

Abstract
Misfolding and aggregation of prion proteins is linked to a number of neurodegenerative disorders such as Creutzfeldt-Jacob disease (CJD) and its variants: Kuru, Gerstmann-Straussler-Scheinker syndrome and fatal familial insomnia. In prion diseases, infectious particles are proteins that propagate by transmitting a misfolded state of a protein, leading to the formation of aggregates and ultimately to neurodegeneration. Prion phenomenon is not restricted to humans. There are a number of prion-related diseases in a variety of mammals, including bovine spongiform encephalopathy (BSE, also known as “mad cow disease”) in cattle. All known prion diseases, collectively called transmissible spongiform encephalopathies (TSEs), are untreatable and fatal. Prion proteins were also found in some fungi where they are responsible for heritable traits. Prion proteins in fungi are easily accessible and provide a powerful model for understanding the general principles of prion phenomenon and molecular mechanisms of mammalian prion diseases. Presently, several fundamental questions related to prions remain unanswered. For example, it is not clear how prions cause the disease. Other unknowns include the nature and structure of infectious agent and how prions replicate. Generally, the phenomenon of misfolding of the prion protein into infectious conformations that have the ability to propagate their properties via aggregation is of significant interest. Despite the crucial importance of misfolding and aggregation, very little is currently known about the molecular mechanisms of these processes. While there is an apparent critical need to study molecular mechanisms underlying misfolding and aggregation, the detailed characterization of these single molecule processes is hindered by the limitation of conventional methods. Although some issues remain unresolved, much progress has been recently made primarily due to the application of nanoimaging tools. The use of nanoimaging methods shows great promise for understanding the molecular mechanisms of prion phenomenon, possibly leading toward early diagnosis and effective treatment of these devastating diseases. This review article summarizes recent reports which advanced our understanding of the prion phenomenon through the use of nanoimaging methods.
Key words: protein misfolding, prion, atomic force microscopy, nanomedicine, force spectroscopy
Structure of Amyloid
Significant progress has been made in understanding the structure of aggregated species at the atomic level using both computational modeling and experimental studies such as X-ray crystallography and solid-state NMR. It is widely accepted that the infectious form of prions is self-propagating fibrillar or amyloid PrP.1 The formation of amyloid has been tightly linked to the development of the disease. Amyloids share common structural features and biophysical properties, including binding of specific dyes, elongated morphology and nucleation-dependent kinetics of aggregation. Based on solid-state NMR and X-ray crystallography data, the underlying structure of prion amyloids was found to share common structural features. They contain similarly arranged parallel β-sheets packed in register for such different prion proteins as Ure2p1-89 (the basis of the [URE3] prion),2 Ure2p10-39,3 Sup35p ([PSI+] prion),4 Rnq1p ([PIN+] prion).5 Generally, beta-strands of a protein molecule run perpendicular to the axis of the fibril and hydrogen bonding in such structures occurs along the fibril axis. Other models, however, need to be considered as some reports suggest that the parallel in-register structural model of amyloids failed to explain experimental observation of the diversity of strain-specific morphologies of the Sup35 amyloid fibrils.6
Small Elements Govern Prion Aggregation
The above-described theme of structural organization of fibrils seems rather common in the formation of amyloid. Short segments of other non-prion amyloid proteins—amyloid-β,7,8 α-synuclein,9,10 islet amyloid polypeptide (IAPP),11 lysozyme12 and β2-microglobulin13—are capable of forming amyloid fibrils. It is reasonable to assume that the nature of amyloidogenic regions may be deduced from the repeating folding motif in the amyloid fibril. Increasing evidence indicates more and more that the building blocks of amyloid fibrils are short fragments of the entire protein sequence. This suggests that the aggregation may begin with only a short segment of the entire protein sequence. Any change in environmental condition that: (1) opens a native protein conformation, exposing this part of the sequence, and/or (2) stimulates conformational change to an aggregation-prone state, could facilitate the aggregation process. The above-mentioned examples support recently proposed amyloid stretch hypothesis. The hypothesis suggests that a highly amyloidogenic short amino acid stretch may trigger the self-assembly process of a protein into aggregates.14,15 Interactions between specific protein regions facilitate the next step in the formation of a misfolded conformation. These regions are known as aggregation “hot regions”,16,17 which are short stretches of a primary protein sequence.
An elegant approach to recognize such small elements of a primary sequence that nucleate and promote aggregation of prion protein Sup35 was recently utilized.18 An array of overlapping peptides 20 amino acid in length derived from the sequence of the prion Sup35 were covalently attached to a glass slide. Peptides were printed onto the surface 10–12 nm apart from each other, providing high resolution and restricting self-interactions. These peptide arrays were incubated with fluorescently labeled full length NM Sup35. The accumulation of fluorescence was observed after 2 hours of incubation at a very small set of imprinted peptides amino acids 9–39 and 90–120. These sequences lie within the regions which were previously identified to be in self-contact within mature fibrils.19 Such a nanoarray approach indicated that these small regions are not only sites of intermolecular contact in mature fibrils, but they also govern self-recognition within soluble non-prion conformers with high specificity. Therefore, small stretches of protein sequence are sufficient to drive the conversion of full-length NM from non-prion state into self-templating amyloid conformation.18 Sometimes a complex interaction among small recognition elements is required for prion propagation. For example, four QN rich domains of Rnq1 can independently transmit the prion state but act cooperatively to attain the final prion conformation.20 The existence of transmission barriers also is affected by the cooperative action of all four prion domains of Rnq1.20 Several other lines of evidence also point to complex interactions within prion protein for the formation of strains. Ohhashi et al.21 recently reported that transient non-native interactions outside the recognition elements are important in determining temperature-dependent strain conformations of Sup35.
Direct Observation of Fibril Growth
Although convincing structural models of amyloid fibrils have recently emerged, the knowledge of precise kinetic mechanisms of fibril formation would greatly benefit our understanding of the prion phenomenon. Novel tools which enable the direct observation of individual fibril growth are important not only for obtaining further insight into the mechanism of protein self-association but also for identifying compounds that inhibit fibril growth. Recent advances in single molecule methods offered by the atomic force microscopy and single molecule fluorescence microscopy allowed the monitoring of aggregation at the level of individual fibrils providing information about the rate and direction of growth.
The single-fibril growth of NM Sup35 was examined using total internal reflection fluorescence microscopy (TIRF).22 Cy5-labeled seeds were attached to a glass slide via biotin-streptavidin linkage. The Cy3-labeled monomer NM was added to the solution. The addition of individual Cy3-labeled molecules could be readily detected at the ends of Cy5-labeled fibrils. Using such an experimental setup, where measurements could follow the fluorescence intensities of the addition events, made it possible to determine how many molecules are added during a single event. The data established that the fibril growth proceeds via addition of NM monomers onto fibril ends, providing additional evidence for the mechanism of fibril growth. However, such experiments required labeling of proteins with dye molecules, and the introduction of the labels might interfere with aggregate formation and/or measurement.
Another method has been introduced to monitor the formation process of amyloid fibrils without labeling of a protein molecule.23 The use of amyloid-fibril specific dye Thioflavin T (ThT) fluorescence in combination with total internal reflection fluorescence microscopy (TIRFM) enabled a direct observation of the amyloid fibril growth in real time.23–25 No additional fluorophore labeling was required to visualize seed-dependent amyloid fibril growth of β-2m,23 or Aβ1–40,24,25 at the single fibril level.
Another useful tool for direct observation of amyloid fibril growth which has been widely used is AFM—atomic force microscopy. The main advantage of AFM is its ability to monitor ongoing processes under various solution conditions without any extra labeling required. Recently, AFM was successfully used to monitor the self-assembly of several amyloidogenic proteins,26,27 including amylin and amyloid-beta.28,29 An assembly of individual fibrils formed by amyloid beta-25–35 was studied by a modified application of the atomic force microscopy, scanning force kymography.30 This method eliminated the need to scan the whole image by scanning fibrils only. Such an improvement significantly increased the temporal resolution with which the observation of single fibril elongation process is measured. Using this method, Kellermayer et al.30 have determined that the assembly of the fibril is polarized and discontinuous. The amyloid assembly proceeded via rapid bursts of growth that extended the fibril by 7 nm or its integer multiples.30 The growth phases were followed by pauses in a stepwise manner, suggesting a fluctuation of amyloid states in which growth may be either permitted or inhibited.
A single fiber growth assay was used to examine the heterogeneity of amyloid fibrils formed by the yeast Sup35 prion protein. This assay used two variants of the Sup35 that were differentially labeled and distinguished by AFM. The two variants differed in their ability to bind an antibody, providing a means of identification of one type of the protein by AFM because binding the antibody increases thickness of the fiber 3-fold. This study found that Sup35 spontaneously forms multiple and distinct fibril types. These types differ by the degree of polarity and overall growth rate, suggesting a great variation in properties of the fibril. The observed diversity seems to be well-suited to account for the range of prion strain phenotypes.31
Polymorphism and Prion Strains
Amyloid fibrils generally exhibit similar appearance in a form of elongated, unbranched structures. Several unrelated proteins such as insulin, amyloid beta and amylin can form almost identical morphologies. Aggregated prion proteins also exhibit similar amyloid fibril appearances. Despite the general similarity of aggregated species, often a mixture of fibrils with various morphologies is formed. Moreover, fibrils of the same polypeptide sequence can form distinct morphologies exhibiting a great wealth of polymorphism.
Figure 1 shows AFM images of fibrils formed by the CGNNQQNY peptide in different environmental conditions. Simple variations in the environment such as pH and ionic strength of solution, temperature and mechanical stress (for example, agitation) can result in the formation of fibrils with different characteristics. Moreover, prion proteins are capable of forming aggregates which differ in their patterns of protease resistance and transmissibility. The differences in these properties of aggregates is generally referred to as prion strains. In transmissible spongiform encephalopathies, distinct phenotypes of a disease correspond to different prion strains.32
Figure 1.

Scheme illustrating that a protein molecule can adopt several conformational states which may result in aggregates of different morphologies. AFM images of CGNNQQNY peptide from Sup35 yeast prion protein aggregated with the formation of fibrils of distinct morphologies at different conditions. Scale bar is 500 nm.
The phenomenon of prion strains implies that there exists an ensemble of distinct propagating conformations for a single polypeptide sequence. Therefore, aggregation of polypeptide might produce several distinct polymorphs depending on initial misfolded conformation. The dynamic nature of the conformational space, the complexity of relationship between strains and the potential to generate a large number of new prion strains poses a critical need for addressing these issues. The great wealth of polymorphism and diversity of prion strains present a serious risk for public health.33
Individual fibrils are generally considered to be structurally uniform, maintaining the same structure over the length of the fibrils. It is believed that the morphology and structure of preformed seeds is propagated in a template-like manner, passing the characteristics of template—morphology, structure and infectivity—to the next generation of aggregates.34–36 Such heritability is so strong that seeds propagate their characteristics even when conditions are changed to favor different conformational states.37
Recent reports, however, suggest that fibrillar structures are composed of protein molecules that have different secondary structures.38,39 A critical need for identifying (probing) the conformations within heterogeneous samples has been recently highlighted.39 It is becoming clear that there is a coexistence of multiple strains or subtypes of the disease-related proteins in aggregates. Such conformational heterogeneity may result in “various diseases.”
A novel immunoconformational assay (or dual color assay) based on specific antibodies labeled with different dyes has been utilized to reveal structural heterogeneity of PrP prion protein within a single fibril.39 This study showed the existence of the conformational heterogeneity across the population of fibrils as well as within individual fibrils.
Recent X-ray crystallography and NMR structural studies emerged to suggest strongly the origin of polymorphic variation. Multiple layers of beta-sheets are closely packed to create amyloid structures. Differences in the packing of these beta-sheets, originating from side-chain packing, register or topology of beta-sheets, may explain morphological variants of fibrillar structure. Solid-state NMR studies clearly showed the coexistence of three distinct conformations of the GNNQQNY peptide within a single fibril.38 Different packing arrangements of peptides within a fibril were proposed to be responsible for the observed differences in NMR chemical shifts. These coexisting conformations differ by the degree of local secondary structure (alpha helical versus beta sheet).38 Recent crystallographic studies have identified several (8) classes of so-called steric zippers.9 Despite the fundamental similarities in the structure with extended protein strands that are perpendicular to the axis of beta sheets, these classes vary in the basic steric zipper structure. Such variation involves mainly orientation of peptides and beta-sheets with respect to each other. It was found that some peptides were capable of forming different polymorphs with regard to their basic steric zipper structure, offering a possible explanation for amyloid polymorphism and prion strains.9
Due to the difficulty of determining the structure of amyloids and the potential for structural heterogeneity within a single fibril, one appreciates the development of novel methods that could provide information about the morphology of aggregates as well as their underlying conformational differences. These methods are currently emerging as a combination of AFM and vibrational spectroscopy (infrared absorption or Raman spectroscopy) capable of recognizing the secondary structure of proteins. IR and Raman scattering have recently found a wide application in characterization of amyloids.40–43 One of the biggest advantages of such combined methods is that vibrational properties can be observed simultaneously with the topographic image. An additional advantage is that the probing of heterogeneous sample is possible.
One recent illustrative example is a combination of AFM and near-field infrared microscopy.44,45 Apertureless near-field scanning infrared microscopy (ANSIM) was utilized to study the morphology and secondary structure of individual amyloid fibrils of beta-2 microglobulin. Coupling near-field infrared spectroscopy with AFM allowed the correlation of fibril morphology and underlying secondary structures. It was observed that the sample exhibited a heterogeneous population of fibrils: some fibrils had parallel and some anti-parallel beta sheet arrangements.
One disadvantage of ASNIM is its limitation to only a few selected wavelengths. This can yield to misleading interpretations if an image contrast of only one or two selected wavelengths is considered. Overlapping features of unexpected bands, slight bands with changes or shifts can therefore easily spoil an interpretation. In order to overcome this shortcoming, a full spectrum is required, but this is only possible with sophisticated equipment at synchrotron sources.
A related approach that allows studies of the secondary structure of fibrils is realized by tip-enhanced Raman scattering (TERS).46–48 In TERS, an AFM or STM (scanning tunneling microscope) is combined with a standard Raman microscope in order to exceed the diffraction limit and to increase the sensitivity of conventional Raman spectroscopy. This is realized by attaching a single silver or gold nanoparticle to the apex of an AFM tip or by utilizing an etched wire. Similar to the ASNIM approach, vibrational modes of molecules under the tip experience a signal enhancement of several orders of magnitude. For an overview, see ref. 49. Given that TERS is a scattering type of spectroscopy, a single excitation frequency yields a full spectrum. Hence, overlapping bands of unexpected compounds can be distinguished easily by multivariate statistics, for instance. This renders TERS a highly sensitive method, with even single-molecule sensitivity.50,51 Simultaneously to spectra acquisition with a lateral resolution determined by the nanoparticle dimensions, the topography of the sample also is mapped with a spatial resolution comparable to standard AFM technology. Furthermore, TERS operates label-free and non-destructive, which means that the samples can be used for a subsequent analysis.
Figure 2A shows a typical TERS setup. Three spectra of a single amyloid fibril formed from a CGNNQQNY peptide (fragment of Sup35 yeast prion protein) at pH 5.6 are selected and shown in Figure 2B. The spectra were recorded at equidistant (7 nm) positions perpendicular to the main fibril axis. Since the peptide had only a short sequence of eight amino acids, all spectra in Figure 2B are similar. The specific positions of the amide I and amide III bands provide evidence of a β-sheet structure that is stable in this area of the fibril. For fibrils with varying secondary structures, TERS enables the precise localization and assignment of different conformations.
Figure 2.

(A) Sketch of a back reflection TERS setup, (B) TERS spectra of a fibril formed by CGNNQQNY peptide from Sup35 yeast prion protein on adjacent points separated by 7 nm.
From previous TERS studies on amino acids it is known that distinct amino acids are distinguishable by specific spectral features.52,53 In the present experiment, an assignment of cysteine (C) and tyrosine (Y) is possible and corresponding signals are marked in the spectrum. Such an assignment can deliver valuable information if the fibril was generated from various or even unknown peptides.
The Nature of an Infectious Agent
The relationship between prion propagation phenomenon and the formation of amyloid fibrils is poorly understood. Most known amyloid-forming proteins are not prions and even amyloids of prion proteins are not always transmissible.54–56 There is still a poor understanding of why changes in the conformation of misfoded proteins can alter their physiological effects. Recently proposed hypothesis of “frangibilty” of amyloid fibrils offered an explanation that might clarify this complicated issue.57 The mechanical properties of amyloid and amyloid's propensity to break, generating new seeds, seem to define their physiological impact.57 Tanaka et al.57 looked at the differences in three conformations (strains) of prion protein Sup35 formed at 4°C (Sc4), 37°C (Sc37) and room temperature (SCS). These three prion strains differ by their polymerization rates and cause distinct color phenotypes in the ADE color assay. An AFM-based single-fiber growth assay was used to measure fiber growth rates for these three distinct conformations. It was found that despite the fact that Sc4 leads to the strongest strain phenotype, it has the slowest intrinsic fiber growth of the three conformers. The physical strength of the conformers turned out to be different, too. The slower growth of Sc4 is accompanied by a greater degree of fragmentation upon stirring. AFM imaging revealed that stirring shattered Sc4 amyloids into many small fibrils (less than 100 nm). Such treatment also increased seeding efficiency consistent with larger number of fibril ends. The variability in the brittleness of the fibrils was proposed to represent a major mechanism by which the strength of prion strain phenotypes is determined by conformational state of the protein.57 It appears that, in vivo, the Hsp104 chaperone helps to fragment fibrils of Sup35 prion protein, and it is also responsible for disaggregating large protein deposits. This chaperone may play an important role in propagation of prion because deletion of Hsp104 has been demonstrated to eliminate the infectivity of prions (reviewed in ref. 58).
Recent studies established a strong correlation between the incubation period of prion disease and conformational stability of synthetic prion.59,60 The conformational stability of a synthetic prion formed from a full-length mammalian prion protein was also found to correlate with the smallest possible size of the fragments.61 Less stable conformations produced smaller pieces of fibrils upon fragmentation, providing additional evidence that fragmentation may be important in transmissibility of prion aggregates and prion disease development. Sun et al.61 also showed that αB-crystallin, a heat shock protein (sHsp), is capable of fragmenting rPrP fibrils.61 An alternative hypothesis for the origin of fibrils by their interactions with a surface of cellular membrane has been proposed.61
Protein aggregation is a complex process which proceeds via many intermediate states, including oligomers, protofibrils and fibrils. There is increasing evidence that the end product of aggregation—the formation of fibrils and big aggregates—is not the main cause of the diseases. There exists an uncertainty about what species in this complex aggregation pathway are responsible for this toxicity. Although the infectivity of prions is associated with a wide range of aggregated states, small oligomers of aggregated prion proteins have been suggested as minimal infectious particles. A recent study revealed that the infectivity of PrP is the greatest for particles 17–27 nm in size, corresponding to 300–600 kDa.62 These oligomeric particles should consist of 14 to 28 prion monomers. The infectivity is decreased for larger aggregates and virtually absent for oligomeric fraction of <5 molecules.62
The above-described experimental observations support even more strongly the new emerging view that infectious and toxic species of prion might have different identities1 with distinct biophysical properties.55 Although amyloid forms of PrP are infectious, they might not be toxic.55 An increasingly accumulating body of evidence suggests that oligomeric assemblies of amyloid proteins are toxic species,63–67 while large aggregates and fibrils are rather inert or even protective.68
Characterization of oligomeric species with conventional, ensemble based methods is challenging due to dynamic and transient nature of oligomers. Single molecule methods can provide unique information on the structure, energetics and dynamics of intermediate transient species formed during initial stages of aggregation. Recently, a dual-color, single-molecule fluorescence technique has been utilized to look at the early stages of aggregation of the SH3 domain of PI3 kinase during aggregation lag and growth phases.69 The study found that the oligomeric species formed in the aggregation reaction comprise a heterogeneous ensemble of oligomers with the median size of 38 ± 10 molecules. Interestingly, this stage of aggregation reaction has been previously found to be maximally cytotoxic. The innovative character of the study is the application of TCCD—two-color coincidence detection—to study the protein self-assembly process. Protein molecules were labeled with two different dyes (Alexa Fluor 488 and Alexa Fluor 647) that can be detected separately. The formation of oligomers proceeded via assembly of proteins incorporating both types of dyes into aggregated species. The presence of oligomers is manifested by simultaneous fluorescence bursts in both red (Alexa Fluor 647) and blue (Alexa Fluor 488) channels. This allowed resolving the individual species present during the initial stages of aggregation process. Interestingly, the study also found that the stability of oligomeric species increases with time while the size of oligomers stays virtually the same. One possible explanation for this effect is that the internal conformational reorganization of disordered oligomers leads to enhanced stability during fibril formation.69 Such a conclusion is in the line with previously proposed nucleated conformational conversion (NCC) model for Sup35.70 Importantly, a recent study of β-lactoglobulin aggregation proposed a similar mechanism of fibril formation.69 Oligomers should undergo a critical conformational change to become nucleating oligomers that seed amyloid formation. The size of such nucleating oligomers has been proposed to be as small as 16 molecules.71
The above-described studies emphasize the advantage of nanotools to understand the mechanisms underlying dynamic early events of protein self-association that involve formation of transient oligomeric species. Additionally, single-molecule methods avoid averaging of information over the ensemble, addressing such important issues as the heterogeneity of the formed species.
Species Barrier
The limited ability to transmit the infectious state between prion proteins of different species is referred to as “species barrier.” The molecular basis of this process is currently poorly understood. However, it is hypothesized that the species barrier is controlled by the misfolding of the prion protein and its conformational properties.72,73 Nevertheless, the species barrier is believed to be closely related to the specificity of interactions between the template and the substrate.
Among other factors, primary structure (amino acid sequence) plays a critical role in defining the ability of prion to propagate through a species barrier. It generally has been believed that the efficiency of cross-species prion transmission is directly related to the degree of homology in the primary sequences of prions for two species.74 Some reports indicate, however, that interspecies transmission is a much more complex phenomenon than simple transmission between closely related species. Depending on the species and the direction of prion transmission, the species barrier of prion infection varies significantly. For example, fibrillization of mouse prion protein PrP (23–144) can be seeded with fibrils of both mouse prion and hamster prion proteins which have 94% homology in sequence.73 On the other hand, the aggregation of hamster prion protein could only be seeded by hamster amyloid fibrils but not mouse. Remarkably, mouse amyloid fibrils which were seeded by hamster amyloid fibrils could seed fibrillization of the hamster monomer. Alteration of phenotypic patterns was also observed in the cross-species reverse transmission of Sup35 protein of Saccharomyces cerevisiae [PSI+] to S. paradoxus or S. bayanus and back to S. cerevisiae.75 A strong variant of S. cerevisiae prion was reversible when propagated via a protein with S. paradoxus prion domain, but the transmission via S. bayanus prion domain altered the phenotype of isolated [PSI+], resulting in a weaker strain.75
Previous studies demonstrated that a single amino acid substitution within a short sequence stretch, located in a prion domain, drastically changes patterns of cross-species prion conversion.75
A new idea has emerged, suggesting that a primary structure alters the spectrum of preferred conformations for a protein, thereby affecting transmission specificity.35 The range of such preferred conformations might be sensitive not only to a primary sequence but also to environmental conditions. It seems that the existence of prion strains and “species barrier” are closely related phenomena. A distinct strain conformation of Sup35 has recently been identified that is capable of transmission from S. cerevisiae to the highly divergent Candida albicans.35 Interestingly, the newly generated prion strain could be transmitted back to yeast, stimulating aggregation of Sup35 in S. cerevisiae. These results suggest that even strong species barrier (only ∼40% similarity in sequence of prion domains between Sc and Ca) can be bridged and therefore no species barrier can be considered absolute.35
Generally, the seeding process is viewed as propagation of morphology and the structure of preformed seeds. This process is believed to overcome sequence- and/or condition-based structural preferences, resulting in fibrils that inherit the characteristics of the template (reviewed in ref. 74). On a similar note, transmission through a cross-species barrier is considered to produce the same strain of the prion. However, strain switching has been observed for several prion systems. Recent studies illustrated that an amyloid can accommodate a significant conformational switch within a single fibril.37 Such a conformational switch was shown to occur when the amino acid sequence of the precursor did not match the sequence of the template. The fibril formation of mouse PrP was seeded with preformed fibrils of hamster PrP. Despite the mismatch in amino acid sequence, individual fibrils were able to recruit the heterologous recombinant prion protein. The elongation of the fibril in this case proceeded through an adaptation of a different conformational state.37
Recent reports also add to the complexity of the species barrier phenomenon by showing that the lack of prion coaggregation for two species does not correlate with decreased cross-species prion transmission.76 This observation, therefore, suggests that prion transmission specificity is controlled at the level of conformational transition rather than coaggregation.
Molecular Mechanisms of Protein Misfolding
There is an increasing amount of evidence showing that many proteins (if not all) have the ability to form fibrils under appropriate conditions.77,78 It seems that although they are very diverse in length, function and location in the organism, aggregates and fibrils have similar morphologies and common properties. Despite the diversity of structural peculiarities of aggregation precursors, they may be rich in β-sheets, α-helices or even natively unfolded proteins; the aggregation is accompanied by destabilization of the native structure and simultaneous formation of β-sheets. β-sheets are formed between alternating peptide strands which run perpendicular to the long axis of the fibrils (reviewed in ref. 79). These structures may be stabilized either by the network of hydrogen bonding between both main-chain amides and by side chain residues, which turn them into so-called “polar zippers”80 or by hydrophobic interactions.81 It has become certain that the aggregation process and β-sheet acquisition are concerted events.82 The aggregation induced by the formation of β-sheet-rich structure is a universal phenomenon, not restricted to a special category of proteins. This suggests that common molecular mechanisms most likely underlie the aggregation of various proteins into highly ordered fibrillar aggregates.83–85
According to molecular dynamics (MD) simulations, it has been suggested that the relative stability of a monomer's beta-prone state determines the fibrillogenesis of a peptide.86 Increasing relative stability of beta-prone state results in changing the aggregation pattern form disordered aggregation to fibril formation, with on-pathway intermediates and finally to fibrillogenesis without any intermediates.
Atomic force microscopy (AFM) was also instrumental in elucidating the molecular origin of fibril properties by providing insight into what determines such material as an amyloid fibril.87 Several peptides which readily form amyloid fibrils with low sequence similarity were analyzed in terms of bending rigidity of the formed fibrils. The mechanical properties of fibrillar structures were characterized by AFM topographic data which enabled the analysis of the decay of tangent along the fibril. Such decay accurately reflects the mechanical properties of the fibrils—some of which appear to be flexible and some are very stiff. Bending rigidities for different fibrils vary by as much as 4 orders of magnitude. It was shown that the major contribution to the fibril's rigidity comes from the generic hydrogen-bonding network of backbone-backbone interactions, which is modulated by side-chain interactions.87 The above-described finding only reinforces the hypothesis that the ability to form amyloid fibril is a generic property because these structures are stabilized by main-chain interactions of the polypeptide backbone. It also explains why so many unrelated proteins form such fibrillar aggregates.74
It is becoming clear that currently available widely used ensemble-based methods bear limitations that prevent full characterization and understanding of protein misfolding and aggregation phenomena. Such methods provide a general picture of a system in which the information is averaged over the ensemble of all conformations present in the system. However, misfolding and early events in protein aggregation are too quick and involve species that are too small for experimental probing by most currently available techniques. The limitations of the current methods force scientists to look for alternative tools to study mechanisms of misfolding and aggregation. Given the inherent nanoscale of these single-molecule events, it is inevitable that existing and emerging nanoimaging tools will be applied to solve these challenging problems by opening new possibilities to understand the mechanisms of protein diseases, to improve diagnosis and to develop effective therapeutic approaches. Current trends and advances in nanoscience indicate that single-molecule methods might help to overcome the limitations of currently used methods.
For example, recent studies using single-molecule fluorescence resonance energy transfer (SM-FRET) and fluorescence correlation spectroscopy investigated the structure and dynamics of monomeric prion yeast protein Sup35.88 The results of this study demonstrated that the NM part (N-terminal and Middle part) of the monomeric protein accommodates an ensemble of conformations in physiological buffers. The ensemble consists of rapidly interconverting compact conformations indicating the highly dynamic nature of protein conformational space.
This study reinforced the idea that new experimental tools are required to fully characterize protein misfolding and aggregation at the level of single molecules. Such tools will allow unambiguous measurements of the kinetics of interconversion between different protein conformations and distinguish conformational changes in individual protein molecules prior to aggregation as well as changes induced by interactions of protein with another protein or other factors. It is also important to note that some conformations (misfolded states) might be more important in aggregation pathways than others even though they are only transiently populated. The transient nature of misfolded conformations makes them not amenable to traditional methods. New tools are emerging which are capable of addressing the misfolding phenomenon at the level of a single molecule.
The transient nature of misfolded conformations and self-propagating nature of amyloids highlights the importance of understanding what interactions between protein molecules drive the formation of a stable nucleus of aggregation. Misfolded aggregation-prone conformations of a protein molecule differ from other conformations by their enhanced propensity to associate with each other, promoting formation of nano-aggregates.89 A monomeric protein should undergo structural reorganization to adopt an intermediate conformational state required for the initiation of aggregation.90 The propensity for such adaptation could be modulated by a primary sequence or external factors such as environmental conditions, interactions with other proteins, toxins, cellular components and/or cellular membrane.
Recently, atomic force spectroscopy single-molecule unfolding methodology has been applied to study the amyloidogenic properties of β2-microglobulin91 and the conformational diversity of alpha-synuclein—a protein involved in Parkinson's disease.92 A single-protein molecule was flanked by three I27 domains, which acted as molecular handles as well as a well-identifiable fingerprint in the measured force curves allowing clear recognition of signals from mechanical stretching of single alpha-synuclein molecule. Such an approach probes the full conformational space of the protein, identifying even poorly populated conformers of alpha-synuclein. Three main distinct conformations were identified and classified as random coil, mechanically weak and beta-like structures. The latter are directly related to aggregation of alpha-synuclein.92 Interestingly, pathological conditions which are known to increase propensity of alpha-synuclein to aggregate, such as high ionic strength and presence of Cu2+, shifted conformational equilibrium towards beta-like structures. Also, this approach was used to characterize pathological mutants of alpha-synuclein—A30P, A53T and E46K.93 The study revealed significant differences in the conformational behaviors of the mutants compared to the wild-type protein. All mutants had higher population of beta-like structures, indicating their higher propensity to aggregate.93
The above-described studies reinforced the hypothesis proposed earlier that misfolded conformations which are capable of triggering protein aggregation are characterized by elevated propensity to interact with one another.94 The use of AFM force spectroscopy has been proposed to probe the properties of interacting individual molecules of the aggregating proteins.94,95 The strategy for such characterization is described in detail elsewhere.89,96,97 Briefly, individual molecules are covalently attached to AFM tip and the probed surface using recently developed surface chemistry.89 The dimer can form when the tip approaches the substrate and the interacting forces between molecules within dimeric contact are measured upon tip retraction. McAllister et al.94 have used AFM to study misfolding, inducing interactions in three different and structurally distinctive proteins: α-synuclein, amyloid β-peptide (Aβ) and lysozyme. The strength of interprotein interaction was measured at various pH values, using pH as a stimulating factor for conformational changes in the proteins. Low pH values are known to induce misfolding of these proteins promoting aggregation and simultaneously conformational transition to β-type structures.98 The propensity of a protein for aggregation, its acquisition of β-sheet conformation and strength of interprotein interactions were found to correlate very well, suggesting intimate relationships among all three.94 The results of this study illustrate the power of force spectroscopy in addressing molecular mechanisms of protein misfolding and aggregation by measuring the strength of interprotein interactions.89,94 A recent application of this single-molecule approach to alpha synuclein97 led to a finding that the formation of a dimer of misfolded alpha-synuclein dramatically increases the lifetime of the protein misfolded state. Such stabilization of misfolded protein in a dimeric form enormously increases the propensity to aggregate further. This is a fundamental finding suggesting that dimerization of amyloidogenic protein triggers the formation of the disease-prone assemblies and thus initiating the disease.
We have used this powerful force spectroscopy approach to analyze interactions between individual molecules of the short peptide CGNNQQNY with a cysteine incorporated to facilitate immobilization of the peptide for the force spectroscopy studies. This peptide is a part of Sup35 yeast prion protein and was found to be amyloidogenic, seeding aggregation of the entire protein.99 The aggregation of this peptide results in aggregates of fibrillar morphology in a wide range of environmental conditions. The aggregation kinetics for this peptide was observed to be the fastest at pH 5.6 among all tested pH values.100 We have chosen pH = 5.6 to measure the interactions between individual peptides. The peptide monomers were covalently attached to AFM tip and mica surface using terminal cysteine residue with surface chemistry previously developed and tested.89 Measurements of rupture forces over a range of loading rates resulted in a dynamic force spectrum (DFS). Figure 3A shows the DFS plot obtained for interactions between CGNNQQNY peptide at pH 5.6. The plot has very distinct parts which divide DFS into several slopes. Dissociation of intermolecular contact that involves overcoming more than one activation barrier results in a dynamic force spectrum with several distinguishable linear regimes characterized by different slopes.101 If the dynamic force spectrum is divided into two parts, assuming two linear regimes in the range of loading rates between 200 and 120,000 pN/sec, these two slopes of DFS plot for the peptide-peptide dimeric contact are translated into two activation energy barriers. A very steep linear plot characterizes the rupture force dependence at high loading rates. This slope produces the off-rate dissociation constant as high as 147 s−1 when fitted with Bell's model.101 The second regime at low loading rates is characterized by a much smaller value of off-rate constant, koff = 1.8 s−1. The dimer lifetime corresponding to this value of off rate constant was found to be 0.6 seconds, suggesting high stability of dimeric species compared to the dynamic characteristics of the protein in monomeric state.100 The dimers, thus, may serve as stable nuclei for the formation of multimeric and aggregated forms of the peptide. This observation is in line with a previous study that suggested a critical role of dimerization in the aggregation of a-synuclein.97 Moreover, a similar pattern was observed for Aβ-40 peptide.100 Thus, all data lead to a new model of the protein aggregation in which the very first stage, the formation of dimers, is a key step triggering the entire self-assembly process.
Figure 3.
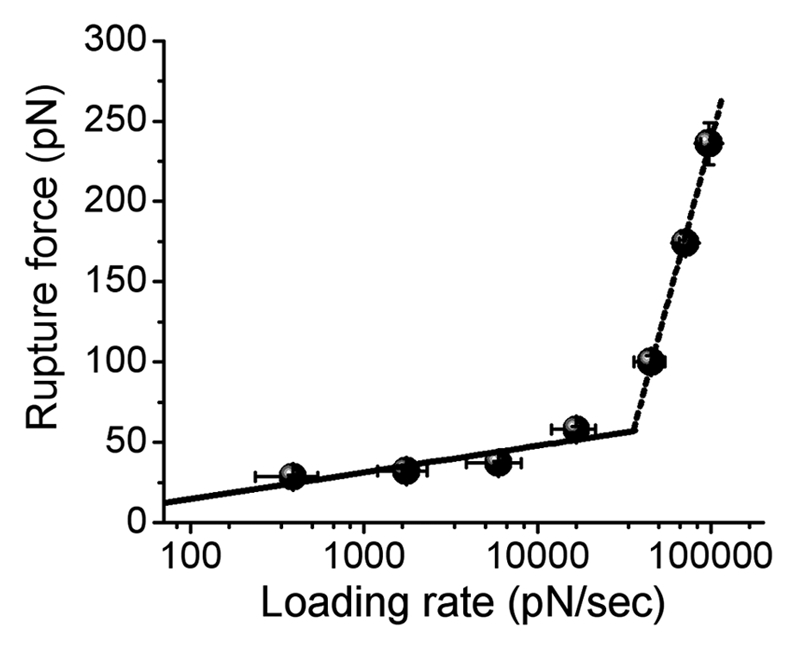
Dynamic force spectrum of CGNNQQNY peptide interactions measured at pH 5.6.
In conclusion, we believe that further exploitation of single-molecule approaches will provide new important insights into the mechanism of protein misfolding and aggregation. We anticipate that single-molecule methods will advance our understanding of such phenomena as misfolding, aggregation and nature of toxic species. Such knowledge is critical for controlling these processes and the development of effective therapeutic agents to prevent related neurodegenerative diseases.
Acknowledgements
The authors thank Luda S. Shlyakhtenko for her help with AFM imaging.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/13125
Financial Support
The work was supported by the following grants: DOE (DE-FG02-08ER64579) and NATO (CBN.NR.NRSFP 983204) (all to Y.L.L.).
References
- 1.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 2.Baxa U, Wickner RB, Steven AC, Anderson DE, Marekov LN, Yau WM, et al. Characterization of beta-sheet structure in Ure2p1–89 yeast prion fibrils by solid-state nuclear magnetic resonance. Biochemistry. 2007;46:13149–162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 3.Chan JC, Oyler NA, Yau WM, Tycko R. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel beta-sheet structure. Proc Natl Acad Sci USA. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wickner RB, Dyda F, Tycko R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register beta-sheet structure. Proc Natl Acad Sci USA. 2008;105:2403. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz-Avalos R, King CY, Wall J, Simon M, Caspar DL. Strain-specific morphologies of yeast prion amyloid fibrils. Proc Natl Acad Sci USA. 2005;102:10165–10170. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, et al. Supramolecular structure in full-length Alzheimer's beta-amyloid fibrils: evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance. Biophys J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 9.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 10.Madine J, Doig AJ, Kitmitto A, Middleton DA. Studies of the aggregation of an amyloidogenic alpha-synuclein peptide fragment. Biochem Soc Trans. 2005;33:1113–1115. doi: 10.1042/BST20051113. [DOI] [PubMed] [Google Scholar]
- 11.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, et al. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J Mol Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 12.Krebs MR, Wilkins DK, Chung EW, Pitkeathly MC, Chamberlain AK, Zurdo J, et al. Formation and seeding of amyloid fibrils from wild-type hen lysozyme and a peptide fragment from the beta-domain. J Mol Biol. 2000;300:541–549. doi: 10.1006/jmbi.2000.3862. [DOI] [PubMed] [Google Scholar]
- 13.Ivanova MI, Sawaya MR, Gingery M, Attinger A, Eisenberg D. An amyloid-forming segment of beta2-microglobulin suggests a molecular model for the fibril. Proc Natl Acad Sci USA. 2004;101:10584–10589. doi: 10.1073/pnas.0403756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez de la Paz M, Serrano L. Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci USA. 2004;101:87–92. doi: 10.1073/pnas.2634884100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esteras-Chopo A, Serrano L, Lopez de la Paz M. The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc Natl Acad Sci USA. 2005;102:16672–16677. doi: 10.1073/pnas.0505905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keskin O, Ma B, Nussinov R. Hot regions in protein—protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez de Groot N, Pallares I, Aviles FX, Vendrell J, Ventura S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol. 2005;5:18. doi: 10.1186/1472-6807-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–561. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, et al. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadnar ML, Articov G, Derkatch IL. Distinct type of transmission barrier revealed by study of multiple prion determinants of Rnq1. PLoS Genet. 2010;6:1000824. doi: 10.1371/journal.pgen.1000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohhashi Y, Ito K, Toyama BH, Weissman JS, Tanaka M. Differences in prion strain conformations result from non-native interactions in a nucleus. Nat Chem Biol. 2010;6:225–230. doi: 10.1038/nchembio.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem. 2003;278:16462–16465. doi: 10.1074/jbc.C300049200. [DOI] [PubMed] [Google Scholar]
- 24.Ban T, Goto Y. Direct observation of amyloid growth monitored by total internal reflection fluorescence microscopy. Methods Enzymol. 2006;413:91–102. doi: 10.1016/S0076-6879(06)13005-0. [DOI] [PubMed] [Google Scholar]
- 25.Ban T, Hoshino M, Takahashi S, Hamada D, Hasegawa K, Naiki H, et al. Direct observation of Abeta amyloid fibril growth and inhibition. J Mol Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 26.Hoyer W, Cherny D, Subramaniam V, Jovin TM. Rapid self-assembly of alpha-synuclein observed by in situ atomic force microscopy. J Mol Biol. 2004;340:127–139. doi: 10.1016/j.jmb.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 27.Kowalewski T, Holtzman DM. In situ atomic force microscopy study of Alzheimer's beta-amyloid peptide on different substrates: new insights into mechanism of beta-sheet formation. Proc Natl Acad Sci USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldsbury C, Kistler J, Aebi U, Arvinte T, Cooper GJ. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J Mol Biol. 1999;285:33–39. doi: 10.1006/jmbi.1998.2299. [DOI] [PubMed] [Google Scholar]
- 29.Goldsbury C, Green J. Time-lapse atomic force microscopy in the characterization of amyloid-like fibril assembly and oligomeric intermediates. Methods Mol Biol. 2005;299:103–128. doi: 10.1385/1-59259-874-9:103. [DOI] [PubMed] [Google Scholar]
- 30.Kellermayer MS, Karsai A, Benke M, Soos K, Penke B. Stepwise dynamics of epitaxially growing single amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:141–144. doi: 10.1073/pnas.0704305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DePace AH, Weissman JS. Origins and kinetic consequences of diversity in Sup35 yeast prion fibers. Nat Struct Biol. 2002;9:389–396. doi: 10.1038/nsb786. [DOI] [PubMed] [Google Scholar]
- 32.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta. 2007;1772:681–691. doi: 10.1016/j.bbadis.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka M, Chien P, Yonekura K, Weissman JS. Mechanism of cross-species prion transmission: an infectious conformation compatible with two highly divergent yeast prion proteins. Cell. 2005;121:49–62. doi: 10.1016/j.cell.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Jones EM, Surewicz WK. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell. 2005;121:63–72. doi: 10.1016/j.cell.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 37.Makarava N, Ostapchenko VG, Savtchenko R, Baskakov IV. Conformational switching within individual amyloid fibrils. J Biol Chem. 2009;284:14386–14395. doi: 10.1074/jbc.M900533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Wel PC, Lewandowski JR, Griffin RG. Solid-state NMR study of amyloid nanocrystals and fibrils formed by the peptide GNNQQNY from yeast prion protein Sup35p. J Am Chem Soc. 2007;129:5117–5130. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- 39.Novitskaya V, Makarava N, Bellon A, Bocharova OV, Bronstein IB, Williamson RA, et al. Probing the conformation of the prion protein within a single amyloid fibril using a novel immunoconformational assay. J Biol Chem. 2006;281:15536–15545. doi: 10.1074/jbc.M601349200. [DOI] [PubMed] [Google Scholar]
- 40.Petty SA, Decatur SM. Intersheet rearrangement of polypeptides during nucleation of {beta}-sheet aggregates. Proc Natl Acad Sci USA. 2005;102:14272–14277. doi: 10.1073/pnas.0502804102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shim SH, Gupta R, Ling YL, Strasfeld DB, Raleigh DP, Zanni MT. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. P Natl Acad Sci USA. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shashilov V, Xu M, Ermolenkov VV, Fredriksen L, Lednev IK. Probing a fibrillation nucleus directly by deep ultraviolet Raman spectroscopy. J Am Chem Soc. 2007;129:6972–6973. doi: 10.1021/ja070038c. [DOI] [PubMed] [Google Scholar]
- 43.Shashilov VA, Lednev IK. 2D correlation deep UV resonance raman spectroscopy of early events of lysozyme fibrillation: kinetic mechanism and potential interpretation pitfalls. J Am Chem Soc. 2008;130:309–317. doi: 10.1021/ja076225s. [DOI] [PubMed] [Google Scholar]
- 44.Paulite M, Fakhraai Z, Gunari N, Tanur A, GW Different Individual Amyloid Fibrils Exhibit Different Beta Sheet Secondary Structures via Near-field Infrared Spectroscopy. Biophys J. 2009;96:87. [Google Scholar]
- 45.Paulite M, Fakhraai Z, Akhremitchev BB, Mueller K, Walker GC. Assembly, tuning and use of an apertureless near field infrared microscope for protein imaging. J Vis Exp. 2009 doi: 10.3791/1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stockle RM, Suh YD, Deckert V, Zenobi R. Nanoscale chemical analysis by tip-enhanced Raman spectroscopy. Chem Phys Lett. 2000;318:131–136. [Google Scholar]
- 47.Hayazawa N, Inouye Y, Sekkat Z, Kawata S. Metallized tip amplification of near-field Raman scattering. Optics Communications. 2000;183:333–336. [Google Scholar]
- 48.Pettinger B, Picardi G, Schuster G, Ertl G. Surface enhanced Raman spectroscopy: towards single molecule spectroscopy. Electrochem. 2000;68:942–949. [Google Scholar]
- 49.Bailo E, Deckert V. Tip-enhanced Raman scattering. Chem Soc Rev. 2008;37:921–930. doi: 10.1039/b705967c. [DOI] [PubMed] [Google Scholar]
- 50.Neacsu CC, Dreyer J, Behr N, Raschke MB. Reply to Comment on “Scanning-probe Raman spectroscopy with single-molecule sensitivity”. Phys Rev B. 2007;75:236402. [Google Scholar]
- 51.Bailo E, Deckert V. Tip-enhanced Raman spectroscopy of single RNA strands: towards a novel direct-sequencing method. Angew Chem Int Ed Engl. 2008;47:1658–1661. doi: 10.1002/anie.200704054. [DOI] [PubMed] [Google Scholar]
- 52.Deckert-Gaudig T, Deckert V. Ultraflat transparent gold nanoplates—ideal substrates for tip-enhanced Raman scattering experiments. Small. 2009;5:432–436. doi: 10.1002/smll.200801237. [DOI] [PubMed] [Google Scholar]
- 53.Deckert-Gaudig T, Rauls E, Deckert V. Aromatic amino acid monolayers sandwiched between gold and silver: a combined tip-enhanced raman and theoretical approach. J Phys Chem C. 2010;114:7412–7420. [Google Scholar]
- 54.Shorter J, Lindquist S. Prions as adaptive conduits of memory and inheritance. Nat Rev Genet. 2005;6:435–450. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 55.Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 56.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 58.True HL. The battle of the fold: chaperones take on prions. Trends Genet. 2006;22:110–117. doi: 10.1016/j.tig.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 59.Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci USA. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Makarava N, Lee CI, Laksanalamai P, Robb FT, Baskakov IV. Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J Mol Biol. 2008;376:1155–1167. doi: 10.1016/j.jmb.2007.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, et al. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, et al. Accelerated oligomerization by Parkinson's disease linked alpha-synuclein mutants. Ann NY Acad Sci. 2000;920:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 65.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 66.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 67.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 68.Goldberg MS, Lansbury PT., Jr Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson's disease? Nat Cell Biol. 2000;2:115–119. doi: 10.1038/35017124. [DOI] [PubMed] [Google Scholar]
- 69.Orte A, Birkett NR, Clarke RW, Devlin GL, Dobson CM, Klenerman D. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. P Natl Acad Sci USA. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 71.He X, Giurleo JT, Talaga DS. Role of small oligomers on the amyloidogenic aggregation free-energy landscape. J Mol Biol. 2009;395:134–154. doi: 10.1016/j.jmb.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moore RA, Vorberg I, Priola SA. Species barriers in prion diseases—brief review. Arch Virol. 2005:187–202. doi: 10.1007/3-211-29981-5_15. [DOI] [PubMed] [Google Scholar]
- 73.Vanik DL, Surewicz KA, Surewicz WK. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell. 2004;14:139–145. doi: 10.1016/s1097-2765(04)00155-8. [DOI] [PubMed] [Google Scholar]
- 74.Chiti F, Dobson CM. Protein misfolding, functional amyloid and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 75.Chen B, Bruce KL, Newnam GP, Gyoneva S, Romanyuk AV, Chernoff YO. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol Microbiol. 2010;76:1483–1499. doi: 10.1111/j.1365-2958.2010.07177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen B, Newnam GP, Chernoff YO. Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc Natl Acad Sci USA. 2007;104:2791–2796. doi: 10.1073/pnas.0611158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 78.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 79.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, et al. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watson D, Castano E, Kokjohn TA, Kuo YM, Lyubchenko Y, Pinsky D, et al. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease. Neurol Res. 2005;27:869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 82.Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci USA. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, et al. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bader R, Bamford R, Zurdo J, Luisi BF, Dobson CM. Probing the mechanism of amyloidogenesis through a tandem repeat of the PI3-SH3 domain suggests a generic model for protein aggregation and fibril formation. J Mol Biol. 2006;356:189–208. doi: 10.1016/j.jmb.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 86.Pellarin R, Caflisch A. Interpreting the aggregation kinetics of amyloid peptides. J Mol Biol. 2006;360:882–892. doi: 10.1016/j.jmb.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 87.Knowles TP, Fitzpatrick AW, Meehan S, Mott HR, Vendruscolo M, Dobson CM, et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 88.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kransnoslobodtsev AV, Shlyakhtenko LS, Ukraintsev E, Zaikova TO, Keana JF, Lyubchenko YL. Nanomedicine and protein misfolding diseases. Nanomedicine. 2005;1:300–305. doi: 10.1016/j.nano.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rubinstein A, Lyubchenko YL, Sherman S. Dynamic properties of pH-dependent structural organization of the amyloidogenic beta-protein (1–40) Prion. 2009;3:31–43. doi: 10.4161/pri.3.1.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sorce B, Sabella S, Sandal M, Samori B, Santino A, Cingolani R, et al. Single-molecule mechanical unfolding of amyloidogenic beta(2)-microglobulin: The force-spectroscopy approach. Chemphyschem. 2009;10:1471–1477. doi: 10.1002/cphc.200900220. [DOI] [PubMed] [Google Scholar]
- 92.Sandal M, Valle F, Tessari I, Mammi S, Bergantino E, Musiani F, et al. Conformational equilibria in monomeric alpha-synuclein at the single-molecule level. PLoS Biol. 2008;6:6. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brucale M, Sandal M, Di Maio S, Rampioni A, Tessari I, Tosatto L, et al. Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers. Chembiochem. 2009;10:176–183. doi: 10.1002/cbic.200800581. [DOI] [PubMed] [Google Scholar]
- 94.McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, et al. Protein interactions and misfolding analyzed by AFM force spectroscopy. J Mol Biol. 2005;354:1028–1042. doi: 10.1016/j.jmb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 95.Lyubchenko YL, Sherman S, Shlyakhtenko LS, Uversky VN. Nanoimaging for protein misfolding and related diseases. J Cell Biochem. 2006;99:53–70. doi: 10.1002/jcb.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yu J, Lyubchenko YL. Early stages for Parkinson's development: alpha-synuclein misfolding and aggregation. J Neuroimmune Pharm. 2009;4:10–16. doi: 10.1007/s11481-008-9115-5. [DOI] [PubMed] [Google Scholar]
- 97.Yu J, Malkova S, Lyubchenko YL. alpha-Synuclein misfolding: single molecule AFM force spectroscopy study. J Mol Biol. 2008;384:992–1001. doi: 10.1016/j.jmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci USA. 2002;99:5596–5600. doi: 10.1073/pnas.042681599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Balbirnie M, Grothe R, Eisenberg DS. An amyloid-forming peptide from the yeast prion Sup35 reveals a dehydrated beta-sheet structure for amyloid. Proc Natl Acad Sci USA. 2001;98:2375–2380. doi: 10.1073/pnas.041617698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lyubchenko Y, Kim BH, Krasnoslobodtsev A, Yu J. Nanoimaging for protein misfolding diseases. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2010 doi: 10.1002/wnan.102. [DOI] [PubMed] [Google Scholar]
- 101.Merkel R, Nassoy P, Leung A, Ritchie K, Evans E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]