

Abstract
The protozoan parasite Trypanosoma brucei switches its variant surface glycoprotein (VSG) to subvert its mammalian hosts’ immune responses. The T. brucei genome contains as many as 1600 VSG genes (VSGs), but most are silent noncoding pseudogenes. Only one functional VSG, located in a telomere-linked expression site, is transcribed at a time. Silent VSGs are copied into a VSG expression site through gene conversion. Truncated gene conversion events can generate new mosaic VSGs with segments of sequence identity to other VSGs. To examine the VSG family sub-structure within which these events occur, we combined the available VSG sequences and annotations with scripted BLAST searches to map the relationships among VSGs in the T. brucei genome. Clusters of related VSGs were visualized in 2- and 3-dimensions for different N- and C-terminal regions. Five types of N-termini (N1 – N5) were observed, within which gene recombinational events are likely to occur, often with fully-coding ‘functional’ or ‘atypical’ VSGs centrally located between more dissimilar VSGs. Members of types N1, N3 and N4 are most closely related in the middle of the N-terminal region, whereas type N2 members are more similar near the N-terminus. Some preference occurs in pairing between specific N- and C- terminal types. Statistical analyses indicated no overall tendency for more related VSGs to be located closer in the genome than less related VSGs, although exceptions were noted. Many potential mosaic gene formation events within each N-terminal type were identified, contrasted by only one possible mosaic gene formation between N-terminal types (N1 and N2). These data suggest that mosaic gene formation is a major contributor to the overall VSG diversity, even though gene recombinational events between members of different N-terminal types occur only rarely.
Keywords: African trypanosome, variant surface glycoprotein, antigenic variation, mosaic gene, sequence alignment
1. Introduction
Trypanosoma brucei cycles between tsetse flies and the mammalian bloodstream, remaining extracellular throughout its life cycle. Bloodstream trypanosomes are covered with about 107 copies of a single glycosylphosphatidylinositol (GPI)-anchored protein called the variant surface glycoprotein (VSG), which constitutes about 5% of the total protein of the organism – a high percentage for a surface protein. Individual trypanosomes switch from one VSG to another at a rate of 10−2 to 10−7 switches per generation time of 5 – 10 hours [1] in a process called antigenic variation. The only known function of this VSG monolayer is to protect other invariant constituents of the membrane from immune attack [2]. Mature VSGs are 400 – 450 amino acids in length with the GPI anchor linked to their C-terminus. Immunologically distinct VSGs typically have less than 25% amino acid identity [3]. The three-dimensional structures of two VSG N-terminal regions (the first 80% of the protein) have been determined by X-ray crystallography [4]. Despite little primary sequence similarity, they share a nearly identical dumbbell-like shape that permits the VSGs to pack tightly on the parasite’s surface. Two large α-helices form a coiled coil that acts as a backbone for other smaller helices and loops of the structure. The solution structure of a VSG C-terminal region (the last 20%) has been determined by NMR spectroscopy and molecular modeling indicates that all of the semi-conserved VSG C-terminal domains share highly similar tertiary structures [5]. Thus, all VSGs are thought to adopt very similar tertiary structures mediated by similar α-helical backbones and disulfide bridges. This monolayer of closely packed VSGs confronts the immune system with a highly repetitive (107) set of epitopes on the surface of each trypanosome.
VSG genes (VSGs) occur on all three chromosomal size classes of T. brucei, i.e., the mega-base, intermediate-sized and mini-chromosomes [6]. The eleven mega-base chromosomes, which have been sequenced [7], possess nearly 1000 VSGs and the entire archive of VSGs, including those on the yet-to-be-sequenced intermediate and mini-chromosomes, has been estimated at 1600 [8], or about 20% of the protein-coding capacity of the genome. Only about 7% of the nearly 1000 sequenced VSGs actually encode ‘functional’ VSGs, i.e., VSGs with conserved cysteines and semi-conserved C-terminal domains. Most of the remaining (66%) are pseudogenes (with frameshifts or in-frame stop codons), some (9%) are atypical genes (that do not encode all of the expected cysteines and/or C-terminal similarities) and the rest (18%) are gene fragments, most of which encode C-terminal domains. These VSGs occur in transcriptionally silent arrays of 3 to 250 members located in subtelomeric regions that are 100 kb or more from the telomere repeats. Functional VSGs are scattered amongst the pseudo-VSGs in the arrays in no discernible pat tern. The expressed or active VSG is not located within the arrays, but is always immediately adjacent to a telomere of one of the mega-base or intermediate chromosomes. The most common VSG switching mechanism is a gene conversion event in which a duplicated copy of a VSG in a silent array replaces the active VSG in a telomere-linked expression site (reviewed in [9–11]).
Since VSGs may enter a telomeric expression site via homologous recombination, there is potential for the formation of new VSGs that are mosaics of existing VSGs. This potential for a fragmented exchange of sequence information within the VSG repertoire led us to use BLAST, which is suitable for identifying fragments of similarity, to generate graphs of relations among the T. brucei VSGs. Previous comparisons of VSG primary sequences have categorized their N-terminal domains into three types, A, B and C, and their C-terminal domains into six types, 1 – 6, on the basis of cysteine positions and sequence similarities [12]. Different N-terminal domains can associate with different C-terminal domains [3]. An online database (www.vsgdb.net) of VSGs located on the eleven mega-base chromosomes is available [12]. We used this database and alternative methodologies to re-examine the N- and C-terminal domain categories and to search for the presence of potential mosaic VSGs created via multiple cross-over events within VSG coding regions. These methods were used to assign clusters of VSGs to categories and sub-divide categories of more closely related VSGs. We find that the original N-terminal type A domain can be sub-divided into three additional groupings for a total of five N-terminal domain categories and that mosaic VSG formation between VSGs of the same N-terminal type is likely a regular occurrence,
2. Materials and methods
2.1 VSG sequence database
Sequences and annotations were obtained from VSGdb, an online resource of annotated VSG sequences (www.vsgdb.net) created by Marcello et al [12]. VSGdb provides separate information on the VSG N- and C-terminal domains, as well as subtypes, and on predicted functional annotations (i.e., functional, atypical, or pseudogene VSG). Chromosomal locations for the VSGs were derived from the annotated genome of T. brucei stock TREU 927 [7] maintained by The Institute for Genomic Research (TIGR) (ftp://ftp.tigr.org/pub/data/Eukaryotic_Projects/t_brucei/). All VSGs analyzed were from this genome unless otherwise indicated. A local database was created to provide the combined VSG information for subsequent steps of the analysis. Sequences and annotations in FASTA format were processed by a pipeline of scripts to visualize networks of relationships among VSGs. Additional sequences, such as additional T. brucei and T. congolense expressed (cDNA) VSG sequences, could be conveniently appended to the FASTA files for comparison in the analysis. The software used to generate clustered plots of nucleotide and amino acid sequences, including a web-based interface for more user-friendly access (see Section 2.2), is available at http://code.google.com/p/visblast/. An overview of the analytical approaches for analyzing the VSG sequences is summarized in Fig. 1.
Fig. 1.
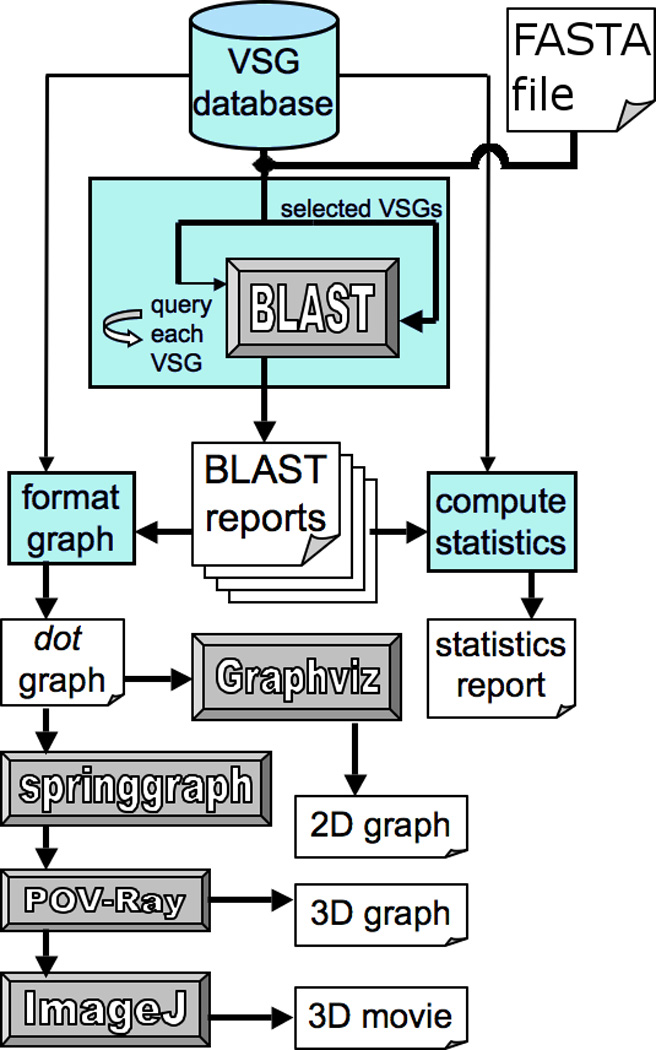
Flow chart of the analyses. From the top, the VSG sequences and annotations were selected for analysis from a locally generated database comprised of sequences and annotations derived from the online T. brucei VSGdb database (www.vsgdb.net) [12], and the sequenced T. brucei genome [7]. More generally, the analysis approach can be applied to any group of sequences input in FASTA format. Blue modules represent scripts written to organize and direct data through the analysis, including one to execute BLAST searches between all selected VSGs. BLAST reports generated from the searches were used to compute statistics about the relations between VSGs, or to generate graphs to visualize the network of similarity among VSGs. Network graphs were clustered and processed into images using software represented by the gray modules.
2.2. A web-based interface to simplify the visualization of clusters of similar sequences
A web-based interface was created as part of the previously mentioned software to visualize graphical output of reciprocal BLAST searches. This interface minimally accepts FASTA files and a BLAST ‘E-value’ (the probability cutoff for such a match occurring in a random set of sequences of the same size) as inputs. Individual sequences are required to have a unique identification (ID) as the first term in the FASTA header, and optionally, a category label may be used as a second term to color-code the graphical clustering output. The web-based interface returns 2D graphical results of clusters of gene or protein similarities, and provides a layer of abstraction from the Perl-script pipeline used to generate the graphical output. To discover and accordingly color-code clusters of similar VSGs, the Markov clustering algorithm (MCL) [13] is used to perform unsupervised clustering. When MCL is used to partition clusters, the interface will also output individual FASTA files for each cluster, CLUSTALW alignments of each cluster, and profile hidden Markov models (profile-HMMs) [14, 15] generated from the alignments. This software can be generally applied to simply generate and visualize clusters from any set of nucleotide or protein sequences.
2.3. Construction of a network graph of VSG similarities
Reciprocal BLAST searches [16] initiated from Perl scripts using the bioperl interface [17] were conducted among the approximately 800 annotated VSG sequences, in FASTA format, selected from the local database. Graphs could be constructed from N- or C-terminal nucleotide or amino acid sequences using the local database. The similarity of relationships indicated by BLAST hits meeting a given threshold E-value were used to construct an un-weighted non-directional graph of related VSGs. An E-value threshold was used to limit the graph to a set of more closely related VSGs. At high (less stringent) E-values the similarities between more distantly related genes produce a more dense graph of relationships at the risk of more false positives being permitted. When the E-value threshold was lowered (more stringent), only highly similar VSGs remained connected, and sub-graphs became disjointed, allowing the resolution of smaller clusters of more highly related VSGs. The BLAST reports were compiled into a flat file to store the names of every VSG pair matched, which part of the sequence matched, and the E-value of each match.
2.4. Identification of N-terminal and C-terminal VSG sequence types
BLAST searches were performed at either the nucleotide or amino acid level with gap size parameters set to 30 nucleotides or 10 amino acids. The VSG sequences represented in the BLAST searches were labeled with sequence annotations such as chromosomal locations, encoded N-terminal or C-terminal sequences or types, and potential functional significance (i.e., either a functional, atypical or pseudo-VSG). The annotated BLAST report comprises a graph of similarities between VSG sequences. Nodes (or vertices) on these graphs are the VSGs, and BLAST hits make up the edges (or lines) on the graph between nodes. E-value thresholds for BLAST were varied to resolve the graph at multiple scales or densities of BLAST hits. These graphs were used to identify clusters of similar VSGs and produce reports on the statistical relationships between the described types of VSG N- and C-termini and their chromosomal locations. The same graphs were also used for visualizing clusters of similar VSGs.
To validate the visualized 5 N-terminal type clustering scheme (see Section 2.9 for methods), the MCL algorithm was applied to the BLAST results of the N-terminal protein sequences. MCL is an unsupervised clustering algorithm capable of partitioning the domains into sub-families in an unsupervised manner [13]. MCL clustering of BLAST results has been applied to trypanosome, and other eukaryotic, genomes for the purpose of identifying groups of orthologs in the orthoMCL database [18]. The MCL inflation parameter (i-value) affects the granularity, or number, of clusters generated from each graph, so i-values were chosen that would maximize the modularity (Q) of the clustering scheme for each E-value threshold tested. Modularity is the fraction of edges within clusters minus the fraction that would be expected if the edges were distributed at random. This maximization was done to highlight only the most well segregated clusters of VSGs at each E-value. E-value thresholds were tested on a log scale from 1 to 1e-10, with i-values ranging from 1.1 to 19. For each E-value threshold, an i-value that maximized modularity for clusters of more than 8 VSGs members was selected. Maximization resulted in nominal i-values ranging from 1.2 to 2 (Supplemental Table T1). These optimized clusters were subjected to further graph analyses for evidence of inter-cluster mosaic VSG formation and measurements of VSG centrality (see section 2.5) within each cluster.
2.5. “Betweenness centrality” analysis
Betweenness is the number of times any particular node (i.e., VSG) lies on the shortest path between any other two nodes in the cluster [19]. By this measurement, VSGs with a high betweenness will be the most similar VSGs between less related sub-families. Tendencies for atypical and functional type VSGs to have high betweenness centrality were determined to assess their possible importance to the diverse community structure. The average betweenness within individual (MCL identified) clusters of N-terminal VSG amino acid sequences was measured at E-values ranging from 0.1 to 1e-10 clustered by MCL. These clusters are the same as those visualized as colored groups within each scale of E-value (see Fig. 5).
Fig. 5.
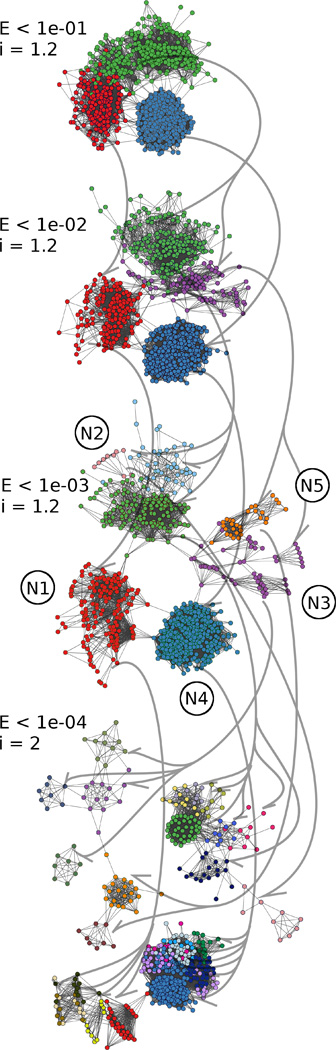
Unsupervised clustering using the MCL algorithm also partitions VSGs into groups of similar sequences at various E-value thresholds. Nodes represent the N-termini of VSG proteins, and connecting edges represent BLAST hits between them at stringencies determined by the E-value listed to the left. As E-value thresholds becomes more stringent, fewer BLAST hits are included, the graphs become sparser and sub-sets of VSGs are separable by MCL. Colors indicate clusters partitioned by the unsupervised MCL algorithm. The coloring scheme for the E < 1e-03 plot is the same as Fig. 2B. Arrowed lines drawn between different scales of E-value indicate how a cluster of VSGs was sub-divided in the next scale. Only clusters with at least 8 members were plotted, so there are fewer nodes plotted as some VSGs no longer shared that level of similarity. E < 1e-03 is the last E-value threshold before large sub-graphs become disconnected.
Statistical significance of the observed average betweenness of functional and atypical genes within the undirected graphs of each of the describe clusters was calculated by comparison with two randomized datasets. One data set (random) was a random graph with the same number of edges and nodes, whereas the other (permutation) was a random selection of VSGs from the same graph in which the atypical or functional VSGs were removed. Additionally, a random set of VSGs of equal size to the set of atypical or functional VSGs was removed prior to the calculation of the observed betweenness in each permutation to maintain an equal number of nodes in the observed and permuted betweenness calculations of each iteration. Both types of analysis were conducted 1000 times to compute p-values (Supplemental Table T2).
2.6. Statistical analysis of distance associations and N- and C-terminal associations
Possible enrichment within a chromosomal region of the same N-terminal type VSGs was calculated by averaging distances between all pairs of VSGs of the same type on the same contiguous region (contig). This average was then compared to a recalculated average after shuffling all VSG type labels. The averages from these permutations were recalculated 1e05 times. For each contig, the number of simulations in which the average distance between same-type VSGs was less than or equal to the observed distance was divided by the number of permutations (1e05) to find the p-value indicating an aggregation of types.
Biases in the associations of N- and C-terminal types were determined by simulating pools of VSGs based on randomly matching the N- and C-termini, then counting the number of associations that exceeded the observations. Randomized sets (1e6) of VSGs were measured empirically to determine p-values of associations.
2.7. Heatmaps
The number of blast hits on a portion of the primary sequence was counted within each N-terminal type, and image files were generated using the matrix2png tool [20]. These BLAST hit overlaps reveal the most common location of BLAST hits occurring within particular N-terminal types. This analysis was used for observing differences in N-terminal types, and determining which portion of the sequence made the greatest contribution to the family clustering. The observation that BLAST hits tended to overlap with cysteine-rich regions of the VSGs was tested for statistical significance by comparing the observed fraction of cysteine residues within BLAST hits in each N-terminal type to 1000 sets of randomly shuffled VSG amino-acid sequences. Only amino acid sequences were shuffled with each iteration; the graph and the start and stop positions of each BLAST hit were held constant.
2.8. Alignments and profile hidden Markov models (profile-HMMs)
The primary amino-acid sequences were aligned for each N-terminal type to identify regions of similarity and conserved cysteine residues using ClustalW [21], and formatted for viewing using the ESPript tool [22]. Secondary structures representative of each N-terminal type were calculated using the psipred [23] secondary structure prediction server [24]. Profile-HMMs representative of the alignments were generated by HMMER software [14, 15]. These models were useful in rapidly identifying VSGs and recapitulating the type definitions determined in this study. Automated generation of profile-HMMs from clustered sequences is a feature of the software developed for this analysis.
The classification accuracy of profile-HMM models created from the N-terminal clustering scheme describe in this report was tested using a leave-one-out approach. Profile-HMMs from each cluster minus one VSG, were built and then tested for classification accuracy as applied to the left-out VSG. This approach was iteratively performed for all VSGs clustered.
2.9. 2D and 3D visualizations of clustered graphs
The 2D and 3D visualizations were generated through a pipeline of Perl scripts. To visualize clusters in either 2D or 3D, all the sequence similarities between VSGs, after being computed as BLAST reports, were compiled into dot format graphs and color-coded according to the annotation information. Each VSG is represented by a single node on the graph. An edge (line) between two nodes indicates a BLAST hit between those two VSGs. In both the 2D and 3D graphs, clustering (node dispersal and edge length) is determined from an un-weighted graph constructed from the presence of BLAST hits (graph edges) between VSGs (graph nodes). The order of edges written into the dot graph was randomized to ensure that the clusters generated were not dependent on the order of inputs. To render the 2D graphs, the dot files were processed by neato, a component of Graphviz (http://www.graphviz.org; http:www.research.att.com/sw/tools/graphviz).
The 3D cluster visualizations were generated by clustering dot files with springgraph (www.chaosreigns.com/code/springgraph/), and rendering the springgraph output with MegaPOV (megapov.inetart.net/), an implementation of POV-ray (www.povray.org), a ray-tracing program. POV-ray can also be used to generate a series of images to be spliced into a 3D movie by ImageJ (rsb.info.nih.gov/ij/). These 3D visualizations reveal substructures not easily visualized in the 2D graphs.
2.10. Detection of mosaic genes
BLAST hits between functional VSGs and other VSGs were used to illustrate regions of possible mosaic gene formation. Multiple sequence alignments from ClustalW were used to examine these BLAST hits and alignment data were used to create illustrations of where sequences in the related genes matched, or did not match. The purpose was to identify short regions at which potential cross-over events between VSGs occurred.
3. Results
3.1 Similarities based on BLAST hits reveal five distinct N-terminal domain types
The N-termini of VSGs (the first 80% of the amino acids; ~330 residues) are much more variable than the semi-conserved C-termini (the last 20%) [7]. Therefore, similar to earlier studies [3, 8, 25], we analyzed these two domains separately. Three general groupings, A, B and C, of T. brucei VSG N-termini, which are based primarily on cysteine positioning and sequence similarity, have been described previously [3]. When the 767 available VSG N-termini whose corresponding chromosomal gene locations are known were visualized in a 2D clustering based on amino acid identity at an E-value < 1e-03 stringency of BLAST hit, five well-clustered groups of N-termini were observed (Fig. 2A). E < 1e-03 was found to be the most stringent E-value that can be used in which the most BLAST hits in the graph remain connected. These five types, called N1 – N5 (Fig. 2B), agree with, and expand on, the previously described three types, A, B and C, and provide a detailed look at sub-clusters of VSG N-termini.
Fig. 2.
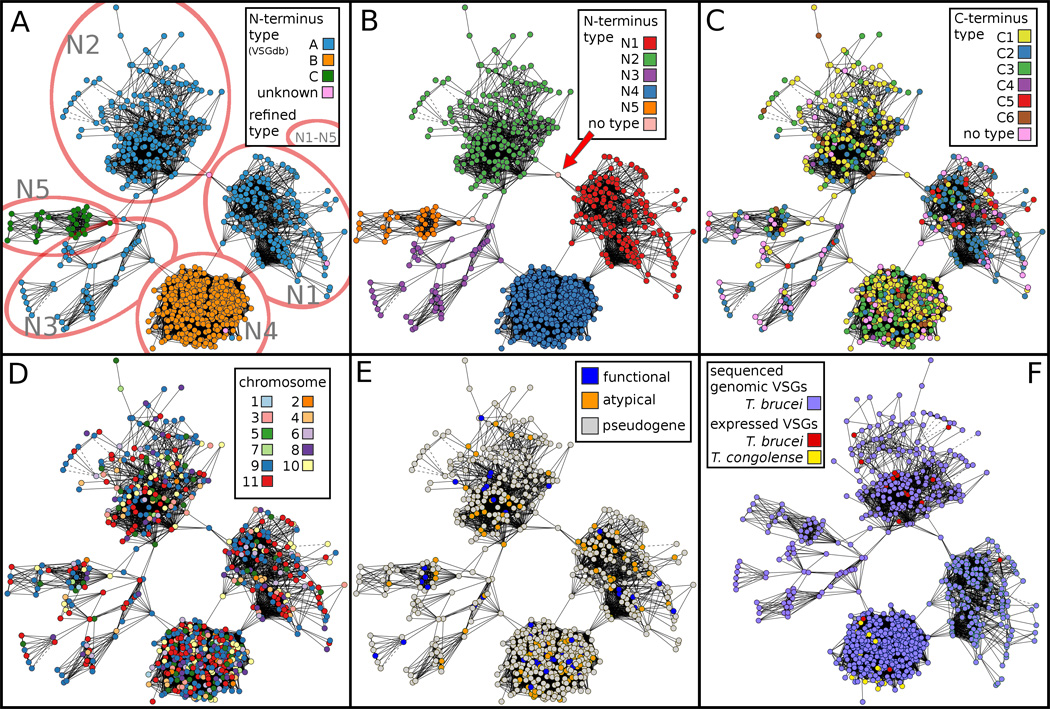
Clustering of N-terminal VSG sequences depicted in 2 dimensions. Panels A–E contain 767 VSGs for which all annotation information is available, including N- and C-terminal sequences, chromosomal gene location and predicted functional annotations (functional, atypical, or pseudogene) [7, 8]. Panel F contains 17 additional expressed (cDNA) N-terminal VSG sequences from other T. brucei isolates [25–28], and 17 expressed (cDNA) N-terminal VSG sequences from T. congolense [29–31], for a total of 801 sequences. All panels were clustered according to similarity between the N-terminal VSG amino acid sequences determined by a BLAST hit of E < 1e-03. Solid lines represent a match from one continuous BLAST hit. Dotted lines represent multiple non-contiguous BLAST hits between two VSGs. (A) VSG N-termini are color-coded according to previously assigned three types A, B and C [25]. The red ovals indicate additional divisions into five distinct types (N1 – N5), three of which (N1 – N3) are subsets of the previous group A [25]. (B) VSGs are color-coded according to these five newly defined N-terminal types. The arrow indicates a VSG discussed in the text that shares similarity with both N1 and N2 types. (C) VSGs are clustered according to their N-terminal type and color-coded according to the C-terminal type (see Fig. 3), illustrating a non-random distribution of some C-terminal types with N-terminal types (see also Table 1). (D) Color coding the clustered N-termini by chromosome does not reveal a tendency for highly similar VSGs to be located near each other within the genome. (E) Color coding according to the VSG annotations as either functional, atypical or pseudogene [7, 8] indicates that functional and atypical VSGs exist within all five N-terminal types. (F) The sampling of additional expressed (cDNA) VSG sequences from T. brucei and the related species T. congolense show that the T. brucei expressed (cDNA) sequences have similarity to types N1, N2, and N4, whereas the available T. congolense expressed (cDNA) sequences only show similarity with type N4.
N-terminal type N4 (equivalent to type B in [25]) is the most clearly resolved and tightly clustered of the five types. Three distinct clusters of VSGs among those previously labeled type A were observed and designated as types N1, N2 and N3. These data also are consistent with the observation [8] that N5 (equivalent to type C) and N3 (part of the original type A) are not as distinctly separated. Nevertheless, profile-HMMs from N3 and N5 can be applied to accurately distinguish between N3 and N5 types (see Section 3.2). Types N1 and N2 display very little similarity with each other based on BLAST analysis (Fig. 2A and B). Only one VSG (Tb09.244.0490) shares any similarity at this E < 1e-03 threshold with both types (red arrow, Fig. 2B). This VSG is actually a truncated pseudogene encoding about 70 amino acids at its extreme N-terminus that are shared with type N1, followed by approximately 70 amino acids highly similar to type N2. Therefore, this particular VSG was left as ‘no type’. Since there is only one nonfunctional potential gene conversion event between these two large clusters of VSGs, and none involving the other large cluster, N4, we speculate that mosaic VSGs formed between these families may be nonfunctional, and that most mosaic combinations between genes encoding different N-terminal types, if they even occur, result in a non-viable VSG. No apparent overall clustering of similar N-terminal coding sequences on specific chromosomes was observed (Fig. 2D); however, a number of examples of similar VSGs of the same N-terminal type occurring on the same chromosome or in proximity on the same contiguous sequence (contig) were observed in the statistical analysis (see below). In addition, VSGs predicted to be nonfunctional pseudogenes or atypical are represented in each of the five types of N-terminal domains (Fig. 2E).
Substructures of related VSG N-terminal types can also be observed within an animation traversing the 3D rendering of the clustered BLAST analysis graph, as illustrated in the video shown in Supplemental Fig. S1. Using an even less stringent E-value of E < 1e-02, the separation between five N-terminal types is clearly observed. The video also illustrates additional features, such as the possibility of subgroups within the N1 cluster.
Residues making the greatest contribution to the intra-family clustering are shown in Fig. 3, which is a heatmap of (blastp) hits overlaid with secondary structure predictions and cysteine residue conservation. This analysis shows that in types N1, N3 and N4 the central portion of the N-terminal domain contributes the greatest number of blast hits to the clustering, whereas sequences proximal to the N-terminus contributes the least. In contrast, type N2 differs in that sequences proximal to the N-terminus of the N-terminal domain most frequently share similarity, whereas sequences proximal to the C-terminus share the least. Type N5 appears to be intermediate between these two extremes of N1, N3 and N4 versus N2, but it also contains the fewest VSGs (32 members), which may compromise its analysis.
Fig. 3.

Locations of BLAST hits within the N-termini, with representative primary and secondary sequence features. VSG families are represented by 161 type N1, 105 type N2, 47 type N3, 349 type N4 and 32 type N5 VSGs. Overall, these N-terminal sequences have an average length of 330 amino acids. An illustration of the predicted secondary structures within the family is shown for each type. Dark blue winding structures indicate a predicted alpha helix. Rectangles indicate a predicted beta strand structure. A ‘black-to-green’ heatmap is also shown for each of the five N-terminal types to display the approximate location of BLAST hits within that type. The lighter green areas depict more BLAST hits, indicating the region of the N-terminal domain making the greatest contribution to the intra-family cluster, and the black areas indicate fewer BLAST hits. Underlying each family is a multiple alignment of the primary amino acid sequence. The cysteine residues that are most conserved have been highlighted in orange and indicated in bold print at the bottom of each sequence alignment for each type.
BLAST hits within N-terminal types show a significant tendency to overlap with cysteine residues (p < 0.05) for N1 – N4, but not N5 (see Section 2.7 for the methods). These results imply those portions of the N-terminal domain may be under different structural constraints, hindering diversifying selection. Comparing the predicted secondary structures between different N-terminal types, types N1 – N3 and N5 were observed to share conserved long predicted alpha helices at the N-terminal end, whereas the corresponding region of type N4 has shorter discontinuous segments of predicted helical structure. It is worth noting, however, that secondary structure predictions are sometimes inaccurate.
3.2 Profile-HMMs accurately classify N-terminal types
The classification accuracy of profile-HMMs generated from the clusters used in this study was tested using an iterative leave-one-out approach. The five-type clustering scheme described in Fig. 2 could accurately classify 773 of 778 N-termini tested (99.4% accuracy). The MCL’s unsupervised clustering at the same scale (E < 1e-03) had the best accuracy of any scale of E-value tested, with correct classification of 756 of 763 VSGs (99.1% accuracy). These results support the conclusion that the selection of E-value threshold and the subsequent clustering scheme of N-termini are distinct and distinguishable groups of sequences. These profile-HMMs can be automatically generated from the web-interface and were useful in identifying the VSG types that T. congolense shared with T. brucei, and support the distinctiveness of the types described (see Section 3.8).
3.3 BLAST hit similarities do not readily separate C-terminal domain types
Within the semi-conserved C-termini, six distinct types, C1 – C6, have been described previously based on conservation of cysteine positions and the ~ 20-amino acid hydrophobic tail that is replaced with a GPI anchor [3, 8, 25]. Thus, we also generated a clustered graph of the relationships among VSG C-terminal domains, excluding the ~20 amino acids replaced by the GPI anchor (Fig. 4). This graph is based on similarities between nucleotide sequences identified by BLAST with an expected (E) value less than 1e-10. When amino acid sequences are used for the clustering, the graph is too dense to resolve any C-terminal types. The clustered VSGs were color-coded by the six C-terminal type definitions in both 3 dimensions (3D; Figs. 4A, B) and 2 dimensions (2D; Figs. 4C, D). Type C2 can be visualized as a cluster with many more intra-cluster edges than inter-cluster edges in both formats, as highlighted by red-lined squares. Types C4 and C6 are not readily discernable (Figs. 4A, C). Viewing a rendering of the clustering in a 3D animation provides better visual resolution of the C2 type (Supplemental Fig. S2); however, the high degree of similarity among most of the C-terminal types and thus the dense graph of BLAST hits among types precludes resolution of all but the most different types. Thus, our methodology and our exclusion of the highly conserved C-terminal hydrophobic tail from the analysis did not result in as clear a resolution of six specific C-terminal regions as previously described; nevertheless, our findings do not disagree with this earlier categorization of the VSG C-terminal domains [3, 8].
Fig. 4.
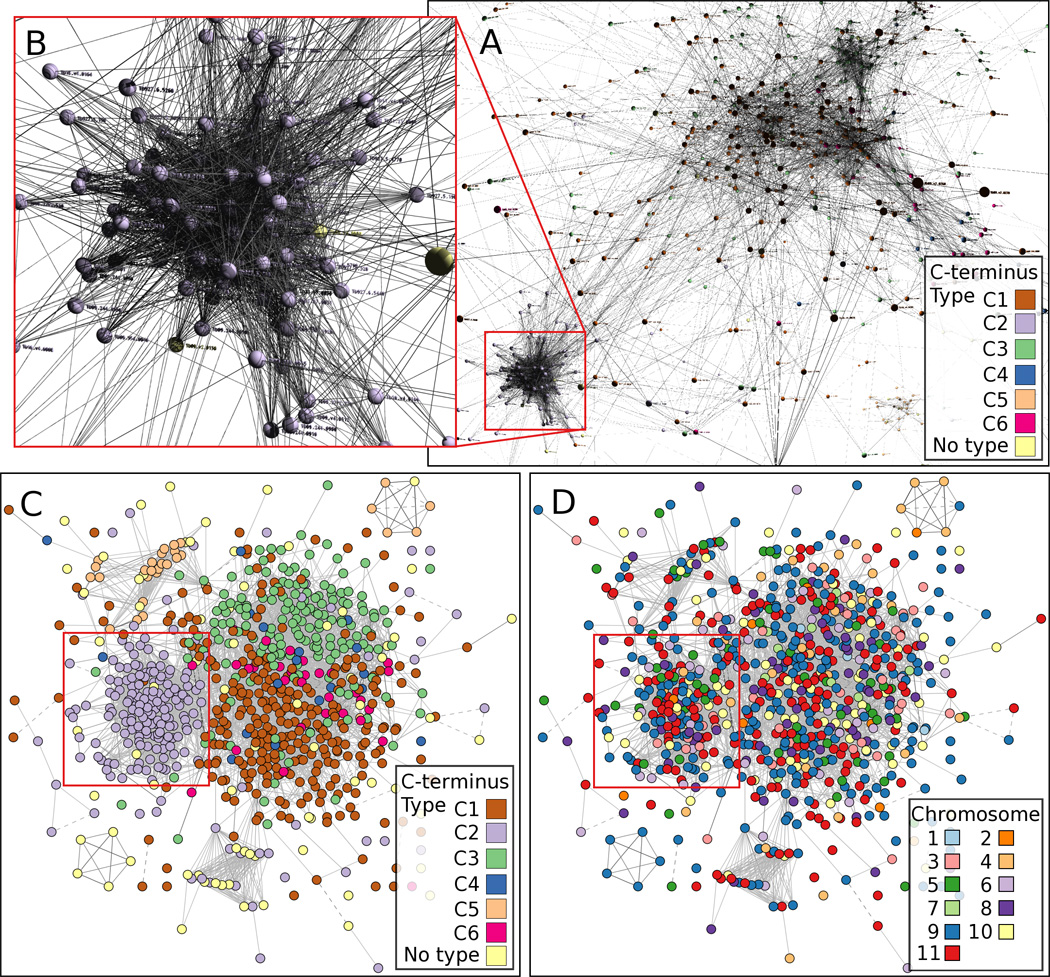
Clustering of C-terminal VSG sequences. Similar sequences among the C-terminal regions of 799 VSGs identified by BLAST between protein sequences with E-values less than 1e-10 are visualized as clusters in 3 dimensions (A,B) and 2 dimensions (C, D). The six reported VSG C-terminal domains (C1 – C6) are represented by nodes color-coded according to the C-terminal type (A, B, C), and lines between nodes represent BLAST hits. Clustering among C-terminal types is especially apparent in the case of type C2, which is outlined by red lines in panels A, C and D, and expanded in B. Similar clustering is not observed when C-termini are color-coded according to the chromosomes on which their genes are located (D). The color coding is difficult to see in panels A and B.
We also color-coded each VSG C-terminus according to its corresponding gene’s chromosome (Fig. 4D) and did not generally observe an overall association between C-terminal type and chromosome distribution. An exception to this can be seen in the bottom left corner of panels 5C and 5D where a cluster of 5 related C-termini are tandemly repeated on chromosome 9. However, these do not have a defined C-terminal type and they do not share significant similarity with other types. A similar cluster of 6 C-termini in the upper right can be observed, which also does not share similarity at this scale of E-value with any other sequences. However, 5 of the 6 are of type C5, and only 4 are tandemly repeated (on chromosome 4).
3.4 Unsupervised MCL clustering agrees with previous clustering and can further sub divide VSG types
The relationships observed between VSG N-terminal sequences at E < 1e-03 were useful in classifying the types and observing differences among them (Fig. 2). As expected, when the stringency of the E-value was increased, graphs became sparser since less similar BLAST hits were excluded. As an alternative clustering approach, unsupervised clusterings with MCL at various thresholds of E-value were conducted (see Section 2.4 for methods), revealing further N-terminal sub-categories and highlighting smaller groups of highly similar VSGs that may indicate a recent expansion or selective process. Fig. 5 shows representative graphs generated by MCL with increasing stringency from E < 1e-01 to 1e-04. At the high E < 1e-01 value, only type N4 is well resolved from the other types. At E < 1e-02, types N1,3, and4 are resolved whereas N3 is not distinguishable from N5, and at E < 1e-03, five clusters are apparent that closely resemble the N1-5 clusters shown in Fig. 2. At a still lower E-value (E < 1.e-04), the five clusters decompose into much smaller sub-divisions. For example, N5 partitioned into two additional sub-families and N2 into multiple sub-families.
3.5 Atypical and functional VSGs display high betweeness centrality
VSGs with a high betweenness centrality are positioned more centrally between less-connected sub-families of VSGs (see Section 2.5). VSGs with high betweenness centrality scores are positioned like ‘bridges’ in sequence similarity between multiple, more divergent, sub-groups. As shown in Supplemental Fig. S3 and Supplemental Table T2, both functional and atypical VSGs have, on average, significantly higher betweenness values than other VSGs in multiple clusters when identified at multiple scales of E-values and compared to either random graphs or random permutations of the same graphs (p < 0.05). These tendencies for atypical and functional VSGs to have high average betweenness in MCL generated clusters were tested at E-value thresholds ranging from 0.1 to 1e-10 (Supplemental Table T1). For the functional VSGs, 42/47 (89%) were determined to be part of a cluster at one of the tested E-value in which the average betweenness was significantly high (p < 0.05). In the case of atypical VSGs, 34/88 (39%) were part of clusters that had significantly high betweenness (Supplemental Table T2). Since these observations were made across multiple scales of E-value, some caution should be taken in the consideration of the p-values; however, no sub-graph studied at any scale of E-value was found to have atypical or functional VSGs with a significantly low average betweenness compared to those measured in random graphs or permuted graphs (p < 0.05). Furthermore, VSGs with a membership in a high atypical or functional betweenness cluster were observed within all five N-terminal types. These results show that both atypical and functional VSGs are positioned as ‘bridges’ in similarity, with a non-random regularity, between more divergent sub-graphs distributed across the VSG repertoire.
3.6 Pairings between N- and C-terminal types show some biases
When the five N-terminal types are considered at the E < 1e-03 threshold, the corresponding six C-terminal types do not specifically assort with a specific N-terminal type, i.e., an N-terminal type can be matched with any C-terminal type (Fig. 2C). However, some pairings are clearly biased for or against, as shown when the observed N- and C-terminal combinations are compared to expected values (Table 1). For example, N1 is much more likely to pair with C2 or C5 than to C1 or C3. N2 pairs with any C-terminal type except C5, for which no pairs were observed. N4 tends to pair with either C1 or C5. N3 and N5 are small enough groups that it is difficult to identify a bias. These tendencies in N- and C-pairing likely provide further evidence for the importance of substructures in the overall VSG tertiary structure.
Table 1.
Based on 710 VSGs containing both N- and C-termini annotations, the biases of each N-terminal type to associate with a particular C-terminal type are shown by the observed (Obs) number of VSGs seen with a particular N-C terminal combination, and the number expected (Exp) if the types were equally likely to pair based the abundance of the N- or C-terminal type. Light gray shaded areas show combinations occurring significantly less than expected (P < 0.001, calculated by empirically simulated data). Dark gray shaded combinations are significantly enriched for a combination, and white are not significantly different than expected. For each N- C-terminus type pair, the first number is the number of VSGs observed. The second number is the number of these N- C- terminal pairs expected if N- and C- termini randomly associated with each other.
| C1 | C2 | C3 | C4 | C5 | C6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Obs | Exp | Obs | Exp | Obs | Exp | Obs | Exp | Obs | Exp | Obs | Exp | |
| N1 | 13 | 57 | 90 | 41 | 5 | 32 | 6 | 4 | 24 | 6 | 1 | 6 |
| N2 | 94 | 70 | 45 | 50 | 40 | 39 | 2 | 5 | 0 | 8 | 8 | 7 |
| N3 | 5 | 17 | 23 | 12 | 1 | 10 | 2 | 1 | 2 | 2 | 0 | 2 |
| N4 | 159 | 123 | 28 | 88 | 108 | 69 | 7 | 8 | 2 | 13 | 19 | 13 |
| N5 | 6 | 11 | 13 | 8 | 3 | 6 | 1 | 1 | 2 | 1 | 1 | 1 |
3.7 Locations of similar VSGs are usually independent of their chromosomal positions
We considered whether two or more VSGs of the same N-terminal type were more likely to occur on the same chromosome in closer physical proximity than expected by random chance. The presence of some similar VSGs located in close physical proximity to one another is supported by statistical analysis (Table 2). When analyzing all VSGs on the same contig, some VSGs of the same N-terminal type were more likely to be located physically close to other VSGs of the same type than to VSGs of different N-terminal types, a finding consistent with the possibility that some members of the same N-terminal type are likely to be generated by tandem gene duplication. However, this statistical observation was only seen on 2 of 19 contigs [chromosome (chr) 4 and Tryp_IXb-68a05.-2k3050], indicating that it does not occur ubiquitously. Some of these tandem duplication events appear to be atypical VSGs that may actually be VSG-like genes. On chr 4 (Tb927_04_v4), which contains 29 VSGs, significant clustering of same-type VSGs (p < 0.01) was observed. However, if an eight-member locus of VSGs comprised of 4 repeated atypical VSGs, 3 pseudogenes, and 1 functional VSG, is excluded, this significant co-localization is lost (p < 0.81).
Table 2.
For each genomic sequence fragment (contig) containing VSGs, the number of each VSG type is listed. To compute a p-value indicating a significant co-localization of same-type VSGs, the average distance between VSGs encoding the same N-terminal type was compared to simulated repertoires in which the type labels were permuted. Two of the contigs examined have same-type VSGs occurring significantly close and are highlighted; the others did not show significant co-localization of same-type VSGs.
| Name of Contig (chr= chromosome) |
Number of VSGs |
Number of N1,N2,N3,N4 and N5 types |
p value for a smaller than expected average distance between same-type VSGs based on 100k permutations |
|---|---|---|---|
| 27P2_V3 | 35 | 6,11,1,17,0 | 0.73867 |
| Tb927_01_v4 (chr 1) | 5 | 0,1,0,3,1 | 0.66382 |
| Tb927_02_v4 (chr 2) | 9 | 1,1,0,6,1 | 0.77749 |
| Tb927_03_v4 (chr 3) | 34 | 10,10,0,14,0 | 0.37428 |
| Tb927_04_v4 (chr 4) | 29 | 7,8,3,11,0 | 0.00925 |
| Tb927_05_v4 (chr 5) | 63 | 8,21,2,29,3 | 0.63349 |
| Tb927_06_v4 (chr 6) | 46 | 13,10,2,20,1 | 0.40205 |
| Tb927_07_v4 (chr 7) | 9 | 0,3,0,5,1 | 0.65537 |
| Tb927_08_v4 (chr 8) | 30 | 6,5,2,14,2 | 0.84335 |
| Tb927_09_v4 (chr 9) | 196 | 39,50,15,81,11 | 0.30494 |
| Tb927_10_v4 (chr 10) | 26 | 4,6,2,12,2 | 0.05658 |
| Tb927_11_01_v4 (chr 11) | 116 | 32,18,8,54,4 | 0.77996 |
| Tb927_11_02_v4 (chr 11) | 60 | 7,27,8,26,2 | 0.73117 |
| Tryp_IXb-217g08.q1c | 6 | 0,1,1,4,0 | 0.83799 |
| Tryp_IXb-218d07.p1c | 29 | 5,5,0,17,2 | 0.30265 |
| Tryp_IXb-68a05.p2k3050 | 13 | 0,2,4,2,5 | 0.03094 |
| Tryp_X-254c10.q1c | 16 | 3,5,1,6,1 | 0.10299 |
| Tryp_X-302f11.q1ca | 9 | 0,2,0,7,0 | 0.65412 |
| Tryp_X-324h11.p1k | 24 | 6,6,0,12,0 | 0.14332 |
3.8 N-termini of Expressed Functional VSGs of T. brucei and T. congolense
The above analyses were conducted on VSGs that occur in the T. brucei stock TREU 927 genome [7], most of which are unexpressed pseudogenes located in subtelomeric regions. To extend these analyses to functional VSGs known to be expressed by bloodstream-stage T. brucei, we also compared the N-termini of 17 VSGs from several different T. brucei isolates that are derived from complete VSG cDNA sequences available in the literature [25–28]. These 17 VSGs were found to occur in the three most abundant N-terminal types, N1, N2 and N4, with the majority occurring in N2 (Fig. 2F red coloring). The presence of these three types in actual expressed VSGs along with the existence of similar ‘functional’ genomic VSGs in each of the five types suggest that all N-terminal types have the capacity to produce functional VSGs.
We also examined whether functional expressed VSGs of another African trypanosome species, Trypanosoma congolense, [29–31] fall into similar N-terminal types. Using an E < 1e-03 threshold of similarity, 17 available T. congolense VSGs (as determined from cDNAs) occurred in the N4 N-terminal type (Fig. 2F, yellow coloring). At higher levels of stringency, however, this relationship between the T. congolense and T. brucei VSGs is lost (not shown). When hidden Markov models representative of VSG N-terminal types were used to search the unpublished T. congolense genomic sequences available online from the Sanger Centre [http://www.sanger.ac.uk/Projects/T_congolense/], both type N2 and N4 functional VSGs could be identified in the T. congolense genomic sequences (data not shown). Thus, as more T. congolense genomic sequence becomes available and annotated, this will be a fruitful area of comparison between these two African trypanosome species.
3.9 Evidence for mosaic gene conversion events
Several examples have been reported of mosaic VSG formation during the duplicative transposition (i.e., gene conversion) of a VSG into a VSG expression site [32–34]. In the current analysis, N-terminal VSG sequences with highly similar or identical segments are excellent potential candidates for recent mosaic gene conversion since their regions of high similarity or identity imply there may have been a recent duplication or gene conversion event. We examined those N-terminal VSGs with substantial similarity, i.e., E < 1e-20, at both the amino acid and nucleotide level for potential gene cross-over regions. We then further narrowed the selected set by focusing on functional VSGs and their relatives since functional VSGs contain complete coding sequences and may have been involved in a more recent gene conversion event. At the stringent criteria of E < 1e-20 (plot not shown), no similarity between functional VSGs was observed. However, many relationships were detected between functional VSGs and the atypical VSGs and pseudogenes, of which eleven examples are shown in the upper panels of Fig. 6.
Fig. 6.
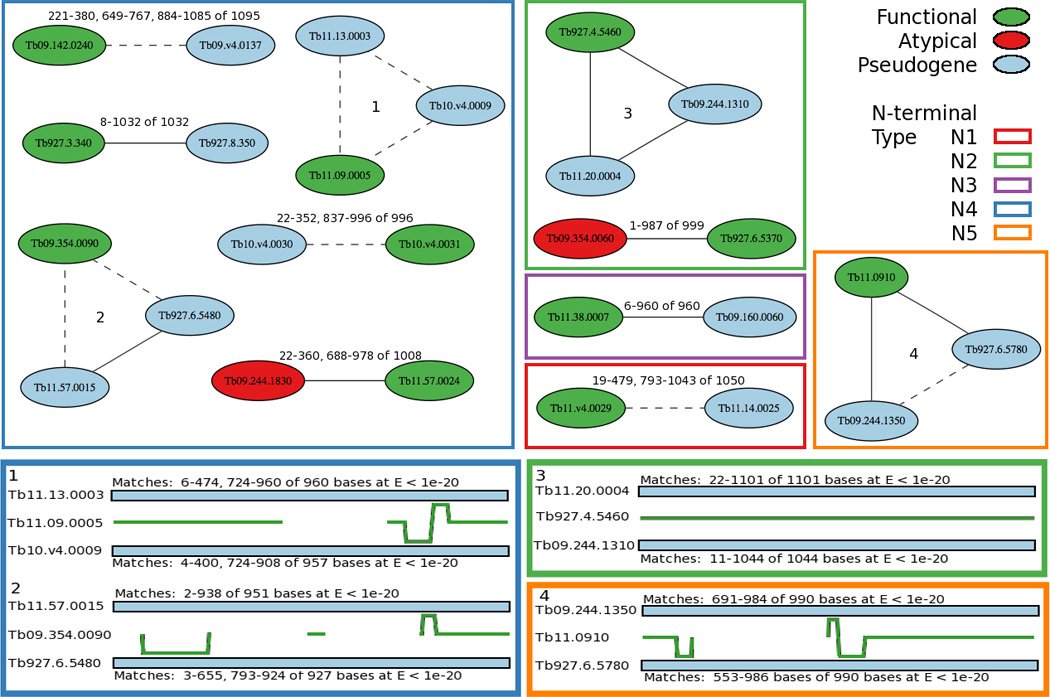
Evidence for mosaic gene conversion between similar VSG N-terminal domains. (Top panels). BLAST hits between functional (green) VSGs and their related pseudogene (blue) or atypical (red) sequences at E < 1e-20, were examined for evidence of mosaic gene conversion. Nodes are labeled by VSG name, and lines connecting nodes representing the BLAST hits are labeled with the range of the sequence matches and the total number of nucleotides in the query sequence. Solid lines represent a match from one continuous BLAST hit and dashed lines represent multiple non-contiguous BLAST hits between two VSGs. Borders of each square or rectangle are color-coded according to the indicated N-terminal type. Examples numbered 1 – 4 in the upper panels correspond to examples 1 – 4 in the lower panels. (Lower panels). Four examples of the relationships between a functional VSG gene and two related VSG pseudogenes are illustrated where the green line represents the indicated functional VSG N-terminal sequence. The blue bars above and below the green line represent two related VSG pseudogene N-terminal sequences, and the output of the BLAST hit between that VSG sequence and the functional VSG is given for each match. In regions where the functional VSG sequence is identical or nearly identical to both related VSG sequences, the green line is in the center. In regions where the functional sequence is more similar to one of the two related sequences, the green line is offset, and when the functional sequence is unrelated to either of two sequences, there is no green line.
In these examples, green colored VSGs indicate functional VSGs, blue colored are pseudogenes, red colored are atypical genes and lines represent a BLAST hit between two similar VSGs. The four examples numbered 1 – 4 in the upper panels of Fig. 6 are groups of three genes shown in more detail in the lower panels. In example 1, functional VSG Tb11.09.0005 matches both of its pseudogene relatives for the first several hundred nucleotides. This segment of identity is followed sequentially by (i) a region of no similarity with other VSGs, (ii) a short region of similarity with both pseudogenes, (iii) a region in which the functional gene shares sequence with pseudogene Tb10.v4.0009, (iv) a region with identity to pseudogene Tb11.13.0003, and finally (v) a region highly similar to both pseudogenes. This pattern suggests that multiple cross-over events have transpired between these three genes and likely at least one other genomic sequence that is not in any of the annotated VSGs.
In example 2, functional VSG Tb09.354.0090 matches one of its pseudogene relatives near the beginning of the N-terminus and matches both of its pseudogene relatives at the end of N-terminal domain. The region or gap in between has little or no similarity with either pseudogene relative. Example 3 shows a functional VSG bearing extensive similarity with two pseudogenes across the entire N-terminal domain, whereas example 4 shows functional VSG Tb11.0910 whose N-terminal domain has only small segments of similarity with one or the other of its pseudogene relatives followed by a large region of identity with both pseudogenes.
The gaps in identity and the regions of greater identity to differing pseudogenes could both be taken as evidence suggesting mosaic gene formation. It is notable that the gaps in BLAST search can identify regions without similarity (highly dissimilar); however, the BLAST search was is not sensitive in identifying regions where differences are due to only small sequence changes. This reduces our ability to apply this method to identify mosaic gene conversion events when the gene conversion events occur between highly similar sequences, as may often be the case. This BLAST method of similarity mapping has the advantage of being able to identify discontinuous regions of identity, and also can be efficiently applied to describe the relationships between a large number of genes.
4. Discussion
About 20% of the protein-coding capacity of the T. brucei genome is consumed by the estimated 1600 VSG sequences [7, 8]. Although only one VSG is transcribed at a time, the sequentially-expressed functional VSG proteins must be both structurally similar and antigenically diverse in order to pack closely on the parasite surface and achieve the parasite’s goal of evading the immune system. By examining the large repertoire of VSGs, it is possible to describe the clusters of relationships between similar VSGs and their annotated locations. We utilized a previously created VSG database [12] to re-examine these VSG relationships in ways that allowed them to be clustered and visualized graphically in 2D or 3D form (Figs. 1, 2, 4, S1, S2), and to be searched for multiple, non-contiguous segments of sequence similarity (Fig. 6). We detected differences between these gene families and observed how subsets of expressed VSG sequences are related to the genomic sequences. We also identified gaps in similarity between VSG sequences and found extensive evidence for potential cross-over events among multiple VSGs.
Despite the need for antigenic diversity, the VSG N-terminal domains display amino acid similarities, which have been used to classify them into groupings or types. Previous analyses have described three VSG N-terminal types [3, 8] and we report here the existence of five N-terminal domain types, N1 – N5, of which the first three, N1 – N3, are derived from one of the three originally described types. These five N-terminal domain types are based on amino acid identities meeting an E < 1e-03 stringency where separation of the five clusters is most apparent (Figs. 2 and S1). As the E-value is changed to higher stringencies, increasing numbers of subtypes are detected within each of the five types (Fig. 5). Furthermore, the region of maximal similarity within the ~330 amino acid N-terminal domain differs among the five types. Members of types N1, N3 and N4 are most similar in the middle of the domain, type N2 members are most similar near the N-terminus of the domain and N5 members are more uniformly similar along the entire domain (Fig. 3). These differences do not correlate with the overall extent of similarity among members within a type, i.e., the compactness of the five domain clusters observed in Fig. 2. For example, of the five types the N4 family members are the most similar to each other (most intra-cluster BLAST hits, forming the most compact cluster), consistent with an earlier finding [8], and N3 members are the least similar (Fig. 2A). It is likely these differences in regions of maximal similarity among the five types reflect both evolutionary expansion of the family and structural constraints imposed on the functional VSGs themselves. The N4 family is also the largest family, suggesting it may have been more prone to recent expansion events that the other types. The N4 family also has a somewhat different predicted alpha-helical structure than the other four types (Fig. 3), although secondary structure predictions must be treated with caution. No restrictions lie in the association of a given N-terminal and C-terminal domain type, although some N- and C-terminal pairings are preferred (Table 1), again likely reflecting possible structural constraints on the VSG protein to ensure close packing of the trypanosome surface, and highlighting the utility in analyzing the N-terminal domain independently from the C-terminal domain.
Varying the threshold of similarity determined by the E-value in BLAST provided a means to resolve different-sized clusters of similar VSGs. The unsupervised MCL algorithm was applied to generate these clusters, within which there was a tendency for the functional and atypical VSGs to be more centrally connected (similar) to divergent sub-groups than are pseudogenes, suggesting these VSGs might be important, either as the progenitors of different sub-groups of degenerating pseudogenes, and/or the product of an ancient mosaic between two more diverse clusters. The central positioning of atypical VSGs is especially interesting since they do not appear to be, in general, diverging in form from the rest of the VSG repertoire, but rather may be important for maintaining the diverse community structure indicated by the different VSG types described.
Experimental evidence for mosaic VSG formation dates back to the 1980s [30, 32], and has indicated that mosaic VSGs are more likely to appear late, rather than early, in infection [8, 32]. The data described here show that mosaic VSG formation is confined almost entirely to donor VSGs from within the same N-terminal type. Despite the different N-terminal types being dispersed throughout the genome, only one clear example was detected of a mosaic VSG containing sequences derived from two N-terminal types (N1 and N2) (red arrow in Fig. 2B). This result is consistent with the likelihood that recombination between highly similar segments of family members within an N-terminal type is responsible at least in part for formation of mosaic VSGs, and the lack of inter-type mosaics suggests those mosaics may be non-functional, possibly due to improper folding. This interpretation is supported by a close examination of the four examples of mosaic functional VSGs shown in Fig. 6B. In each of these cases, the boundaries of potential recombinational cross-over points that could be examined occur in regions of highly similar sequences. Left unexplained are the origins of sequences within the mosaic functional VSGs of Fig. 8B that do not have a counterpart sequence in a known donor VSG, but these sequences could be derived from the several hundred VSGs on minichromosomes or intermediate chromosomes whose sequences have yet to be determined [6].
The methods described here provide a means to explore the communities of related VSGs at varying degrees of similarity. As we have shown with the T. congolense VSG cDNAs, profile-HMMs generated from these sets of VSGs can be useful for comparing and contrasting the composition of the VSG repertoire between species. These approaches can be readily applied to compare and contrast the VSG repertoires in different trypanosome isolates. As the genome sequences of additional species and strains of the Trypanosomatid protozoa and other microorganisms are emerging, these methods can be applied to comparisons between families of genes to assess the diversity or relatedness of virulence factors in almost any genera. Correlation of these data with differences in clinical presentation or disease severity may add new dimensions to our ability to discern whether microbial diversity might account for diverse manifestations of different microbial infectious diseases.
Supplementary Material
Video traversing a three-dimensional rendering of the VSG N-terminal clusters at E < 1e-02, color-coded according to Fig. 2B.
Video traversing the three-dimensional rendering of the VSG C terminal clusters shown in Fig. 4A.
VSGs clustering schemes generated by MCL at E-values from 1e-01 to 1e-10.
Atypical and functional VSGs that are members of clusters calculated to have an average betweenness higher than would be observed in random or permuted graphs (p < 0.05).
Atypical (Tb927.5.4730) and functional (Tb09.160.0110) VSGs were selected from Supplemental Table T2 to represent VSGs from among MCL-identified clusters with significantly high average betweenness for functional or atypical VSGs. These high average betweenness clusters are shown at multiple scales of E-value. Pseudogenes are color-coded as gray, atypical are orange and functional are blue. As graphs become sparser at higher E-values, the tendency for an atypical or functional VSG to occur between a less connected sub-graph becomes more visibly apparent, illustrating the meaning of high betweenness.
{kind=link}
Acknowledgements
This work was supported in part by a Merit Review grant and an OEF-OIF program grant from the Department of Veterans’ Affairs. It was supported in part by NIH grants NIAID R01 AI045540 (MEW), R01AI067874 (MEW), R01 AI076233 (MEW) and R01 AI059451 (JED, MEW). It was performed in part during the tenure of JW on NIH training grants T32 GM008629 and T32 GM082729. We thank Sabarish Babu in the Department of Computer Science at the University of Iowa, and now at Clemson University, for helpful discussions.
Abbreviations
- VSG
variant surface glycoprotein
- VSG
VSG gene
- GPI
glycosylphosphatidylinositol
- 2D
two dimensional
- 3D
three dimensional
- contig
contiguous sequence
- MCL
Markov clustering algorithm
- profile-HMM
profile hidden Markov model
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Turner CM. The rate of antigenic variation in fly-transmitted and syringe-passaged infections of Trypanosoma brucei. FEMS Microbiol Lett. 1997;153:227–231. doi: 10.1111/j.1574-6968.1997.tb10486.x. [DOI] [PubMed] [Google Scholar]
- 2.Schwede A, Jones N, Engstler M, Carrington M. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol Biochem Parasitol. 2011;175:201–204. doi: 10.1016/j.molbiopara.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchinson OC, Smith W, Jones NG, Chattopadhyay A, Welburn SC, Carrington M. VSG structure: similar N-terminal domains can form functional VSGs with different types of C-terminal domain. Mol Biochem Parasitol. 2003;130:127–131. doi: 10.1016/s0166-6851(03)00144-0. [DOI] [PubMed] [Google Scholar]
- 4.Metcalf P, Down JA, Turner MJ, Wiley DC. Crystallization of amino-terminal domains and domain fragments of variant surface glycoproteins from Trypanosoma brucei brucei. J Biol Chem. 1988;263:17030–17033. [PubMed] [Google Scholar]
- 5.Chattopadhyay A, Jones NG, Nietlispach D, Nielsen PR, Voorheis HP, Mott HR, et al. Structure of the C-terminal domain from Trypanosoma brucei variant surface glycoprotein MITat1.2. J Biol Chem. 2005;280:7228–7235. doi: 10.1074/jbc.M410787200. [DOI] [PubMed] [Google Scholar]
- 6.Melville SE, Leech V, Gerrard CS, Tait A, Blackwell JM. The molecular karyotype of the megabase chromosomes of Trypanosoma brucei and the assignment of chromosome markers. Mol Biochem Parasitol. 1998;94:155–173. doi: 10.1016/s0166-6851(98)00054-1. [DOI] [PubMed] [Google Scholar]
- 7.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 8.Marcello L, Barry JD. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 2007;17:1344–1352. doi: 10.1101/gr.6421207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donelson JE. Antigenic variation and the African trypanosome genome. Acta Trop. 2003;85:391–404. doi: 10.1016/s0001-706x(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 10.Pays E. Regulation of antigen gene expression in Trypanosoma brucei. Trends Parasitol. 2005;21:517–520. doi: 10.1016/j.pt.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Schwede A, Carrington M. Bloodstream form Trypanosome plasma membrane proteins: antigenic variation and invariant antigens. Parasitology. 2010;137:2029–2039. doi: 10.1017/S0031182009992034. [DOI] [PubMed] [Google Scholar]
- 12.Marcello L, Menon S, Ward P, Wilkes JM, Jones NG, Carrington M, et al. VSGdb: a database for trypanosome variant surface glycoproteins, a large and diverse family of coiled coil proteins. BMC Bioinformatics. 2007;8:143. doi: 10.1186/1471-2105-8-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dongen Sv. PhD thesis. University of Utrecht; 2000. Graph Clustering by Flow Simulation. [Google Scholar]
- 14.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 15.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 16.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 17.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen F, Mackey AJ, Stoeckert CJ, Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeman CL. A Set of Measures of Centrality Based on Betweenness. Sociometry. 1977;40:35–41. [Google Scholar]
- 20.Pavlidis P, Noble WS. Matrix2png: a utility for visualizing matrix data. Bioinformatics. 2003;19:295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 21.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 23.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 24.McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 25.Carrington M, Miller N, Blum M, Roditi I, Wiley D, Turner M. Variant specific glycoprotein of Trypanosoma brucei consists of two domains each having an independently conserved pattern of cysteine residues. J Mol Biol. 1991;221:823–835. doi: 10.1016/0022-2836(91)80178-w. [DOI] [PubMed] [Google Scholar]
- 26.Reddy LV, Hall T, Donelson JE. Sequences of three VSG mRNAs expressed in a mixed population of Trypanosoma brucei rhodesiense. Biochem Biophys Res Commun. 1990;169:730–736. doi: 10.1016/0006-291x(90)90392-z. [DOI] [PubMed] [Google Scholar]
- 27.Lenardo MJ, Rice-Ficht AC, Kelly G, Esser KM, Donelson JE. Characterization of the genes specifying two metacyclic variable antigen types in Trypanosoma brucei rhodesiense. Proc Natl Acad Sci U S A. 1984;81:6642–6646. doi: 10.1073/pnas.81.21.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbet AF, Myler PJ, Williams RO, McGuire TC. Shared surface epitopes among trypanosomes of the same serodeme expressing different variable surface glycoprotein genes. Mol Biochem Parasitol. 1989;32:191–199. doi: 10.1016/0166-6851(89)90070-4. [DOI] [PubMed] [Google Scholar]
- 29.Urakawa T, Eshita Y, Majiwa PA. The primary structure of Trypanosoma (Nannomonas) congolese variant surface glycoproteins. Exp Parasitol. 1997;85:215–224. doi: 10.1006/expr.1996.4140. [DOI] [PubMed] [Google Scholar]
- 30.Strickler JE, Binder DA, L'Italien JJ, Shimamoto GT, Wait SW, Dalheim LJ, et al. Trypanosoma congolense: structure and molecular organization of the surface glycoproteins of two early bloodstream variants. Biochemistry. 1987;26:796–805. doi: 10.1021/bi00377a021. [DOI] [PubMed] [Google Scholar]
- 31.Helm JR, Hertz-Fowler C, Aslett M, Berriman M, Sanders M, Quail MA, et al. Analysis of expressed sequence tags from the four main developmental stages of Trypanosoma congolense. Mol Biochem Parasitol. 2009;168:34–42. doi: 10.1016/j.molbiopara.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Longacre S, Eisen H. Expression of whole and hybrid genes in Trypanosoma equiperdum antigenic variation. EMBO J. 1986;5:1057–1063. doi: 10.1002/j.1460-2075.1986.tb04322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamper SM, Barbet AF. Surface epitope variation via mosaic gene formation is potential key to long-term survival of Trypanosoma brucei. Mol Biochem Parasitol. 1992;53:33–44. doi: 10.1016/0166-6851(92)90004-4. [DOI] [PubMed] [Google Scholar]
- 34.Roth C, Bringaud F, Layden RE, Baltz T, Eisen H. Active late-appearing variable surface antigen genes in Trypanosoma equiperdum are constructed entirely from pseudogenes. Proc Natl Acad Sci U S A. 1989;86:9375–9379. doi: 10.1073/pnas.86.23.9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video traversing a three-dimensional rendering of the VSG N-terminal clusters at E < 1e-02, color-coded according to Fig. 2B.
Video traversing the three-dimensional rendering of the VSG C terminal clusters shown in Fig. 4A.
VSGs clustering schemes generated by MCL at E-values from 1e-01 to 1e-10.
Atypical and functional VSGs that are members of clusters calculated to have an average betweenness higher than would be observed in random or permuted graphs (p < 0.05).
Atypical (Tb927.5.4730) and functional (Tb09.160.0110) VSGs were selected from Supplemental Table T2 to represent VSGs from among MCL-identified clusters with significantly high average betweenness for functional or atypical VSGs. These high average betweenness clusters are shown at multiple scales of E-value. Pseudogenes are color-coded as gray, atypical are orange and functional are blue. As graphs become sparser at higher E-values, the tendency for an atypical or functional VSG to occur between a less connected sub-graph becomes more visibly apparent, illustrating the meaning of high betweenness.