

Abstract
The uPA/uPAR system is known to play a critical role in angiogenesis of glioblastoma. Previously, we have shown that shRNA against uPA and uPAR attenuates angiogenesis by blocking nuclear translocation of angiogenin, inhibition of angiopoietin/Tie2 signaling, and regulating several other pro‐angiogenic, angiostatic and anti‐angiogenic molecules. Further analysis revealed that GM‐CSF, a pleiotropic cytokine, was significantly inhibited in U87MG and 4910 co‐cultures with endothelial cells transfected with shRNA against uPA and uPAR. The role of the uPA/uPAR system in this process is not completely understood. Analysis of tumor conditioned medium of U87MG, 4910 and HMECs transfected with shRNA against uPA or uPAR alone or in combination (pU2) revealed inhibition of GM‐CSF‐enhanced secretion of SVEGFR1 as shown by Western blotting and ELISA. Moreover, phosphorylation of JAK2 and STAT5, the downstream effectors of GM‐CSF signaling, was also inhibited in all three cell lines. Phosphorylation at Tyr 166 position of the GM‐CSFRβ subunit, the signal activating subunit of the GM‐CSF receptor, was inhibited in HMEC, U87MG and 4910 cells. Further analysis revealed that shRNA against uPA and/or uPAR increased secretion of TIMP‐1, which is known to enhance SVEGFR1 secretion in endothelial cells. Moreover, addition of purified uPA (with and without GM‐CSF) activated JAK2/STAT5 signaling in HMEC. Exogenous addition of SVEGFR1 to pU2 tumor conditioned medium enhanced inhibition of VEGF‐induced endothelial capillary tube formation as assessed by an in vitro angiogenesis assay. To determine the significance of these events in vivo, nude mice with pre‐established tumors treated with shRNA against uPA and/or uPAR showed decreased levels of GM‐CSF and increased levels of SVEGFR1 and TIMP‐1 when compared with controls. Enhanced secretion of SVEGFR1 by puPA, puPAR and pU2 in endothelial and GBM cells was mediated indirectly by MMP‐7 and augmented by ectodomain shedding of VEGFr1 by tyrosine phosphorylation at the 1213 position. Taken together, these results suggest that the uPA/uPAR system could prove beneficial as an indirect target for inhibition of angiogenesis in glioblastoma.
Keywords: uPA and uPAR, shRNA, Angiogenesis, Glioblastoma
Highlights
First report highlighting the role and mechanism by which shRNA against uPA/uPAR inhibits angiogenesis in glioblastoma.
shRNA against uPA/uPAR enhances secretion of sVEGFr1, a scavenger for VEGF.
sVEGFr1 secretion is independent of GM‐CSF unlike monocytes but dependent on TIMP‐1.
TIMP‐1 secretion is mediated by MMP‐7 which is further regulated by uPA and uPAR.
Abbreviations
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- GM‐CSF
granulocyte macrophage colony stimulating factor
- SVEGFR1
soluble vascular endothelial growth factor receptor 1
- TIMP‐1
tissue inhibitor of matrix metalloproteinase‐1
1. Introduction
Glioblastoma multiforme is characterized by abnormal angiogenesis, leaky vasculature and highly invasive nature (Anderson et al., 2008; Norden et al., 2008). Thus far, many studies have implicated the role of the uPA/uPAR system in different aspects of tumor progression including tumor cell proliferation, adhesion, migration, invasion, metastasis and angiogenesis (Bene et al., 2004; Blasi and Carmeliet, 2002; Choong and Nadesapillai, 2003; Duffy and Duggan, 2004; Irigoyen et al., 1999; Pepper, 2001; Raghu et al., 2010; Ulisse et al., 2009). It is now understood that the roles of uPA and uPAR in angiogenesis are multi‐faceted, and simultaneous transcriptional inactivation of uPA and uPAR in glioblastoma cells and tumor endothelial cells have significant deleterious effects on inhibition and enhancement in secretion of several pro‐angiogenic and anti‐angiogenic molecules in glioblastoma cells (Raghu et al., 2010).
Degradation by proteolysis of the extracellular matrix is the initial step in glioblastoma progression, and the uPA/uPAR system is important for migration and invasion of endothelial cells during angiogenesis (Lakka et al., 2005). Angiogenesis is a complex process that affects proteolysis, migration, proliferation, ECM degradation, and invasion and involves several pro‐angiogenic factors, such as VEGF (Park et al., 1993). Previously, we have demonstrated that uPA/uPAR downregulation in glioblastoma (GBM) inhibits a key pro‐angiogenic factor, VEGF (Gondi et al., 2007). Several studies have implicated uPA and uPAR in regulation of angiogenin, angiopoeitin/Tie2 signaling (Raghu et al., 2010), and Notch signaling (unpublished observations), and thus, GBM pathogenesis.
GM‐CSF is a pleiotropic cytokine known to play important roles in the differentiation of hematopoietic precursor cells to mature granulocytes, macrophages or dendritic cells (Wognum et al., 1994). Studies by Eubank et al. (2004, 2009) have shown GM‐CSF as an anti‐tumor agent in breast cancer both in vitro and in vivo. The function of GM‐CSF in the non‐hematopoietic environment, particularly in relevance to cancer, has not been studied in detail. The role of the uPA/uPAR system, particularly uPA, in mediating GM‐CSF signaling and function is not known.
Tumor endothelial cells express VEGFr1 (Flt1) and VEGFr2 (KDR), and it also known that VEGF‐A signals through VEGFr1 (Matsumoto and Claesson‐Welsh, 2001). Our studies have shown that VEGF is inhibited in pU2‐transfected glioblastoma tumors; however, the exact mechanism is not known. A relationship between SVEGFR1, a known scavenger for VEGF and uPA, is yet to be elucidated. VEGF‐mediated metastasis has also been established in breast tumors (Eubank et al., 2003). In the present study, our results suggest that shRNA against uPA/uPAR reduces GM‐CSF and VEGF by inducing secretion of SVEGFR1 by endothelial and glioblastoma cells, and thus, reducing the VEGF available for angiogenesis. Our studies suggest that increased secretion of SVEGFR1 is probably mediated by the stimulation through TIMP‐1. This study also shows that shRNA against uPA/uPAR inhibits GM‐CSFRβ subunit‐mediated JAK2/STAT5 activation. Our study has revealed a novel mechanism by which shRNA against uPA/uPAR inhibits angiogenesis both in vitro and in vivo by enhancing secretion of SVEGFR1 and TIMP‐1 in endothelial cells. Our model shows the inhibition of angiogenesis by blocking uPA/uPAR in GBM is enhanced by secretion of SVEGFR1 dependent on TIMP‐1 but independent of GM‐CSF.
2. Materials and methods
2.1. Ethics statement
The institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Peoria (Peoria, IL) approved all surgical interventions and post‐operative care. The approved protocol number is 851 and is dated May 12, 2010. No de novo cell lines were used.
2.2. Cells and reagents
U87MG (obtained from ATCC, Manassas, VA), xenograft cell lines (4910 cells kindly provided by Dr. David James at the University of California‐San Francisco) were cultured as previously described (Kunigal et al., 2006). Human microvascular endothelial cells (HMECs) were cultured as per standard protocols established in our laboratory. Antibodies to GM‐CSF, Flotillin1, pJAK2 (pTyr 1007/1008), TJAK2, TSTAT5, pSTAT5 (pTyr 695/699), siGM‐CSF, and TIMP‐1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody for SVEGFR1 was obtained from Abcam (Cambridge, MA). RhGM‐CSF was obtained from Sigma (St. Louis, MO). Antibody against pTyr 766 positions against the β subunit of GM‐CSFR was obtained from LSBIO (Seattle, WA). TIMP‐1 ELISA was obtained from Ray Biotech (Norcross, GA), and ELISA for mSVEGFR1, msGM‐CSF, hGM‐CSF and hSVEGFR1 was obtained from R&D Systems (Minneapolis, MN). Purified uPA was obtained from American Diagnostica (Stanford, CT) and recombinant TIMP‐1 was obtained from Prospecbio (Rehovot, Israel).
2.3. uPA and uPAR shRNA constructs
shRNA sequences targeting uPAR and uPA were constructed as described previously (Subramanian et al., 2006).
2.4. Transfection with shRNA constructs
1.5×105 cells were plated in 100‐mm dishes for each transfection experiment. The cells were transfected in serum‐free L‐15 media using 10μg of Fugene reagent (Roche, Indianapolis, Indiana) as per the manufacturer's instructions. The following constructs were used for transfection: puPA, puPAR, pU2 (shRNA against uPA and uPAR), and pSV. No plasmids were introduced in the control dishes. After 12h of transfection, the serum‐free media were replaced with serum‐containing media, and cells were left in the incubator at 37°C for 48h. The media were then replaced with serum‐free media, and conditioned media were collected 12h later. Cells were harvested for isolation of total RNA or total cell lysate. Conditioned media were used for ELISA.
2.5. In vitro angiogenesis assay
Angiogenesis assay was performed as described earlier (Gondi et al., 2004). Briefly, human microvascular endothelial cells (2×104 cells per well) were grown in the presence of tumor conditioned medium (TCM) of pU2‐treated U87MG cells, left untreated, or treated with SVEGFR1, VEGF alone, VEGF with SVEGFR1, or TIMP‐1 in 48‐well plates and incubated for 48h at 37°C. The formation of capillary‐like structures was captured with an Olympus 1× 71 digital fluorescent microscope after staining with Hema‐3 stain.
2.6. Western blotting
HMEC, U87MG, and 4910 cells were left untreated or transfected with SV, puPA, puPAR, or pU2. siGM‐CSF was transfected in HMEC according to the manufacturer's instructions. Cells were collected and whole cell lysates were prepared by lysing cells in RIPA lysis buffer containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Equal amounts of protein fractions, immunoprecipitates or lysates were resolved by SDS PAGE and transferred to a polyvinylidene difluoride membrane. Proteins were detected with appropriate primary antibodies followed by HRP‐conjugated secondary antibodies. Comparable loading of proteins was verified by reprobing the blots with an antibody specific for the housekeeping gene product, GAPDH.
2.7. hGM‐CSF, hSVEGFR1, hTIMP‐1, mSVEGFR1, and msGM‐CSF quantification by ELISA
Conditioned medium from mock, pSV‐, puPA‐, puPAR‐ or pU2‐transfected U87, 4910, and HMEC were subjected to ELISA for hGM‐CSF, hSVEGFR1, TIMP‐1, and serum from animals, which were left untreated or transfected with pSV, puPA, puPAR or pU2, were subjected to ELISA for msGM‐CSF and mSVEGFR1 (R&D Systems, Minneapolis, MN) as per the manufacturer's instructions.
2.8. Animal experiments
U87 MG cells (2×106) were injected into the brains of nude mice using a stereotactic frame. After 8–10 days, the mice were treated with pSV, puPAR, puPA or pU2. The in vivo intracranial delivery of the vectors was performed using Alzet mini‐osmotic pumps at the rate of 0.25μL/h: mock (PBS), 150μg of vector DNA, 150μg of puPA, 150μg of puPAR, and 150μg of pU2. All experiments were performed in compliance with institutional guidelines set by the University of Illinois College of Medicine at Peoria. After five weeks, or when the mice started showing symptoms, mice were euthanized by cardiac perfusion with formaldehyde. The brains were then removed and embedded in paraffin as per standard protocols. 1–2mL of blood was collected near the region of the brain and also by cardiac puncture. Blood was centrifuged, and the serum was separated and processed to check for mSVEGFR1, msTIMP‐1 and msGM‐CSF levels by ELISA.
3. Results
3.1. shRNA against uPA and uPAR alone or in combination inhibits secretion of GM‐CSF
Previously, we reported that simultaneous downregulation of uPA and uPAR inhibited angiogenesis in vitro and in vivo in glioblastoma cells and xenografts (Raghu et al., 2010). Among the different molecules downregulated by pU2 (bicistronic construct against uPA and uPAR) in conditioned medium of glioblastoma cell co‐cultured with endothelial cells, we found that GM‐CSF was significantly inhibited in U87MG and 4910 co‐cultures. To confirm inhibition of GM‐CSF in monocultures of U87MG, 4910 and HMEC, which were left untreated or transfected with scrambled vector, puPA, puPAR or pU2, we performed ELISA on the conditioned medium from the three cell lines. As shown by ELISA, GM‐CSF levels were downregulated in puPA‐, puPAR‐ and pU2‐transfected cells. Down regulation of GM‐CSF was enhanced upto three folds in puPA‐ and 3.5 folds in pU2‐transfected cells and by two folds in puPAR‐transfected cells (Figure 1A–C) as compared to mock and scrambled vector‐transfected cells. Results from ELISA were confirmed by Western blotting of the conditioned medium of all three cell lines (Figure 1D and E). The conditioned medium samples were loaded after normalization of protein content. As shown in our previous studies (Raghu et al., 2010), puPA, puPAR and pU2 inhibited the expression of uPA and uPAR at the protein and mRNA levels in all the three cell lines examined. Taken together, these results suggest that shRNA against uPA and/or uPAR inhibits the secretion of GM‐CSF when compared with controls in glioblastoma and endothelial cells.
Figure 1.
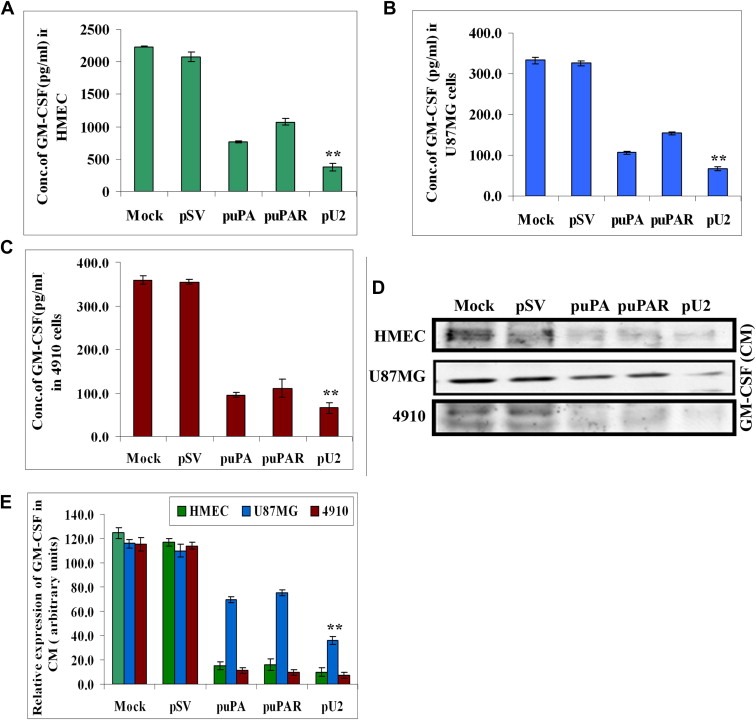
shRNA against uPA and uPAR inhibits secreted levels of GM‐CSF in cancer cells and endothelial cells. (A–C) 1.5×105 HMEC, U87MG and 4910 cells were transfected as described earlier. Conditioned medium was collected after 72h and assayed for GM‐CSF levels by ELISA as per the manufacturer's instructions. Data represented were the average of three independent experiments. ∗p<0.01; ∗∗p<0.001. (D) HMEC, U87MG and 4910 cells were transfected with shRNA constructs, and conditioned medium was collected. After protein normalization, equal quantity of conditioned medium was loaded and immunoblotted with primary antibody for GM‐CSF and corresponding secondary antibodies. A representative blot of three independent experiments is shown. (E) Quantitative analysis was done by optical densitometry as per standard protocols. Data represented were the average of three independent experiments.
3.2. shRNA against uPA and/or uPAR increases secretion of SVEGFR1 in glioblastoma and endothelial cells
Studies have shown that GM‐CSF induces the expression of SVEGFR1 in human monocytes and inhibits angiogenesis in mice (Eubank et al., 2004). To ascertain if shRNA against uPA and/or uPAR induced the secretion of SVEGFR1, we checked for SVEGFR1 levels in U87MG, 4910 and HMEC, which were left untreated or transfected with pSV, puPA, puPAR or pU2. Surprisingly, ELISA results showed that SVEGFR1 levels in the three examined cell lines were increased significantly in puPA‐, puPAR‐ and pU2‐transfected cells when compared with controls (Figure 2A–C). To confirm the ELISA results, conditioned medium from HMEC, U87MG and 4910 cells were subjected to Western blotting for SVEGFR1 expression; results showed that SVEGFR1 levels were significantly increased in puPA‐, puPAR‐ and pU2‐transfected cells when compared with controls (Figure 2D). There was almost four fold increase in SVEGFR1 levels in pU2‐treated HMEC compared with controls (Figure 2E). Taken together, these results show that shRNA against uPA and/or uPAR enhances the secretion of SVEGFR1 in U87MG, 4910 and HMEC as compared to controls, thereby suggesting that uPA and/or uPAR play an important role in secretion of SVEGFR1, which is a scavenger of VEGF.
Figure 2.
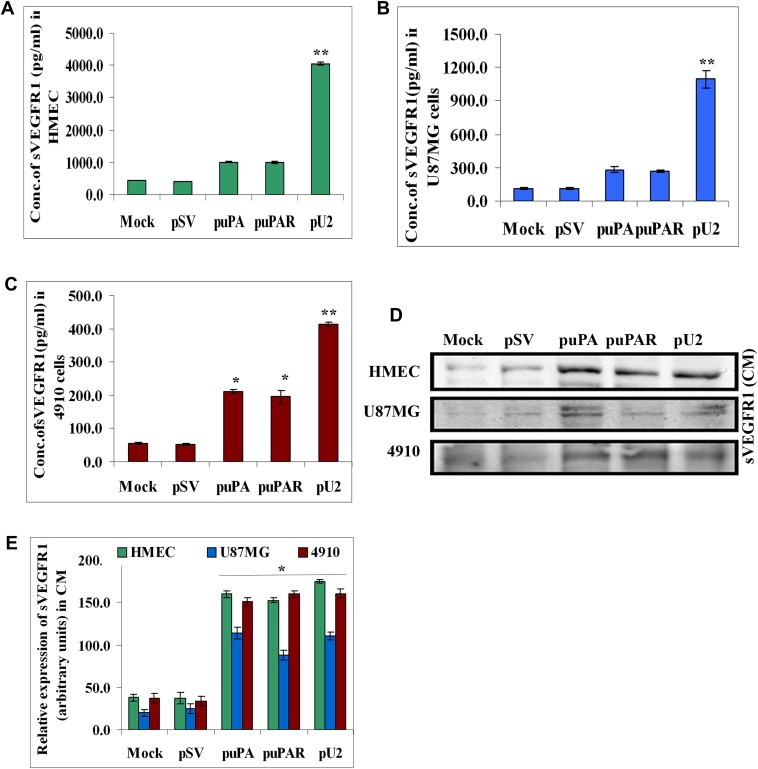
shRNA against uPA and uPAR enhances SVEGFR1 in U87MG, 4910 and endothelial cells. (A–C) 1.5×105 HMEC, U87MG and 4910 were transfected as described earlier. Conditioned medium was collected after 72h and assayed for SVEGFR1 levels by ELISA as per the manufacturer's instructions. Data represented are the average of three independent experiments. ∗p<0.01; ∗∗p<0.001. (D) HMEC, U87MG and 4910 cells were transfected with shRNA constructs and conditioned medium was collected. After protein normalization, equal quantity of conditioned medium was loaded and immunoblotted with primary antibody for SVEGFR1 and corresponding secondary antibodies. (E) Quantitative analysis was done by optical densitometry as per standard protocols. Data represented were the average of three independent experiments.
3.3. shRNA against uPA and/or uPAR inhibits phosphorylation of JAK2 and STAT5 and phosphorylation of GM‐CSFRβ at Tyr 766 in glioblastoma and endothelial cells
GM‐CSF is known to bind to GM‐CSFRα receptor and initiate signaling via the GM‐CSFRβ subunit in endothelial cells and phosphorylate JAK2 (Janus kinase), a member of the cytosolic tyrosine kinase family that is known to mediate several non‐tyrosine kinase receptors (Soldi et al., 1997). The binding of GM‐CSF to its receptor is also known to phosphorylate STAT5 at the pTyr 695/699 position. To ascertain whether shRNA against uPA and/or uPAR inhibits phosphorylation of JAK2 and STAT5, both downstream effectors of GM‐CSF signaling, lysates from HMEC, U87MG and 4910 cells, which were left untreated or transfected with pSV, puPA, puPAR and pU2, were probed for pJAK2 (Tyr 1007/1008) and pSTAT5 (Tyr 695/699), respectively. Results showed that in the three cell lines tested, down regulation of uPA and/or uPAR by transfection with puPA, puPAR or pU2 significantly inhibited the phosphorylation of JAK2 and STAT5 (Figure 3A–C). Overall, these results suggest that uPA and/or uPAR down regulation inhibits JAK2/STAT5 phosphorylation in glioblastoma and endothelial cells. To further decipher whether down regulation of phospho JAK2/STAT5 could be mediated by inhibition of GM‐CSFRβc phosphorylation, we checked for phosphorylation of the GM‐CSFRβ subunit at the Tyr 766 position. As anticipated, we observed a reduction in the phosphorylation of the β subunit of the GM‐CSF receptor, in all the three cell lines tested. Quantitative representation of the results in Figure 3D is represented in Figure 3E. Overall, these results suggest that shRNA against uPA and/or uPAR inhibits GM‐CSF‐mediated signaling in endothelial and glioblastoma cells.
Figure 3.
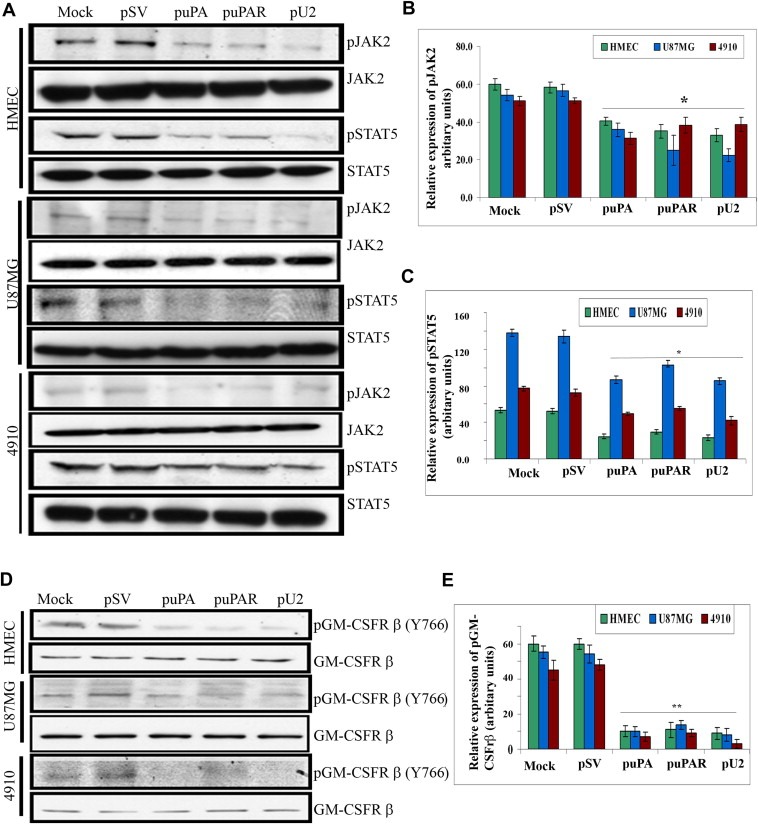
shRNA against uPA and uPAR down regulates phosphorylation of JAK2/STAT5 in glioblastoma and endothelial cells and phosphorylation of GM‐CSFrβ at Tyr 766 position in HMEC, U87MG and 4910 cells. (A) Western blot analysis of shRNA‐transfected HMEC, U87MG, and 4910 cells was carried out with primary antibodies against pJAK2 (Tyr1007/1008) and pSTAT5 (pTyr 695/699) and with total forms of JAK2 and STAT5. A representative blot of three independent experiments is shown. (B and C) Quantitative analysis was done for pJAK2 and pSTAT5 by optical densitometry as per standard protocols. (D) Western blot analysis of shRNA‐transfected HMEC, U87MG, and 4910 cells was carried out with primary antibody against pTyr766 position of the GM‐CSFrβ unit and appropriate secondary antibody. GM‐CSFRβ antibody was also immunoblotted to demonstrate equal loading of lysates. A representative blot of three independent experiments is shown. (E) Quantitative analysis was done by optical densitometry as per standard protocols.
3.4. shRNA against uPA and/or uPAR enhances secretion of TIMP‐1 in endothelial and glioblastoma cells
TIMP‐1 is recognized as a multifunctional protein, and its role in cancer is distinguished both as a tumor growth stimulator and a tumor growth inhibitor (Ikenaka et al., 2003). TIMP‐1 is also known to enhance the secretion of SVEGFR1 from human endothelial cells (Bruegmann et al., 2009). To determine whether TIMP‐1 is involved in SVEGFR1 secretion, we checked for TIMP‐1 expression in the conditioned medium of shRNA‐transfected HMEC, U87MG and 4910 cells. Our results showed that there was increased TIMP‐1 (by 3‐fold) expression in HMEC, U87MG and 4910 cells in shRNA‐transfected cells as compared with pSV‐treated and untreated cells (Figure 4A–C). Western blot results showing increased TIMP‐1 secretion in conditioned medium of shRNA‐treated cells was confirmed by ELISA (Figure 4C). These results suggest that downregulation of uPA and/or uPAR induced TIMP‐1 in GBM and HMECs.
Figure 4.
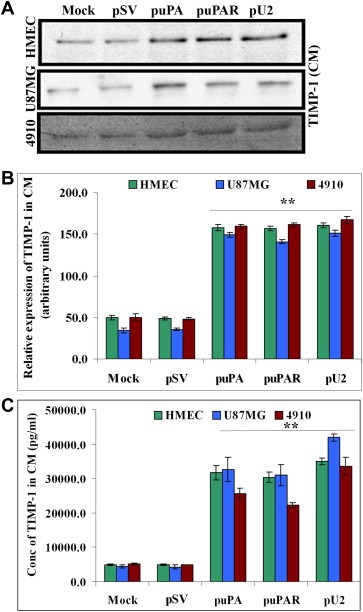
shRNA against uPA and uPAR enhances secretion of TIMP‐1 from HMEC, U87MG and 4910 cells by Western Blot and ELISA. (A) (Upper panel) 1.5×105 HMEC, U87MG and 4910 cells were transfected as described earlier. Conditioned medium was collected after 72h and TIMP‐1 levels determined by Western blotting. Equal amount of conditioned medium was loaded after normalization of protein content. (B) Graph represents quantification of Western blotting results using NIH Image J software. (C) 1.5×105 HMEC, U87MG and 4910 cells were transfected as described earlier. Conditioned medium was collected after 72h and TIMP‐1 levels determined by ELISA as per the manufacturer's instructions. Data represented are the average of three independent experiments. ∗p<0.01; ∗∗p<0.001.
3.5. uPA activates JAK2/STAT5 in HMEC in the absence of GM‐CSF
To further determine whether uPA alone activates JAK2 and STAT5 in the absence of GM‐CSF, HMECs were either left untreated treated with uPA or rhGM‐CSF, or transfected with pSV, puPA, or siGM‐CSF with and without addition of uPA. Next, we immunoblotted for pJAK2 and pSTAT5 expression. Results showed that siGM‐CSF and puPA resulted in similar levels of pJAK2 and pSTAT5 inhibition in HMEC (Figure 5A). Initially we have tested the efficiency of GM‐CSF siRNA in HMEC (Figure 5B). Addition of uPA to siGM‐CSF treated cells slightly increased expression levels of pJAK2 and pSTAT5 (Figure 5A and C). However, simultaneous addition of uPA and rhGM‐CSF together in HMEC did not increase phosphorylation of JAK2 and but showed increase in STAT5 phosphorylation as compared to addition of either uPA or rhGM‐CSF alone (Figure 5D and E). These results suggest that uPA independently activates the GM‐CSF receptor signaling pathway, and our results are consistent with previous studies that show GM‐CSF regulates uPA (Pei et al., 1999).
Figure 5.
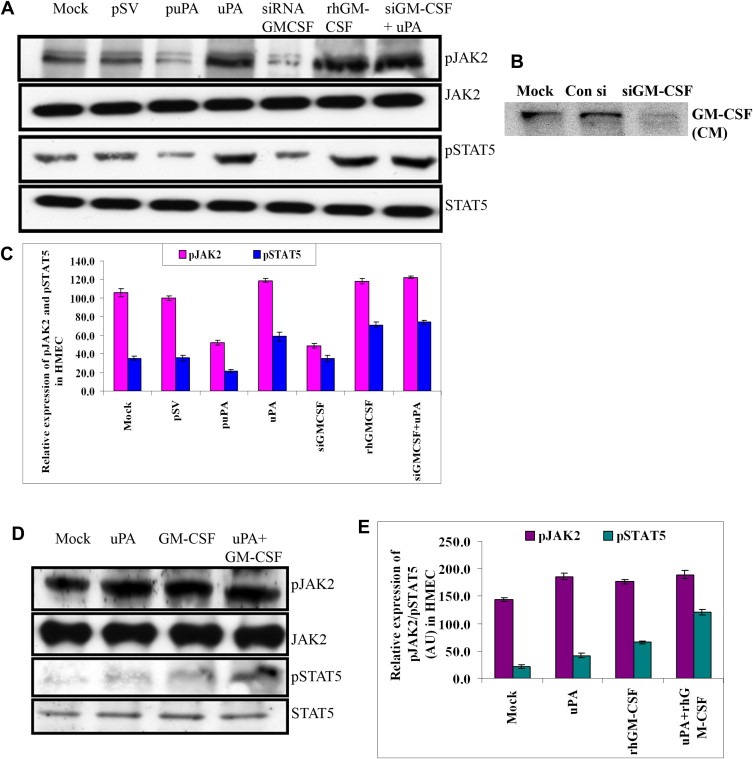
uPA activates JAK2/STAT5 independent of GM‐CSF in HMEC. (A) Lysates were made from HMEC untreated or treated with pSV, puPA, purified uPA, siGM‐CSF, rhGM‐CSF, siGM‐CSF or uPA and immunoblotted for pJAK2, pSTA5, and total forms of JAK2 and STAT5 and with corresponding secondary antibodies. A representative blot of three independent experiments is shown. (B) Conditioned medium from untreated, control si, and siGM‐CSF treated HMEC was immunoblotted with primary antibody to GM‐CSF and corresponding secondary antibody. A representative blot of three independent experiments is shown. (C) Graph represents quantitative representation of the Western blotting results for three independent experiments, p<0.05. (D) Lysates were made from HMEC left untreated or treated with purified uPA, with rh GM‐CSF or both and immunoblotted for phospho and total forms of JAK2 and STAT5. A representative blot of three independent experiments is shown. (E) Graph represents quantitative expression of the Western blotting, p<0.05.
3.6. Secretion of SVEGFR1 by uPA/uPAR downregulation in HMEC/GBM cells is mediated by an indirect MMP‐7‐dependent mechanism and by ectodomain shedding of VEGFr1 due to increased tyrosine phosphorylation at 1213 position
Previous reports (Ito et al., 2009; Rahimi et al., 2009) have suggested that MMP‐7 and phosphorylation of VEGFr1 could enhance SVEGFR1 secretion. To gain further insight into this process, we checked for MMP‐7 levels after addition of rTIMP‐1 in GBM and HMEC. The addition of TIMP‐1 reduced the expression levels of MMP‐7, suggesting that MMP‐7 could have an indirect role in SVEGFR1 secretion. Moreover we observed the reduction in MMP‐7 expression levels up to the 48‐h time point, which suggests that downregulation of MMP‐7 by TIMP‐1, is a stable process (Figure 6A–D). Exogenous TIMP‐1 also enhanced the secretion of SVEGFR1 as shown by Western blotting (Figure 6E–H). To rule out if shRNA against uPA and uPAR results in ectodomain shedding of VEGFr1, we checked for the phosphorylation of VEGFr1 in HMEC, U87MG and 4910 cells. Notably, phosphorylation of VEGFr1 at Y1213 position seems to be enhanced in GBM cells and HMEC in shRNA treatments as compared to controls. This suggests that uPA/uPAR downregulation (Figure 6I and J) augments SVEGFR1 secretion by enhancing pTyr 1213 phosphorylation of VEGFr1 receptor and also indirectly by an MMP‐7‐dependent mechanism. To confirm these results further, we checked the expression of MMP‐7 in HMEC, U87MG and 4910 cells before and after treatment with SV, puPA, puPAR and pU2. Results showed the puPA, puPAR and pU2 inhibited expression of MMP‐7 compared with controls (Figure 6K and L). Taken together, our results suggest that shRNA against uPA and uPAR enhance secretion of SVEGFR1 that is dependent on TIMP‐1 but independent of GM‐CSF.
Figure 6.
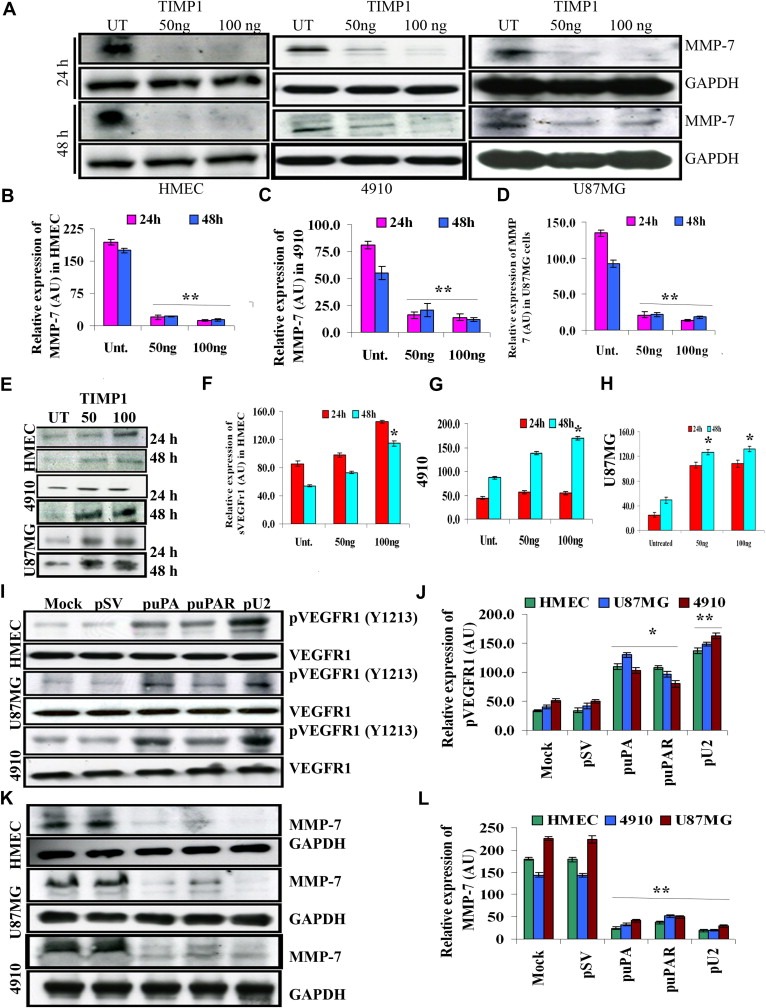
Enhanced secretion of SVEGFR1 by uPA/uPAR downregulation in GBM and HMEC cells is mediated by MMP‐7 and augmented by tyrosine phosphorylation of VEGFr1 at 1213 position. (A) HMEC, U87MG and 4910 cells were left untreated or treated with different concentrations of TIMP‐1 and lysates were made at 24 and 48h and immunoblotted for MMP‐7 with appropriate primary and secondary antibodies. Representative blots from each cell line are shown. The blots were probed for GAPDH to verify equal loading of lysates. (B–D) Graphs represent quantitative representation of the Western blotting results in HMEC, 4910 and U87MG, respectively. (E) Conditioned medium from above‐mentioned conditions were immunoblotted for SVEGFR1. A representative blot from three independent experiments is shown. (F–H) Graphs represent quantitative representation of the Western blotting results in HMEC, U87MG and 4910 cells, respectively. (I) Lysates made from HMEC, U87MG and 4910 cells, which were left untreated or treated with pSV, puPA and pU2 were immunoblotted for pVEGFr1 pTyr 1213 position primary antibody and with appropriate secondary antibodies. A representative blot from three independent experiments is shown. Equal loading of lysates is shown by reprobing the blots and blotting for GAPDH. (J) Graphical representation of quantitative estimation of the Western blotting results. (K) Lysates made from HMEC, U87MG and 4910 cells which were left untreated or treated with pSV, puPA and pU2 were immunoblotted for MMP‐7 primary antibody and with appropriate secondary antibodies. A representative blot from three independent experiments is shown. Equal loading of lysates is shown by reprobing the blots and blotting for GAPDH. (L) Graphical representation of quantitative estimation of the Western blotting results is shown.
3.7. Exogenous addition of SVEGFR1 results in increased inhibition of angiogenesis and inhibits VEGF‐mediated endothelial capillary tube formation
To assess the effect of enhanced secretion of SVEGFR1 on angiogenesis in glioblastoma, tumor conditioned medium (TCM) from pSV‐treated U87MG cells, VEGF alone, pU2‐treated U87MG cells, and pU2 TCM with exogenously added SVEGFR1 and rVEGF were co‐cultured with endothelial cells and evaluated for tube formation by an in vitro angiogenesis assay (Figure 7A). Results show that pSV‐treated cells and VEGF alone treated cells had an increased number of branch points, which led to endothelial tube formation (Figure 7A). In contrast, in the pU2 TCM and pU2 TCM with exogenous SVEGFR1 treatments, tube formation was inhibited (Figure 7A). Addition of rVEGF augmented this inhibition (Figure 7A). The quantitative representation of the percent of HMEC branch points relative to controls is represented in Figure 7B. Taken together, these results suggest that exogenous addition of SVEGFR1 inhibits VEGF‐mediated angiogenesis in glioblastoma and endothelial cells.
Figure 7.
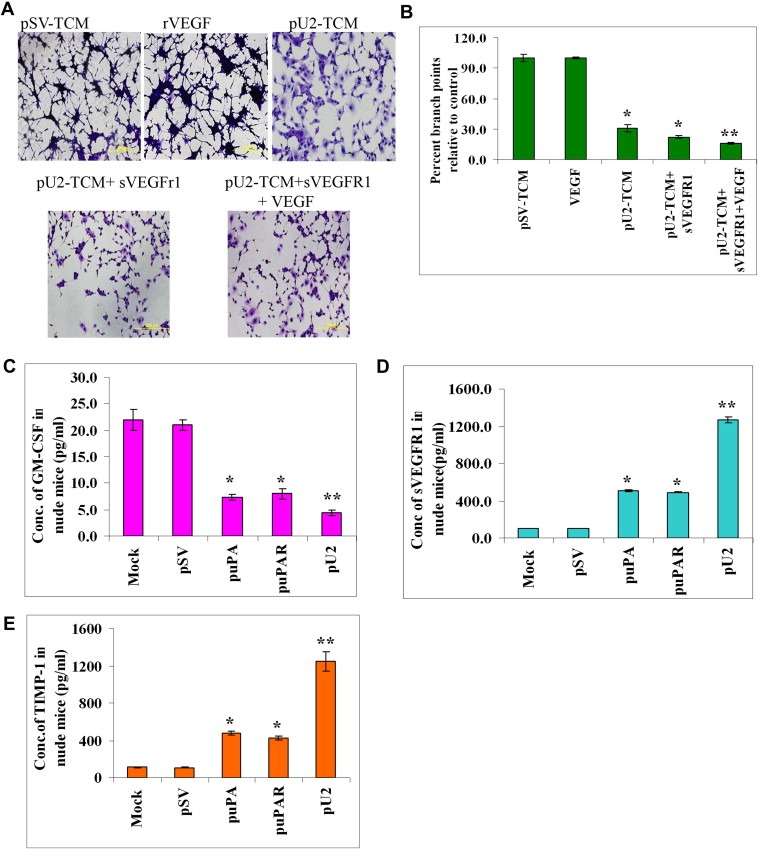
Exogenous addition of SVEGFR1 enhances inhibition of capillary tube formation as shown by in vitro angiogenesis assay. (A) HMECs treated with conditioned media from U87MG cells, which were left untreated or treated with pU2 and/or supplemented with sVEGFR1 and VEGF alone were cultured with the collected conditioned medium in 48‐well plates for 24h. After the incubation period, the medium was removed, and the cells stained with Hema‐3 stain and examined under a microscope. (B) Quantification of angiogenesis in endothelial cells that were left untreated, treated with pU2 conditioned medium, or treated with pU2 conditioned medium supplemented with SVEGFR1. Values are mean±S.D. from three different experiments. (C–E) shRNA against uPA/uPAR inhibits secretion of GM‐CSF and enhances SVEGFR1 and TIMP‐1 secretion in pre‐established intracranial tumors in nude mice. U87MG cells in suspension (2×106 in 10μL serum‐free medium) were injected intracranially in nude mice. One week later, the mice were injected with either pSV or shRNA‐expressing vectors (puPA, puPAR and pU2) using an Alzet mini‐osmotic pump (constructs diluted to 1.5μg/mL in PBS and injected at 0.25μg/h, with 5 mice in each group). After a five‐week follow‐up period, mice were sacrificed, and blood was collected and serum was separated to assay for the levels of mSVEGFR1 (C) and msGM‐CSF (D) and mTIMP‐1 (E) by ELISA according to the manufacturer's instructions.
3.8. shRNA treatments against uPA and/or uPAR inhibit secretion of GM‐CSF and enhance TIMP‐1, SVEGFR1 secretion in pre‐established intracranial tumors in nude mice
We next determined if our in vitro effects would be seen in vivo, so we checked for SVEGFR1, GM‐CSF and TIMP‐1 levels in nude mice with pre‐established intracranial tumors as described previously (Gondi et al., 2004). Mice were sacrificed on the 35th day, and blood was collected to check for levels of GM‐CSF, TIMP‐1 and SVEGFR1. Mice which were given shRNA against uPA and/or uPAR, showed decreased levels of GM‐CSF and increased levels of SVEGFR1 and TIMP‐1 levels in serum at the end of 35th day when compared with the controls. Mice that received the bicistronic construct pU2 showed the highest levels of GM‐CSF inhibition by four folds and increased levels of SVEGFR1 by ten and eight folds of sTIMP‐1. The graph shows the average values of GM‐CSF, SVEGFR1 and TIMP‐1 for each group analyzed (Figure 7C–E). The in vivo results were in accordance with our in vitro results and indicate that shRNA against uPA and/or uPAR could be used as an effective anti‐angiogenesis agent in glioblastoma. The schematic representation of the mechanism by which shRNA against uPA and uPAR inhibits angiogenesis in GBM is described in Figure 8. In depth research to study the role of TIMP‐1, MMP‐7 and sVEGFR1 and its regulation by uPA and uPAR will be carried out in future studies.
Figure 8.
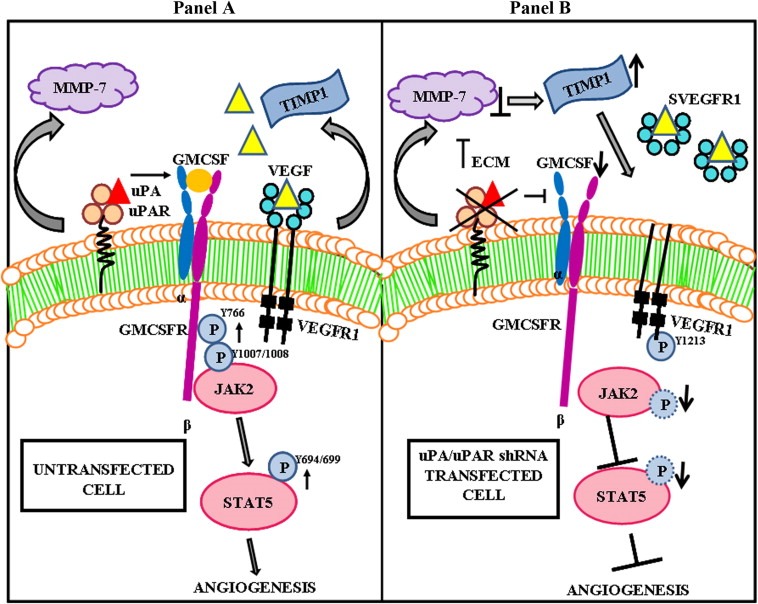
Schematic diagram showing the mechanism by which uPA/uPAR shRNA inhibits angiogenesis in glioblastoma cells and xenografts. Panel (A) Binding of uPA to uPAR co‐ordinates with GM‐CSF receptor to activate JAK2/STAT5 mediated angiogenic signaling pathway. VEGF/VEGFR signaling is also well known to initiate angiogenesis. Panel (B) Knockdown of uPA and uPAR inhibits the angiogenesis signaling induced by both GM‐CSF and VEGF. uPA/uPAR down regulation decreased GM‐CSF secretion resulting in inhibition of JAK2/STAT5 activation. Further knock down of uPA/uPAR increased sVEGFR1 secretion which binds to free bound VEGF and acts as a scavenger inhibiting VEGF‐mediated signaling. sVEGFR1 secretion was mediated by TIMP‐1 and ectodomain shedding of VEGFR1 regulated by phosphorylation at the Tyr1213 of the VEGFR1 receptor.
4. Discussion
This study introduces a novel anti‐angiogenic role for shRNA against uPA and/or uPAR in HMEC, U87MG and 4910 cells. In the present study, we show that the anti‐angiogenic effects of _upa, puPAR and pU2 are mediated by enhanced secretion of SVEGFR1, which is regulated by TIMP‐1. Using both in vitro and in vivo methods, we demonstrate that, unlike in monocytes (Eubank et al., 2004), SVEGFR1 secretion mediated by shRNA against uPA/uPAR in GBM and endothelial cells is independent of GM‐CSF. The rationale for our study originates from our previous study wherein simultaneous downregulation of uPA and uPAR inhibited angiogenesis in GBM and endothelial cells by blocking several factors including GM‐CSF (Raghu et al., 2010). The answer to our question as to whether additional mechanisms are involved in the anti‐angiogenic effects of _upa, puPAR and pU2 led us to screen for SVEGFR1 in tumor conditioned medium. Here, we report that SVEGFR1 levels increased in shRNA‐treated HMEC, U87MG and 4910 cells when compared with untreated and pSV‐treated controls. These data are further supported by the observation of a cumulative increase in SVEGFR1 levels when both uPA and uPAR were downregulated simultaneously rather than individually.
The function of SVEGFR1 as a biomarker in different cancers has been shown in many studies (Hu et al., 2004; Nagaoka et al., 2010; Yamaguchi et al., 2007). A recent study by Harrington et al. (2008) has shown that DLL4, a notch ligand, regulates multiple angiogenic pathways in HUVEC cells, which includes SVEGFR1 that attenuates VEGF‐mediated signaling. Moreover, studies report increased SVEGFR1 levels in colon cancer patients undergoing laparotomy but significance of this finding has not been evaluated (Kirman et al., 2007). Using orthotopic brain tumor models of GBM, direct intracranial delivery of a recombinant adeno‐associated virus (rAAV) serotype 8 vector encoding SVEGFR1/r2 significantly reduced overall tumor volume and increased median survival time (Harding et al., 2006). In accordance with the above‐mentioned studies, our results show that tumor conditioned medium from pU2‐treated U87MG cells prevented the formation of endothelial tube‐like structures and the addition of exogenous SVEGFR1 augmented the inhibitory effect. Further addition of exogenous VEGF did not reverse endothelial tube formation, but enhanced the inhibitory effect, thereby suggesting that SVEGFR1 inhibits VEGF bioavailability. Our in vitro results were similar to our in vivo results where we noticed that the in vivo tumor model in mice treated with shRNA constructs showed reduced level of GM‐CSF in serum and increased levels of SVEGFR1 and sTIMP‐1. Serum samples collected prior to tumor induction did not show any changes in the different levels of SVEGFR1, GM‐CSF and TIMP‐1. Overall, the results of the present study confirm our previous study and those of others, which demonstrate the potential of utilizing uPA and uPAR as indirect targets for inhibition of angiogenesis (Behzadian et al., 2003; Kaneko et al., 2003; Kargiotis et al., 2008a; Mohan et al., 1999; Nagaoka et al., 2010; Subramanian et al., 2006).
So far, many studies have shown that GM‐CSF is known to activate the JAK2/STAT5 pathway in chronic myeloid leukemia and human eosinophils (Stout et al., 2004; Wang et al., 2007). uPAR is known to be associated with the components of the JAK1/STAT1 signaling in human kidney epithelial tumor cell line TCL‐598 (Hampson et al., 1997). Dumler et al. (1998) have shown that uPAR activates the JAK/STAT pathway in human aortic vascular smooth muscle cells. Another study by the same group has shown that urokinase activates JAK1/STAT1 signaling in human vascular endothelial cells (Dumler et al., 1999). As our studies demonstrated the inhibition of GM‐CSFrβ phosphorylation in shRNA‐treated GBM and endothelial cells, our observation that uPA activates JAK2/STAT5 phosphorylation in HMEC in the absence of GM‐CSF suggests an additional novel role for uPA. The exact mechanisms involved in this process are under further investigation as well as whether mTOR (Busch et al., 2009) has a role in this process.
Further analysis of SVEGFR1 secretion by puPA, puPAR and pU2 led us to TIMP‐1 (tissue inhibitor of matrix metalloproteinase‐1), which was increased in shRNA‐treated cells. Previous reports have shown that TIMP‐1 regulates SVEGFR1 in endothelial cells (Bruegmann et al., 2009). These findings led us to hypothesize that puPA, puPAR and pU2 induced TIMP‐1 might be involved in SVEGFR1 secretion in endothelial and glioblastoma cells. Our hypothesis was further confirmed when exogenous rhTIMP‐1 added to serum‐starved HMEC, U87MG and 4910 cells resulted in enhanced secretion of SVEGFR1 at the 24‐h and 48‐h time points.
Tissue inhibitors of metalloproteinases (TIMPs) are described as negative regulators of matrix metalloproteinases (MMPs) that degrade the components of the extracellular matrix and are known to play important roles in invasion, angiogenesis, proliferation, migration and apoptosis (Groft et al., 2001, 1995, 1995, 1994). A study by Nasser et al. (2006) has shown that TIMP‐1 is an inhibitor of high‐grade glioma invasion. These studies further reinforce our observation that puPA/pU2‐induced TIMP‐1 mediates SVEGFR1‐induced VEGF inhibition and angiogenesis in GBM and endothelial cells.
Confirmation of the observed results led us to analyze different mechanisms by which upa/puPAR‐induced TIMP‐1 enhances the secretion of SVEGFR1 in endothelial and glioblastoma cells. MMP‐7, which is involved in cancer invasiveness (Stamenkovic, 2003; Vihinen and Kahari, 2002), degrades SVEGFR1 (Ito et al., 2009). In accordance with previous studies, we observed pU2 TCM‐induced TIMP‐1 inhibited MMP‐7 in HMEC, U87MG and 4910 cells. It may be possible that SVEGFR1 is enhanced by an MMP‐7‐dependent mechanism in GBM and endothelial cells. We plan to carry out an in depth analysis of this mechanism in future studies.
Rahimi et al. (2009) has reported that pro‐angiogenic factors PIGF and VEGF phosphorylate VEGFr1 at Tyr 1213 position, and this tyrosine phosphorylation induces ectodomain shedding resulting in extracellular SVEGFR1 fragment and an intracellular cytoplasmic fragment. In accordance with the present study, shRNA against uPA and uPAR enhanced the phosphorylation of VEGFr1 at Tyr 1213 position suggesting that shRNA against uPA/uPAR could result in ectodomain shedding of VEGFr1.
Growing evidences have shown the importance of uPA/uPAR in regulating angiogenesis both by extracellular proteolytic activity as well as by activation of several intracellular signaling pathways (Binder et al., 2007; D'Alessio and Blasi, 2009; Lakka et al., 2005; Ulisse et al., 2009). The potential of using transcriptional inactivation of uPA and uPAR in the treatment of glioblastoma has been proved in many studies (Behzadian et al., 2003, 2007, 2003, 2008, 2011, 2010). In our previous study Raghu et al. (2010) were able to demonstrate that simultaneous down regulation of uPA and uPAR inhibits angiogenesis in co‐cultures of GBM cells with endothelial cells by inhibition of nuclear localization of angiogenin and angpt1 signaling. uPA/uPAR down regulation in U87 co‐cultures inhibited IL‐6, GM‐CSF, GRO, VEGF, Ang‐1, VEGFR2 and MMP‐2 while in U87SPARC co‐cultures with HMEC, pU2 inhibited ANG, EGF, GRO, IL‐6, MCP‐1, PDGFBB, VEGF, Ang‐1 and VEGFR2. The significance of many of these pro‐angiogenic factors in uPA/uPAR down regulation is being studied in detail in our laboratory. uPA/uPAR down regulation also has an inhibitory effect on invasion of GBM cells and xenografts by a Notch1 mediated mechanism (Raghu et al., 2011). Functional inhibition of uPAR has also been shown to decrease the invasive potential of endothelial cells (Min et al., 1996; Ossowski, 1996). uPA/uPAR down regulation plays important roles in inhibition and angiogenesis of glioblastoma cells and endothelial cells by multiple mechanisms and understanding them will gives us further insight into the potential of this multifunctional proteolytic system.
To the best of our knowledge, this is the first report showing uPA/uPAR shRNA induces SVEGFR1 in GBM and endothelial cells where SVEGFR1 is mediated by TIMP‐1 but is independent of GM‐CSF. At present, we are investigating the involvement of other signaling molecules in this process.
Funding
This research was supported by a grant from National Institutes of Health, CA75557, (to J.S.R.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Shellee Abraham for manuscript preparation and Diana Meister and Sushma Jasti for manuscript review.
Garay Joseph P., Gray Joe W., (2012), uPA and uPAR shRNA inhibit angiogenesis via enhanced secretion of SVEGFR1 independent of GM‐CSF but dependent on TIMP‐1 in endothelial and glioblastoma cells, Molecular Oncology, 6, doi: 10.1016/j.molonc.2011.11.008.
References
- Anderson, J.C. , McFarland, B.C. , Gladson, C.L. , 2008. New molecular targets in angiogenic vessels of glioblastoma tumours. Expert Rev. Mol. Med.. 10, e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behzadian, M.A. , Windsor, L.J. , Ghaly, N. , Liou, G. , Tsai, N.T. , Caldwell, R.B. , 2003. VEGF-induced paracellular permeability in cultured endothelial cells involves urokinase and its receptor. FASEB J.. 17, 752–754. [DOI] [PubMed] [Google Scholar]
- Bene, M.C. , Castoldi, G. , Knapp, W. , Rigolin, G.M. , Escribano, L. , Lemez, P. , 2004. CD87 (urokinase-type plasminogen activator receptor), function and pathology in hematological disorders: a review. Leukemia. 18, 394–400. [DOI] [PubMed] [Google Scholar]
- Binder, B.R. , Mihaly, J. , Prager, G.W. , 2007. uPAR-uPA-PAI-1 interactions and signaling: a vascular biologist's view. Thromb. Haemost.. 97, 336–342. [PubMed] [Google Scholar]
- Blasi, F. , Carmeliet, P. , 2002. uPAR: a versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol.. 3, 932–943. [DOI] [PubMed] [Google Scholar]
- Bruegmann, E. , Gruemmer, R. , Neulen, J. , Motejlek, K. , 2009. Regulation of soluble vascular endothelial growth factor receptor 1 secretion from human endothelial cells by tissue inhibitor of metalloproteinase 1. Mol. Hum. Reprod.. 15, 749–756. [DOI] [PubMed] [Google Scholar]
- Busch, S. , Renaud, S.J. , Schleussner, E. , Graham, C.H. , Markert, U.R. , 2009. mTOR mediates human trophoblast invasion through regulation of matrix-remodeling enzymes and is associated with serine phosphorylation of STAT3. Exp. Cell Res.. 315, 1724–1733. [DOI] [PubMed] [Google Scholar]
- Choong, P.F. , Nadesapillai, A.P. , 2003. Urokinase plasminogen activator system: a multifunctional role in tumor progression and metastasis. Clin. Orthop. Relat. Res.. S46–S58. [DOI] [PubMed] [Google Scholar]
- D'Alessio, S. , Blasi, F. , 2009. The urokinase receptor as an entertainer of signal transduction. Front Biosci.. 14, 4575–4587. [DOI] [PubMed] [Google Scholar]
- Duffy, M.J. , Duggan, C. , 2004. The urokinase plasminogen activator system: a rich source of tumour markers for the individualised management of patients with cancer. Clin. Biochem.. 37, 541–548. [DOI] [PubMed] [Google Scholar]
- Dumler, I. , Kopmann, A. , Weis, A. , Mayboroda, O.A. , Wagner, K. , Gulba, D.C. , 1999. Urokinase activates the Jak/Stat signal transduction pathway in human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol.. 19, 290–297. [DOI] [PubMed] [Google Scholar]
- Dumler, I. , Weis, A. , Mayboroda, O.A. , Maasch, C. , Jerke, U. , Haller, H. , 1998. The Jak/Stat pathway and urokinase receptor signaling in human aortic vascular smooth muscle cells. J. Biol. Chem.. 273, 315–321. [DOI] [PubMed] [Google Scholar]
- Eubank, T.D. , Galloway, M. , Montague, C.M. , Waldman, W.J. , Marsh, C.B. , 2003. M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J. Immunol.. 171, 2637–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubank, T.D. , Roberts, R. , Galloway, M. , Wang, Y. , Cohn, D.E. , Marsh, C.B. , 2004. GM-CSF induces expression of soluble VEGF receptor-1 from human monocytes and inhibits angiogenesis in mice. Immunity. 21, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubank, T.D. , Roberts, R.D. , Khan, M. , Curry, J.M. , Nuovo, G.J. , Kuppusamy, P. , 2009. Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res.. 69, 2133–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondi, C.S. , Lakka, S.S. , Dinh, D.H. , Olivero, W.C. , Gujrati, M. , Rao, J.S. , 2004. RNAi-mediated inhibition of cathepsin B and uPAR leads to decreased cell invasion, angiogenesis and tumor growth in gliomas. Oncogene. 23, 8486–8496. [DOI] [PubMed] [Google Scholar]
- Gondi, C.S. , Lakka, S.S. , Dinh, D.H. , Olivero, W.C. , Gujrati, M. , Rao, J.S. , 2007. Intraperitoneal injection of an hpRNA-expressing plasmid targeting uPAR and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin. Cancer Res.. 13, 4051–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groft, L.L. , Muzik, H. , Rewcastle, N.B. , Johnston, R.N. , Knauper, V. , Lafleur, M.A. , 2001. Differential expression and localization of TIMP-1 and TIMP-4 in human gliomas. Br. J. Cancer. 85, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson, R. , Humbert, O. , Macpherson, P. , Aquilina, G. , Karran, P. , 1997. Mismatch repair defects and O6-methylguanine-DNA methyltransferase expression in acquired resistance to methylating agents in human cells. J. Biol. Chem.. 272, 28596–28606. [DOI] [PubMed] [Google Scholar]
- Harding, T.C. , Lalani, A.S. , Roberts, B.N. , Yendluri, S. , Luan, B. , Koprivnikar, K.E. , 2006. AAV serotype 8-mediated gene delivery of a soluble VEGF receptor to the CNS for the treatment of glioblastoma. Mol. Ther.. 13, 956–966. [DOI] [PubMed] [Google Scholar]
- Harrington, L.S. , Sainson, R.C. , Williams, C.K. , Taylor, J.M. , Shi, W. , Li, J.L. , 2008. Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc. Res.. 75, 144–154. [DOI] [PubMed] [Google Scholar]
- Hu, Q. , Dey, A.L. , Yang, Y. , Shen, Y. , Jilani, I.B. , Estey, E.H. , 2004. Soluble vascular endothelial growth factor receptor 1, and not receptor 2, is an independent prognostic factor in acute myeloid leukemia and myelodysplastic syndromes. Cancer. 100, 1884–1891. [DOI] [PubMed] [Google Scholar]
- Ikenaka, Y. , Yoshiji, H. , Kuriyama, S. , Yoshii, J. , Noguchi, R. , Tsujinoue, H. , 2003. Tissue inhibitor of metalloproteinases-1 (TIMP-1) inhibits tumor growth and angiogenesis in the TIMP-1 transgenic mouse model. Int. J. Cancer. 105, 340–346. [DOI] [PubMed] [Google Scholar]
- Irigoyen, J.P. , Munoz-Canoves, P. , Montero, L. , Koziczak, M. , Nagamine, Y. , 1999. The plasminogen activator system: biology and regulation. Cell Mol. Life Sci.. 56, 104–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, T.K. , Ishii, G. , Saito, S. , Yano, K. , Hoshino, A. , Suzuki, T. , 2009. Degradation of soluble VEGF receptor-1 by MMP-7 allows VEGF access to endothelial cells. Blood. 113, 2363–2369. [DOI] [PubMed] [Google Scholar]
- Kaneko, T. , Konno, H. , Baba, M. , Tanaka, T. , Nakamura, S. , 2003. Urokinase-type plasminogen activator expression correlates with tumor angiogenesis and poor outcome in gastric cancer. Cancer Sci.. 94, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargiotis, O. , Chetty, C. , Gogineni, V. , Gondi, C.S. , Pulukuri, S.M. , Kyritsis, A.P. , 2008. uPA/uPAR downregulation inhibits radiation-induced migration, invasion and angiogenesis in IOMM-Lee meningioma cells and decreases tumor growth in vivo. Int. J. Oncol.. 33, 937–947. [PMC free article] [PubMed] [Google Scholar]
- Kargiotis, O. , Chetty, C. , Gondi, C.S. , Tsung, A.J. , Dinh, D.H. , Gujrati, M. , 2008. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene. 27, 4830–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirman, I. , Belizon, A. , Balik, E. , Feingold, D. , Arnell, T. , Horst, P. , 2007. Perioperative sargramostim (recombinant human GM-CSF) induces an increase in the level of soluble VEGFR1 in colon cancer patients undergoing minimally invasive surgery. Eur. J. Surg. Oncol.. 33, 1169–1176. [DOI] [PubMed] [Google Scholar]
- Kunigal, S. , Gondi, C.S. , Gujrati, M. , Lakka, S.S. , Dinh, D.H. , Olivero, W.C. , 2006. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. Int. J. Oncol.. 29, 1349–1357. [PMC free article] [PubMed] [Google Scholar]
- Lakka, S.S. , Gondi, C.S. , Rao, J.S. , 2005. Proteases and glioma angiogenesis. Brain Pathol.. 15, 327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, T. , Claesson-Welsh, L. , 2001. VEGF receptor signal transduction. Sci. STKE. 2001, RE21 [DOI] [PubMed] [Google Scholar]
- Min, H.Y. , Doyle, L.V. , Vitt, C.R. , Zandonella, C.L. , Stratton-Thomas, J.R. , Shuman, M.A. , 1996. Urokinase receptor antagonists inhibit angiogenesis and primary tumor growth in syngeneic mice. Cancer Res.. 56, 2428–2433. [PubMed] [Google Scholar]
- Mohan, P.M. , Lakka, S.S. , Mohanam, S. , Kin, Y. , Sawaya, R. , Kyritsis, A.P. , 1999. Downregulation of the urokinase-type plasminogen activator receptor through inhibition of translation by antisense oligonucleotide suppresses invasion of human glioblastoma cells. Clin. Exp. Metastasis. 17, 617–621. [DOI] [PubMed] [Google Scholar]
- Mohanam, S. , Wang, S.W. , Rayford, A. , Yamamoto, M. , Sawaya, R. , Nakajima, M. , 1995. Expression of tissue inhibitors of metalloproteinases: negative regulators of human glioblastoma invasion in vivo. Clin. Exp. Metastasis. 13, 57–62. [DOI] [PubMed] [Google Scholar]
- Nagaoka, S. , Yoshida, T. , Akiyoshi, J. , Akiba, J. , Hisamoto, T. , Yoshida, Y. , 2010. The ratio of serum placenta growth factor to soluble vascular endothelial growth factor receptor-1 predicts the prognosis of hepatocellular carcinoma. Oncol. Rep.. 23, 1647–1654. [DOI] [PubMed] [Google Scholar]
- Nakagawa, T. , Kubota, T. , Kabuto, M. , Sato, K. , Arai, Y. , Kodera, T. , 1995. Production of tissue inhibitor of metalloproteinases-1 (TIMP-1) by human astrocytic tumors. Neurol. Med. Chir. (Tokyo). 35, 728–731. [DOI] [PubMed] [Google Scholar]
- Nakagawa, T. , Kubota, T. , Kabuto, M. , Sato, K. , Kawano, H. , Hayakawa, T. , 1994. Production of matrix metalloproteinases and tissue inhibitor of metalloproteinases-1 by human brain tumors. J. Neurosurg.. 81, 69–77. [DOI] [PubMed] [Google Scholar]
- Nasser, J.A. , Falavigna, A. , Ferraz, F. , Duigou, G. , Bruce, J. , 2006. Transcription analysis of TIMP-1 and NM23-H1 genes in glioma cell invasion. Arq. Neuropsiquiatr.. 64, 774–780. [DOI] [PubMed] [Google Scholar]
- Norden, A.D. , Drappatz, J. , Wen, P.Y. , 2008. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol.. 7, 1152–1160. [DOI] [PubMed] [Google Scholar]
- Ossowski, L. , 1996. Effect of antisense inhibition of urokinase receptor on malignancy. Curr. Top. Microbiol. Immunol.. 213, 101–112. [DOI] [PubMed] [Google Scholar]
- Park, J.E. , Keller, G.A. , Ferrara, N. , 1993. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol. Biol. Cell. 4, 1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, X.H. , Nakanishi, Y. , Takayama, K. , Bai, F. , Hara, N. , 1999. Granulocyte, granulocyte-macrophage, and macrophage colony-stimulating factors can stimulate the invasive capacity of human lung cancer cells. Br. J. Cancer. 79, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper, M.S. , 2001. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol.. 21, 1104–1117. [DOI] [PubMed] [Google Scholar]
- Raghu, H. , Gondi, C.S. , Dinh, D.H. , Gujrati, M. , Rao, J.S. , 2011. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch -1 receptor. Mol. Cancer. 10, 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu, H. , Lakka, S.S. , Gondi, C.S. , Mohanam, S. , Dinh, D.H. , Gujrati, M. , 2010. Suppression of uPA and uPAR attenuates angiogenin mediated angiogenesis in endothelial and glioblastoma cell lines. PLoS One. 5, e12458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi, N. , Golde, T.E. , Meyer, R.D. , 2009. Identification of ligand-induced proteolytic cleavage and ectodomain shedding of VEGFR-1/FLT1 in leukemic cancer cells. Cancer Res.. 69, 2607–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldi, R. , Primo, L. , Brizzi, M.F. , Sanavio, F. , Aglietta, M. , Polentarutti, N. , 1997. Activation of JAK2 in human vascular endothelial cells by granulocyte-macrophage colony-stimulating factor. Blood. 89, 863–872. [PubMed] [Google Scholar]
- Stamenkovic, I. , 2003. Extracellular matrix remodelling: the role of matrix metalloproteinases. J. Pathol.. 200, 448–464. [DOI] [PubMed] [Google Scholar]
- Stout, B.A. , Bates, M.E. , Liu, L.Y. , Farrington, N.N. , Bertics, P.J. , 2004. IL-5 and granulocyte-macrophage colony-stimulating factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3 protein expression in human eosinophils. J. Immunol.. 173, 6409–6417. [DOI] [PubMed] [Google Scholar]
- Subramanian, R. , Gondi, C.S. , Lakka, S.S. , Jutla, A. , Rao, J.S. , 2006. siRNA-mediated simultaneous downregulation of uPA and its receptor inhibits angiogenesis and invasiveness triggering apoptosis in breast cancer cells. Int. J. Oncol.. 28, 831–839. [PMC free article] [PubMed] [Google Scholar]
- Ulisse, S. , Baldini, E. , Sorrenti, S. , D'Armiento, M. , 2009. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr. Cancer Drug Targets. 9, 32–71. [DOI] [PubMed] [Google Scholar]
- Vihinen, P. , Kahari, V.M. , 2002. Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int. J. Cancer. 99, 157–166. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Cai, D. , Brendel, C. , Barett, C. , Erben, P. , Manley, P.W. , 2007. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood. 109, 2147–2155. [DOI] [PubMed] [Google Scholar]
- Wognum, A.W. , Westerman, Y. , Visser, T.P. , Wagemaker, G. , 1994. Distribution of receptors for granulocyte-macrophage colony-stimulating factor on immature CD34+ bone marrow cells, differentiating monomyeloid progenitors, and mature blood cell subsets. Blood. 84, 764–774. [PubMed] [Google Scholar]
- Yamaguchi, T. , Bando, H. , Mori, T. , Takahashi, K. , Matsumoto, H. , Yasutome, M. , 2007. Overexpression of soluble vascular endothelial growth factor receptor 1 in colorectal cancer: association with progression and prognosis. Cancer Sci.. 98, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]