the Y-maze. Rodents will perform arm alternations, choosing a novel arm to explore, when given a choice. Deficits in working memory result in a reduction in the number of alternations. Wildtype mice chose to explore an alternate arm 75 % of the time in this test and alternation rates in wildtype mice were not significantly different for Grik4−/− mice (alternations Grik4+/+: 74.7 ± 3.6 %, n = 8; alternations Grik4−/−: 72.0 ± 18.6 %, n = 13; p > 0.05; Fig 2D) or heterozygous Grik4+/− mice (alternations 66.7 ± 6.9 %, n = 9; p > 0.05; Fig 2D). These results suggest that ablation of Grik4 does not impair hippocampal-dependent working memory that underlies behavior in the Y-maze.
3.5 Novelty-induced suppression of feeding
The novelty-induced suppression of feeding test (NSF) assesses anxiety by measuring the latency of an animal to eat food in a novel anxiogenic environment following food deprivation. We found a significant effect of genotype for the latency to bite a food pellet at the center of the arena, with the knockout group having a significantly shorter latency to feed (latency Grik4+/+: 107.0 ± 10.6 s, n = 18; latency Grik4−/−: 81.4 ± 10.2 s, n = 13; p < 0.05, Fig 3A). Grik4−/− mice did not differ from Grik4+/+ mice in their food intake in the homecage during a 5 min period immediately following the latency test (food intake Grik4+/+: 0.14 ± 0.01 g, n = 18; food intake Grik4−/−: 0.16 ± 0.01 g, n = 13, p > 0.05, Fig 3B), nor did they differ in the amount of weight lost during the 24 h food deprivation period (% weight lost Grik4+/+: 7.8 ± 0.9 %, n = 18; % weight lost Grik4−/−: 9.9 ± 0.8 %, n = 13; p > 0.05, Fig 3C) strongly suggesting that reduced latency to feed was directly related to reduced anxiety in Grik4−/− mice.
Figure 3. Anxiety-related latency to feed is reduced in Grik4−/− mice.
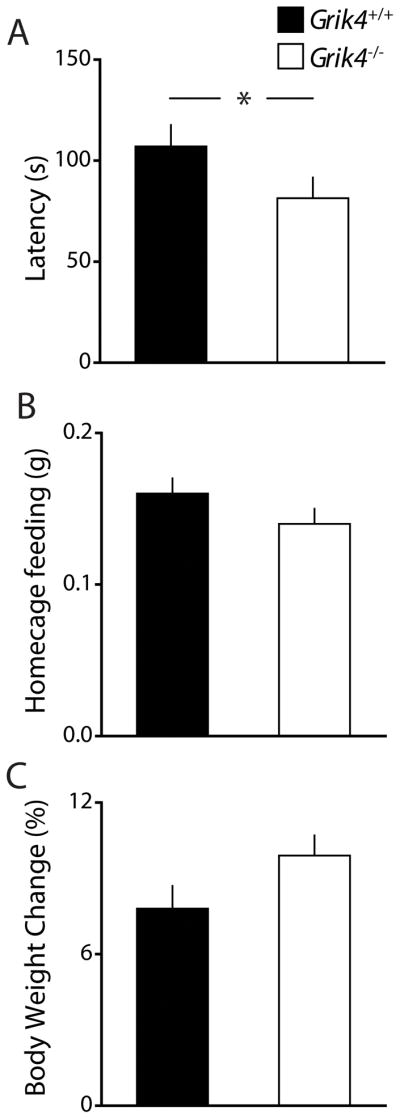
(A) Grouped data of latency to bite food in a novel anxiogenic environment following 24 h food deprivation. (B) Food intake of mice in the home cage during a 5 min period following NSF latency test (C) Weight lost (%) during 24 h food deprivation prior to NSF. Latency to bite was significantly reduced in Grik4−/− mice. There was no genotype dependent effect for homcage feeding following latency or weight lost during food deprivation. (A – C) Data for Grik4+/+ (black bar) and Grik4−/− (white bar). Data are expressed as mean ± S.E.M. * p < 0.05.
3.6 Forced swim test
The forced swim test (FST) is a test for learned helplessness used to test rodent models of depression. Mice that show depressive-like behavior cease to swim and spend more time immobile in the water [37]. We found that during a 10 min test period, both Grik4+/+ and Grik4−/− mice initially displayed equivalent levels of activity. However, as the test progressed the Grik4−/− group diverged from Grik4+/+ demonstrating increased activity and reduced immobility during the final 4 min of testing (immobility time 7–10min Grik4+/+: 175.3 ± 14.6 s; immobility time 7–10 min Grik4−/−: 118.5 ± 16.86 s; n = 11, p < 0.01; Fig 4). There was no effect of genotype in the heterozygous group Grik4+/− (immobility time Grik4+/−: 142.5 ± 15.31 s, n = 11; p > 0.05; Fig 4B). Increased immobility is often interpreted as an antidepressant-like phenotype in rodent models suggesting that ablation of Grik4 results in an antidepressant-like behavior in mice.
Figure 4. Grik4−/− mice display antidepressant-like behavior in test of learned helplessness.

(A) Percentage of time spent immobile during the course of forced swim test (FST). Data are presented in 1 min bins of a 10 min test for Grik4+/+ (closed squares) and Grik4−/− (open squares). (B) Total time spent immobile during the final 4 min of testing. Grik4−/− mice spent significantly less time immobile during final 4 min of testing. Data for Grik4+/+ (black bar), Grik4+/− (gray bar), and Grik4−/− (white bar). Data are expressed as mean ± S.E.M. * p < 0.05, ** p < 0.01.
3.7 Sucrose Preference
The sucrose preference test (SPT) is a model of anhedonia, a core symptom of depression, in which animals presumably fail to elicit pleasure from an activity that is normally enjoyable. Sucrose preference in mice is highly dependent upon the background strain. Therefore, we first tested preference in wildtype mice in our hybrid isogenic strain. Grik4+/+ mice demonstrated a small preference for sucrose with increasing sucrose concentration (0.5 % preference Grik4+/+: 42.1 ± 8.3 %; 2 % preference Grik4+/+: 68.0 ± 6.0 %). Whereas wildtype mice in a congenic C57/Bl6 strain show a very large preference for sucrose (1 % preference C57/Bl6: 82.8 ± 7.0 %, n = 6; p < 0.05, Fig 5A). When we tested knockout mice and a cohort of wildtype littermates in this test (with a 1% solution), we saw a significantly increased preference for sucrose on the Grik4−/− group (preference Grik4+/+: 53.2 ± 3.1 %, n = 21; preference Grik4−/−: 62.9 ± 2.9 %, n = 19; p < 0.05, Fig 5B). The effect was not accounted for by a difference in total fluid intake (fluid intake Grik4+/+: 3.65 ± 0.13 g, n = 21; fluid intake Grik4−/−: 3.78 ± 0.12 g, n = 19; p > 0.05, Fig 5C). The increased preference by Grik4−/− mice for sucrose is consistent with an antidepressant-like phenotype suggested by results in the FST.
Figure 5. Grik4−/− mice demonstrate increased preference for sucrose.
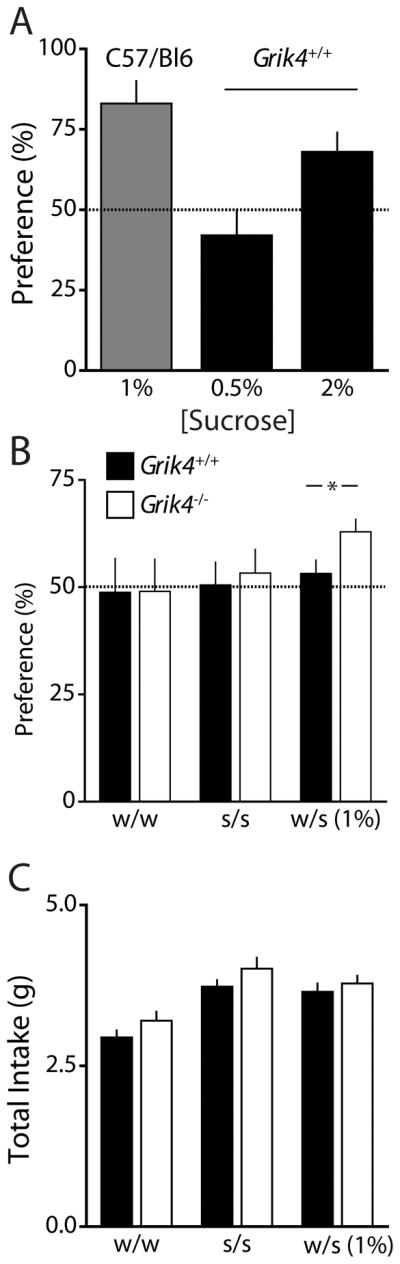
(A) Preference (%) for sucrose for wildtype mice in a congenic C57/Bl6 strain (gray bar; 1 % sucrose) and for isogenic hybrid strain Grik4+/+ (black bars; 0.5 % and 2 % sucrose). (B) Data of preference for bottle “A” over bottle “B” (A/(A + B)) during an eight day sucrose preference test. No preference was observed for each genotype when both bottles contained water on days 1 and 2 (w/w) or when both bottles contained sucrose (1%) on days 3 and 4 (s/s). However, Grik4−/− mice display significantly increased preference for the sucrose bottle during the preference test on days 5 – 8 (s/w). (C) Grouped data of total fluid intake during each stage of the test. There was no effect of genotype on fluid intake during the test. (A – C) Data for C57/Bl6 strain (gray bar), Grik4+/+ (black bar) and Grik4−/− (white bar). Data are expressed as mean ± S.E.M. * p < 0.05.
3.8 Mossy fiber long-term potentiation
The GluK4 receptor subunit is expressed at high levels in the CA3 region of the hippocampus and the dentate gyrus [25]. KARs play roles in plasticity at the mossy fiber synapse in the CA3, and there is a clear deficit in mossy fiber (MF) LTP in Grik2 and Grik3 knockout animals suggesting that GluK2 and GluK3 subunits are required for the proper function of MF KARs [26, 29, 38, 39]. However, no disruption in MF LTP is observed when the high affinity subunit GluK5 is ablated, although this subunit likely contributes to the heterotetrameric KAR complex [40]. We previously demonstrated that short-term plasticity is not impaired in Grik4 knockout mice [22] however it is not known whether loss of the GluK4 receptor subunit impacts MF LTP. Using whole-cell patch-clamp recording from CA3 pyramidal neurons, we found that MF LTP induced by a tetanic 100Hz 1s train was significantly impaired in slices from Grik4 knockout mice (LTP Grik4+/+: 172.9 ± 14.4 %, n = 7; LTP Grik4−/−: 101.6 ± 10.2 %, n = 6; p < 0.01; Fig 6A). Not surprisingly, in additional experiments, we also saw deficits in MF LTP in slices from double high-affinity KAR subunit knockout mice (LTP Grik4−/−/Grik5−/−: 120.5 ± 13.5 %, n = 10; p < 0.05; Fig 6B). The reduction in Grik4−/−/Grik5−/− mice is likely due to ablation of Grik4 as MF LTP is normal in Grik5−/− mice [40]. Previous work has shown that presynaptic axonal KARs impart an associativity to MFs that reduces the induction threshold for LTP [39]. We therefore tested whether KAR mediated modulation of MF axon excitability was altered in Grik4 knockout mice. We recorded the isolated MF presynaptic fiber volley (FV) and determined the effect of application of low concentrations of a KAR agonist. As previously demonstrated, application of 500 nM kainic acid significantly increased the size of the FV in wildtype mice (Fig 6C & D) [41]. However, there was significantly less potentiation of fiber volley responses by kainic acid in Grik4−/− mice (FV potentiation Grik4+/+ mice: 143.3 ± 6.6 %, n = 3; FV potentiation Grik4−/−: 122.7 ± 5.6 %, n = 7; p < 0.05; Fig 6C). These results demonstrate a significant role for the GluK4 receptor subunit in regulating synaptic plasticity in the CA3 region of the hippocampus.
Figure 6. Mossy fiber long-term potentiation in the hippocampus is impaired in Grik4−/− mice.

(A) Time course of mossy fiber (MF) LTP in Grik4+/+ (closed squares) and Grik4−/− (open squares) mice. Black arrow represents induction at time = 0. Inhibition of the EPSC by the group II mGluR agonist DCG-IV at the end of each experiment (black bar) was used to confirm that the EPSCs were of MF origin. Inset shows representative examples of EPSCs in Grik4+/+ (closed square) and Grik4−/− (open square) mice during baseline period (1) and 20 – 30 min post-induction (2). (B) Cumulative probability histogram of tetanus-induced MF LTP in Grik4+/+ (closed squares), Grik4−/− (open squares), and Grik4−/−/Grik5−/− (open triangles) mice. LTP was measured as the potentiation 20 – 30 min post-induction compared with baseline period. MF LTP was significantly impaired in Grik4−/− and Grik4−/−/Grik5−/− mice. (C) Representative recording of the presynaptic MF fiber volley (FV) recorded in the stratum lucidum during stimulation of hilar region. FV in Grik4+/+ (black square) and Grik4−/− (white square) during control period and application of 500 nM kainic acid. (D) Grouped data of potentiation of MF FV by 500 nM kainic acid. Kainate-induced potentiation of MF FV was significantly reduced in Grik4−/− mice. Data for Grik4+/+ (black bar) and Grik4−/− (white bar). Data are expressed as mean ± S.E.M. * p < 0.05.
4. Discussion
The GluK4 kainate receptor subunit gene, GRIK4, has been identified as a susceptibility gene for schizophrenia and mood disorders. Here we performed behavioral analysis of Grik4−/− mice to determine whether loss of this subunit was manifest in a significant behavioral phenotype. In several independent tests that are routinely used to test anxiety in rodents, we found that Grik4−/− mice demonstrated behavior consistent with reduced anxiety and increased risk-taking behavior. Moreover, these mice also displayed an antidepressant-like phenotype in a goal directed test for behavioral despair and a test for hedonic behavior. We did not observe any deficits in general activity or working memory, suggesting that Grik4 ablation has very specific effects on the behavior of the animals. The phenotype was only apparent in homozygous knockout mice and no gene dosage effects were observed in heterozygous mice, likely because heterozygosity has little effect on the function of native heterotetrameric KAR complex.
Human studies of carriers of the haplotype protective against bipolar disorder have found that there are increases in mRNA and GluK4 protein associated with the deletion variant [33, 34]. There are also several studies that have found reduced KAR expression in postmortem tissue from patients with schizophrenia [42–44]. It is difficult to draw direct parallels between findings in knockout mice and altered KAR expression associated with human neuropsychiatric disorders. The biology underlying the involvement of KARs in pathology is likely complex, but studies in mice enable us to begin to address some mechanistic questions such as understanding the circuit and synaptic basis for altered brain function that might contribute to disrupted cognitive processes.
Glutamate receptors have recently received increased scrutiny as possible targets for the development of the next generation of antidepressants. Existing antidepressant drugs target monoamine neuromodulatory neurotransmitter systems, but these treatments have a very long latency to show clinical efficacy and are without any effect in the majority of patients. Both preclinical and clinical studies have demonstrated that NMDA receptor antagonists have a rapid and significant antidepressant action [16, 45, 46]. Similarly, potentiators of AMPA receptors, the other major class of ionotropic glutamate receptors, have also demonstrated efficacy in animals models of depression [47]. While there are no subtype selective ligands for targeting GluK4 containing receptors, our demonstration of a robust antidepressant phenotype in Grik4−/− mice suggests that targeting these receptors with antagonists or negative allosteric modulators warrants further investigation for the development of glutamatergic agents for rapid antidepressant treatment.
Of all the KAR subunits, the GluK4 subunit has one of the most limited expression pattern in the brain, being prominently expressed in CA3 and dentate gyrus [23, 24]. These receptor subunits assemble as heterotetrameric complexes with the other KAR subunits expressed in these regions to form a unique population of receptors with particular functional properties [22]. Knockout mice of the more ubiquitously expressed subunits have also been generated and in some cases display overlapping behavioral phenotypes with Grik4−/− mice. GluK2 is the most abundantly expressed KAR subunit, and it is likely that all GluK4 receptors assemble with this subunit in the hippocampus [22]. Grik2−/− mice also exhibit less anxiety-like behavior in the elevated plus-maze and have an antidepressant-like phenotype in the FST [48]. However, Grik2−/− mice also display mania-like symptoms, including hyperactivity and increased aggressive behavior, which were alleviated by chronic lithium administration [48]. Interestingly, genome-wide linkage studies have identified the GRIK2 (the gene encoding GluK2) gene locus as having a significant susceptibility for bipolar disorder [49, 50]. In contrast to the co-localization of GluK2 and GluK4 subunits in the hippocampus and the overlapping phenotype in knockout mice, the other major KAR subunit GluK1 is largely excluded from principal neurons in the hippocampus [24]. Polymorphisms in GRIK1 have been linked to schizophrenia [51] but in contrast to Grik2 and Grik4 knockout mice, Grik1−/− mice demonstrate increased anxiety-like behavior, which can be replicated by local administration of GluK1 antagonists into the amygdala [52]. The behavioral abnormalities seen in Grik2−/− and Grik4−/− mice indicate that hippocampal KARs are a potential therapeutic target to decrease despair and anxiety in the treatment of depression.
The neural basis for depression has not been fully defined and is likely complex involving diverse genetic and circuit level disruptions in the brain. Circuit dysfunction in limbic regions, particularly the hippocampus, has received special interest based upon observations that both rodent models of depression and antidepressant administration impact adult neurogenesis in the dentate gyrus [53–56]. While the current study does not directly speak to the involvement of GluK4-containing KARs in processes that regulate neurogenesis, it is important to note that the GluK4 subunit is highly, and almost exclusively, expressed in CA3 pyramidal and dentate gyrus granule cells. The role of GluK4-containing receptors in regulating synaptic and circuit function in this area has not been exhaustively studied. However, we previously demonstrated that the postsynaptic KAR component of the MF EPSC was reduced in amplitude and had significantly slowed kinetics [22]. Mossy fiber short-term plasticity which is regulated by KARs [26, 27, 29, 57] was not impaired in these mice [22]. Here we reassessed the involvement of GluK4 receptor subunits focusing on MF LTP. Induction of LTP at the MF-CA3 synapses is NMDAR-independent [58], while expression is presynaptic, manifesting as a persistent increase in glutamate release probability [59]. While not required for induction, KARs alter the threshold for induction of MF LTP [39], and MF LTP is impaired in GluK2 and GluK3 knockout mice [26, 29, 38]. We found that MF LTP is impaired in Grik4−/− mice, and that KAR mediated enhancement of MF axon excitability is also reduced, suggesting a clear role for the GluK4 receptor subunit in modulating MF LTP. While these results do not directly provide a causative link between plasticity in the CA3 region and the behavioral alterations in these knockout mice, they do begin to determine the cellular and circuit roles of GluK4-containing receptors in these important limbic regions. Interestingly, recent work has demonstrated that chronic treatment with the antidepressant fluoxetine results in a de-maturation of dentate granule cells and affects plasticity of their MF outputs to the CA3 [60].
In summary, we provide evidence that strongly links the GluK4 KAR subunit to anxiety and depressive-like behavior. These studies support genetic, molecular and human imaging studies that have provided a basis for considering a role for KARs in mood disorders.
Highlights.
Ablation of Grik4 results in an antidepressant-like phenotype
Grik4−/− mice demonstrate reduced anxiety
LTP at hippocampal mossy fiber synapses is impaired in Grik4−/− mice
Acknowledgments
Role of the funding source
This work was supported by grants from NIH/NINDS (R01NS058894 to AC) and a John Nicholson Fellowship (to JSC). No funding source played a role in study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication.
We are grateful to Dr Geoffrey Swanson for discussions. We also acknowledge Dr Craig Weiss and members of the Northwestern University Behavioral Phenotyping Core for consultation on the development and interpretation of behavioral assays.
Footnotes
Disclosure Statement
The authors have no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Justin S. Catches, Email: justincatches2008@u.northwestern.edu.
Jian Xu, Email: jian-xu@northwestern.edu.
References
- 1.Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77. doi: 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duman RS, Li N, Liu R-J, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62:35–41. doi: 10.1016/j.neuropharm.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altamura CA, Mauri MC, Ferrara A, Moro AR, D’Andrea G, Zamberlan F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am J Psychiatry. 1993;150:1731–3. doi: 10.1176/ajp.150.11.1731. [DOI] [PubMed] [Google Scholar]
- 4.Kim JS, Schmid-Burgk W, Claus D, Kornhuber HH. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr. 1982;232:299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- 5.Mauri MC, Ferrara A, Boscati L, Bravin S, Zamberlan F, Alecci M, et al. Plasma and Platelet Amino Acid Concentrations in Patients Affected by Major Depression and under Fluvoxamine Treatment. Neuropsychobiology. 1998;37:124–9. doi: 10.1159/000026491. [DOI] [PubMed] [Google Scholar]
- 6.Mitani H, Shirayama Y, Yamada T, Maeda K, Ashby CR, Jr, Kawahara R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1155–8. doi: 10.1016/j.pnpbp.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 7.Ajilore O, Haroon E, Kumaran S, Darwin C, Binesh N, Mintz J, et al. Measurement of Brain Metabolites in Patients with type 2 Diabetes and Major Depression Using Proton Magnetic Resonance Spectroscopy. Neuropsychopharmacology. 2007;32:1224–31. doi: 10.1038/sj.npp.1301248. [DOI] [PubMed] [Google Scholar]
- 8.Taylor MJ, Mannie ZN, Norbury R, Near J, Cowen PJ. Elevated cortical glutamate in young people at increased familial risk of depression. The International Journal of Neuropsychopharmacology. 2011;14:255–9. doi: 10.1017/S1461145710001094. [DOI] [PubMed] [Google Scholar]
- 9.Auer DP, Pütz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. 2000;47:305–13. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- 10.Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced Prefrontal Glutamate/Glutamine and {gamma}-Aminobutyric Acid Levels in Major Depression Determined Using Proton Magnetic Resonance Spectroscopy. Arch Gen Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- 11.Sanacora G, Gueorguieva R, Epperson CN, Wu Y-T, Appel M, Rothman DL, et al. Subtype-Specific Alterations of {gamma}-Aminobutyric Acid and Glutamate in Patients With Major Depression. Arch Gen Psychiatry. 2004;61:705–13. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 12.Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–11. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 13.Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P-f, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–5. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Goffer Y, Xu D, Tukey DS, Shamir DB, Eberle SE, et al. A single subanesthetic dose of ketamine relieves depression-like behaviors induced by neuropathic pain in rats. Anesthesiology. 2011;115:812–21. doi: 10.1097/ALN.0b013e31822f16ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quan MN, Zhang N, Wang YY, Zhang T, Yang Z. Possible antidepressant effects and mechanisms of memantine in behaviors and synaptic plasticity of a depression rat model. Neuroscience. 2011;182:88–97. doi: 10.1016/j.neuroscience.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 16.Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science. 2010;329:959–64. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 18.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A Randomized Trial of an N-methyl-D-aspartate Antagonist in Treatment-Resistant Major Depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 19.Jane DE, Lodge D, Collingridge GL. Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Matute C. Therapeutic Potential of Kainate Receptors. CNS Neurosci Ther. 2010:no–no. doi: 10.1111/j.1755-5949.2010.00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contractor A, Mulle C, Swanson GT. Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011;34:154–63. doi: 10.1016/j.tins.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandes H, Catches J, Petralia R, Copits B, Xu J, Russell T, et al. High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron. 2009;63:818–29. doi: 10.1016/j.neuron.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werner P, Voigt M, Keinanen K, Wisden W, Seeburg PH. Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature. 1991;351:742–4. doi: 10.1038/351742a0. [DOI] [PubMed] [Google Scholar]
- 24.Wisden W, Seeburg PH. A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci. 1993;13:3582–98. doi: 10.1523/JNEUROSCI.13-08-03582.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–47. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinheiro PS, Perrais D, Coussen F, Barhanin J, Bettler B, Mann JR, et al. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2007;104:12181–6. doi: 10.1073/pnas.0608891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitz D, Mellor J, Nicoll RA. Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science. 2001;291:1972–6. doi: 10.1126/science.1057105. [DOI] [PubMed] [Google Scholar]
- 28.Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, McBain CJ. Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci. 2004;24:9658–68. doi: 10.1523/JNEUROSCI.2973-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–16. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 30.Paddock S, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJ, et al. Association of GRIK4 with outcome of antidepressant treatment in the STAR*D cohort. Am J Psychiatry. 2007;164:1181–8. doi: 10.1176/appi.ajp.2007.06111790. [DOI] [PubMed] [Google Scholar]
- 31.Pickard BS, Malloy MP, Christoforou A, Thomson PA, Evans KL, Morris SW, et al. Cytogenetic and genetic evidence supports a role for the kainate-type glutamate receptor gene, GRIK4, in schizophrenia and bipolar disorder. Mol Psychiatry. 2006;11:847–57. doi: 10.1038/sj.mp.4001867. [DOI] [PubMed] [Google Scholar]
- 32.Gejman PV, Sanders AR, Kendler KS. Genetics of Schizophrenia: New Findings and Challenges. Annu Rev Genomics Hum Genet. 2011;12:121–44. doi: 10.1146/annurev-genom-082410-101459. [DOI] [PubMed] [Google Scholar]
- 33.Knight HM, Walker R, James R, Porteous DJ, Muir WJ, Blackwood DHR, et al. GRIK4/KA1 protein expression in human brain and correlation with bipolar disorder risk variant status. Am J Med Genet B Neuropsychiatr Genet. 2011:n/a–n/a. doi: 10.1002/ajmg.b.31248. [DOI] [PubMed] [Google Scholar]
- 34.Pickard BS, Knight HM, Hamilton RS, Soares DC, Walker R, Boyd JK, et al. A common variant in the 3′UTR of the GRIK4 glutamate receptor gene affects transcript abundance and protects against bipolar disorder. Proc Natl Acad Sci U S A. 2008;105:14940–5. doi: 10.1073/pnas.0800643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whalley HC, Pickard BS, McIntosh AM, Zuliani R, Johnstone EC, Blackwood DH, et al. A GRIK4 variant conferring protection against bipolar disorder modulates hippocampal function. Mol Psychiatry. 2009;14:467–8. doi: 10.1038/mp.2009.7. [DOI] [PubMed] [Google Scholar]
- 36.Njung’e K, Handley SL. Evaluation of marble-burying behavior as a model of anxiety. Pharmacol Biochem Behav. 1991;38:63–7. doi: 10.1016/0091-3057(91)90590-x. [DOI] [PubMed] [Google Scholar]
- 37.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Archives Internationales de Pharmacodynamie et de Therapie. 1977;229:327–36. [PubMed] [Google Scholar]
- 38.Breustedt J, Schmitz D. Assessing the role of GLUK5 and GLUK6 at hippocampal mossy fiber synapses. J Neurosci. 2004;24:10093–8. doi: 10.1523/JNEUROSCI.3078-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitz D, Mellor J, Breustedt J, Nicoll RA. Presynaptic kainate receptors impart an associative property to hippocampal mossy fiber long-term potentiation. Nat Neurosci. 2003;6:1058. doi: 10.1038/nn1116. [DOI] [PubMed] [Google Scholar]
- 40.Contractor A, Sailer AW, Darstein M, Maron C, Xu J, Swanson GT, et al. Loss of kainate receptor-mediated heterosynaptic facilitation of mossy-fiber synapses in KA2−/− mice. J Neurosci. 2003;23:422–9. doi: 10.1523/JNEUROSCI.23-02-00422.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitz D, Frerking M, Nicoll RA. Synaptic activation of presynaptic kainate receptors on hippocampal mossy fiber synapses. Neuron. 2000;27:327–38. doi: 10.1016/s0896-6273(00)00040-4. [DOI] [PubMed] [Google Scholar]
- 42.Sokolov BP. Expression of NMDAR1, GluR1, GluR7, and KA1 Glutamate Receptor mRNAs Is Decreased in Frontal Cortex of “Neuroleptic-Free” Schizophrenics: Evidence on Reversible Up-Regulation by Typical Neuroleptics. J Neurochem. 1998;71:2454–64. doi: 10.1046/j.1471-4159.1998.71062454.x. [DOI] [PubMed] [Google Scholar]
- 43.Porter RH, Eastwood SL, Harrison PJ. Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 1997;751:217–31. doi: 10.1016/s0006-8993(96)01404-7. [DOI] [PubMed] [Google Scholar]
- 44.Scarr E, Beneyto M, Meador-Woodruff JH, Dean B. Cortical Glutamatergic Markers in Schizophrenia. Neuropsychopharmacology. 2005;30:1521–31. doi: 10.1038/sj.npp.1300758. [DOI] [PubMed] [Google Scholar]
- 45.Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163:153–5. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Liu R-J, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol Psychiatry. 2011;69:754–61. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098) Neuropharmacology. 2001;40:1028–33. doi: 10.1016/s0028-3908(00)00194-5. [DOI] [PubMed] [Google Scholar]
- 48.Shaltiel G, Maeng S, Malkesman O, Pearson B, Schloesser RJ, Tragon T, et al. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry. 2008;13:858–72. doi: 10.1038/mp.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, et al. Genomewide Linkage Analyses of Bipolar Disorder: A New Sample of 250 Pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet. 2003;73:107–14. doi: 10.1086/376562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulze TG, Buervenich S, Badner JA, Steele CJM, Detera-Wadleigh SD, Dick D, et al. Loci on chromosomes 6q and 6p interact to increase susceptibility to bipolar affective disorder in the national institute of mental health genetics initiative pedigrees. Biol Psychiatry. 2004;56:18–23. doi: 10.1016/j.biopsych.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Shibata H, Joo A, Fujii Y, Tani A, Makino C, Hirata N, et al. Association study of polymorphisms in the GluR5 kainate receptor gene (GRIK1) with schizophrenia. Psychiatr Genet. 2001;11:139–44. doi: 10.1097/00041444-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 52.Wu LJ, Ko SW, Toyoda H, Zhao MG, Xu H, Vadakkan KI, et al. Increased anxiety-like behavior and enhanced synaptic efficacy in the amygdala of GluR5 knockout mice. PLoS ONE. 2007;2:e167. doi: 10.1371/journal.pone.0000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic Antidepressant Treatment Increases Neurogenesis in Adult Rat Hippocampus. J Neurosci. 2000;20:9104–10. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kodama M, Fujioka T, Duman RS. Chronic olanzapine or fluoxetine administration increases cell proliferation in hippocampus and prefrontal cortex of adult rat. Biol Psychiatry. 2004;56:570–80. doi: 10.1016/j.biopsych.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Pham K, Nacher J, Hof PR, McEwen BS. Repeated restraint stress suppresses neurogenesis and induces biphasic PSA-NCAM expression in the adult rat dentate gyrus. Eur J Neurosci. 2003;17:879–86. doi: 10.1046/j.1460-9568.2003.02513.x. [DOI] [PubMed] [Google Scholar]
- 56.Hanson ND, Nemeroff CB, Owens MJ. Lithium, but Not Fluoxetine or the Corticotropin-Releasing Factor Receptor 1 Receptor Antagonist R121919, Increases Cell Proliferation in the Adult Dentate Gyrus. J Pharmacol Exp Ther. 2011;337:180–6. doi: 10.1124/jpet.110.175372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT, et al. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001;32:697–709. doi: 10.1016/s0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- 58.Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl D-aspartate antagonists. Neurosci Lett. 1986;70:132–7. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- 59.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–24. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 60.Kobayashi K, Ikeda Y, Sakai A, Yamasaki N, Haneda E, Miyakawa T, et al. Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc Natl Acad Sci U S A. 2010;107:8434–9. doi: 10.1073/pnas.0912690107. [DOI] [PMC free article] [PubMed] [Google Scholar]