

SUMMARY
Although transcription factors that repress gene expression play critical roles in nervous system development, their mechanism of action remains to be understood. Here we report that the Olig-related transcription factor Bhlhb5 (also known as Bhlhe22) forms a repressor complex with the PR/SET domain protein, Prdm8. We find that Bhlhb5 binds to sequence-specific DNA elements and then recruits Prdm8, which mediates the repression of target genes. This interaction is critical for repressor function since mice lacking either Bhlhb5 or Prdm8 have strikingly similar cellular and behavioral phenotypes, including axonal mistargeting by neurons of the dorsal telencephalon and abnormal itch-like behavior. We provide evidence that Cadherin-11 functions as a target of the Prdm8/Bhlhb5 repressor complex that must be repressed for proper neuronal development to occur. These findings suggest that Prdm8 is an obligate partner of Bhlhb5, forming a repressor complex that directs neural development in part through the precise regulation of Cadherin-11.
INTRODUCTION
A key unresolved issue in neurobiology is the nature of the molecular programs that regulate the differentiation of neural precursors into specialized neurons with appropriate connectivity. One family that has emerged as important in this regard is the basic helix-loop-helix (bHLH) containing transcription factors (Bertrand et al., 2002; Ross et al., 2003). For instance, studies investigating neocortical development have revealed that members of the bHLH family, including the Neurogenins and NeuroD family members, orchestrate the formation of glutamatergic neurons (Schuurmans and Guillemot, 2002) and many of these are sufficient to activate a pan-neuronal program of gene expression that drives the differentiation of neural precursors into neurons (Farah et al., 2000; Lee et al., 1995; Ma et al., 1996).
Like many other bHLH transcription factors, Bhlhb5 is broadly expressed in excitatory neurons in the dorsal telencephalon. However, in contrast to the Neurogenins and NeuroD family members, Bhlhb5 appears not to be a transcriptional activator and does not mediate early steps in neuronal differentiation. Rather, Bhlhb5 belongs to a subfamily of bHLH factors including Bhlhb4 (also known as Bhlhe23) and the Olig proteins (Olig1–3) that function predominantly as transcriptional repressors. As an example, when Olig2 is fused to the repressor domain of Engrailed, this fusion protein, but not an activating Olig2-VP16 fusion protein, recapitulates the function of native Olig2 by specifying neural fate in the chick spinal cord (Zhou and Anderson, 2002). Bhlhb5 and Bhlhb4 are likewise thought to mediate repression based on their ability to inhibit the transactivation of NeuroD-responsive target genes in luciferase assays (Bramblett et al., 2002; Peyton et al., 1996; Xu et al., 2002). However, while these findings suggest that the Oligs, ff, and Bhlhb5 form a sub-family of bHLH factors that mediate transcriptional repression, the manner in which these repressors function endogenously to repress transcription and orchestrate neural development remains obscure.
Studies in the spinal cord have provided a framework for understanding the cellular function of Olig proteins, and these studies suggest that a common function of the Oligs is to confer the neuronal identity of neural progenitors. For instance, Olig1 and Olig2 are expressed in select progenitors of the ventral spinal cord and, in the absence of these factors, neural precursors are re-specified to an alternate fate: instead of forming motor neurons and oligodendrocytes, these pMN progenitors inappropriately generate V2 interneurons and astrocytes (Lu et al., 2002; Takebayashi et al., 2002; Zhou and Anderson, 2002). It is thought that this type of mis-specification occurs because the Oligs function, at least in part, to promote the generation of one subtype of neuron over another by inhibiting the expression of transcription factors that mediate the alternative cell fate choices (Marquardt and Pfaff, 2001).
Though Bhlhb4 and Bhlhb5 are closely related to the Oligs, their expression is almost exclusively limited to post-mitotic neurons rather than proliferating neural progenitors, hinting at the possibility that Bhlhb4 and Bhlhb5 regulate later aspects of neuronal differentiation (Bramblett et al., 2002; Joshi et al., 2008; Ross et al. 2010). Further evidence in support of this idea comes from studies in the retina where loss of either Bhlhb4 or Bhlhb5 results not in the mis-specification of retinal progenitors to alternate fates but rather the loss of subsets of neurons, presumably due to apoptosis. Thus, mice lacking Bhlhb4 have an absence of rod bipolar cells, whereas Bhlhb5 mutants are lacking cone bipolar and selective amacrine cells (Bramblett et al., 2004; Feng et al., 2006). Similarly, in the spinal cord, disruption of Bhlhb5 function results in the selective apoptosis of a specific population of inhibitory interneurons in the superficial dorsal horn that are required for the normal sensation of itch (Ross et al., 2010). Together, these studies raise the possibility that Bhlhb4 and Bhlhb5 are involved in late aspects of neuronal differentiation, such as neural circuit assembly, that may be essential for neuronal survival. However, why certain neurons die in the absence of these transcriptional repressors is unknown, and this gap in knowledge stems in part from a lack of mechanistic understanding of how these transcription factors function in neural development.
A possible clue to this puzzle comes from studies of Bhlhb5 in the dorsal telencephalon. In mice lacking Bhlhb5, neurons of the dorsal telencephalon survive and send out axons, but these projections fail to reach their targets. For instance, corticospinal motor neurons terminate prematurely along the pyramidal tract in the ventral hindbrain and none extend into the spinal cord (Joshi et al., 2008). Moreover, as we report here, Bhlhb5 mutants also show a complete absence of the three fiber tracts that connect the cerebral hemispheres, suggesting that axonal mistargeting in the absence of Bhlhb5 is a widespread phenomenon. Since mice lacking Bhlhb5 show axon targeting defects, we reasoned that one of the roles of Bhlhb5 in the dorsal telencephalon may be to regulate neuronal connectivity, perhaps by repressing specific genes until they are needed, thereby ensuring that genes are expressed at the right time and place for correct neural circuit assembly. To investigate this possibility, we identified the targets of the Bhlhb5 transcriptional repressor, hoping to elucidate how this transcription factor functions at a mechanistic level.
Here we show that Bhlhb5 functions by binding to specific DNA sequence elements and then recruiting the PR/SET domain-containing protein, Pr-domain 8 (Prdm8) to mediate the repression of target genes. Our observations suggest that Bhlhb5 and Prdm8 are obligate partners for key aspects of neuronal development and, consistent with this idea, we find that mice lacking either Bhlhb5 or Prdm8 have strikingly similar cellular and behavioral abnormalities. We use genetic rescue experiments to demonstrate that one important target of the Prdm8/Bhlhb5 repressor complex is Cadherin-11 (Cdh11), a cell-cell adhesion molecule involved in neural circuit assembly. Taken together these experiments have revealed how a bHLH transcription factor associates with a PR/SET-domain repressor protein to regulate genes involved in neural development.
RESULTS
Prdm8 and Bhlhb5 regulate common aspects of neural development
To gain insight into how Bhlhb5 functions to regulate axon targeting, we sought to determine possible Bhlhb5 target genes by identifying genes that are misexpressed in Bhlhb5 mutant mice during the early development of the dorsal telencephalon (E13.5, E15.5 and E17.5). By expression profiling we found a total of eight transcripts that were significantly misregulated (FDR < 0.05) when Bhlhb5 is disrupted. Notably, all of these genes were upregulated in Bhlhb5 mutant mice, consistent with the idea that Bhlhb5 may act directly to inhibit their expression. Of these, Prdm8 and Cdh11 were the most significantly misregulated genes, and we selected these for follow-up in the present study (Figures 1A and 7A). Other significantly misregulated genes that we identified are the gap junction protein Connexin 36; the MAGE family proteins Necdin and Magel2, which are inactivated in Prader-Willi syndrome (Nicholls and Knepper, 2001); the neurotrophin receptor p75 NTR; the neuropeptide, Neurexophilin 3; and the actin-binding protein Fmnl1 (Figure S1). Of note, several of these genes, including Cdh11, p75 NTR, Necdin, and MageL2, are known to mediate axon extension (Lee et al., 2005a; Marthiens et al., 2005; Yamashita et al., 1999), consistent with the idea that Bhlhb5 may control a program of gene expression that mediates aspects of neural development including axonal outgrowth.
Figure 1. Screen for Bhlhb5 target genes identifies the PR/SET-domain protein, Prdm8.

A) Affymetrix microarray-based gene profiling was performed to identify genes that are misexpressed in the dorsal telencephalon of Bhlhb5−/− mice. Three embryonic stages (E13.5, E15.5 and E17.5) were investigated, and each time point was analyzed using three independent pairs of littermate mice. Using a false discovery rate (FDR) of 0.05 or less, we identified a total of eight genes, and all of these genes were up-regulated in Bhlhb5−/− mice (also see Figures 7A and S1). Of these, Prdm8 (shown) was the most significantly misregulated. Data are mean +/− SEM, with wild type (WT) in black and Bhlhb5−/− in red, as indicated. B) Quantitative PCR confirms that Prdm8 mRNA transcripts are upregulated in the dorsal telencephalon of Bhlhb5−/− mice relative to WT littermates at E17.5. Data are normalized to WT and are presented as mean +/− SEM of biological replicates. * indicates significant difference relative to controls (p < 0.05, t-test). C) Prdm8 protein is upregulated in the cortex of Bhlhb5−/− mice at E16.5. Matched coronal sections were stained with a Prdm8 antibody raised against a GST-Prdm8 fusion protein. Also see Figure S4 for details on the generation of Prdm8 antibodies and Figure S6 for further analysis of the upregulation of Prdm8 in other regions of the nervous system and at other times during development.
Figure 7. Cdh11 is a key target of the Bhlhb5/Prdm8 repressor complex.

A) Affymetrix microarray-based gene profiling identifies Cdh11 as a gene that is significantly upregulated in the dorsal telencephalon of Bhlhb5−/− mice (FDR < 0.05) from E13.5 – E17.5. Data are mean +/− SEM of biological replicates, with WT in black and Bhlhb5−/− in red, as indicated. B) Cdh11 mRNA is also upregulated in the dorsal telencephalon of Prdm8−/− mice relative to WT littermates at E14.5, as shown by qPCR. Data are normalized to WT and are presented as mean +/− SEM of biological replicates. * indicates significant difference relative to controls (p < 0.05, t-test). C–D) Cdh11 protein is upregulated in the dorsal telencephalon of Bhlhb5−/− mice. Sagittal sections from mice of the indicated genotype at E16.5 were stained with antibodies to Cdh11. Note the high Cdh11 expression in the axons of corticofugal fibers in the internal capsule (white arrows). Boxed insets are enlarged in D, which also shows the corresponding immunostaining with antibodies to Bhlhb5. E) Sagittal schematic of the brain illustrating the path of the corticospinal tract. Regions I and II (gray) indicate the location of sections in the coronal plane that were used to quantify the area of the corticospinal tract in Bhlhb5−/− mice and Bhlhb5−/−; Cdh11−/− double mutant mice (F). F) Quantification of the axon area in regions I and II. Relative to WT mice, there is a dramatic loss of axon area in the Bhlhb5−/− mice (black bars) and this loss is partially rescued in Bhlhb5−/−;Cdh11−/− double mutant mice (red bars). Five pairs of adult littermate mice were analyzed. * indicates significant (p < 0.05, t-test). G) Representative images showing the corticospinal tract stained with PKCγ in WT, Cdh11−/−, Bhlhb5−/−, and Bhlhb5−/−;Cdh11−/− mice. Matched cervical sections (corresponding to region II) from adult littermate mice are shown. The corticospinal tract is almost absent in the cervical spinal cord of Bhlhb5−/− mice but a partial rescue is seen in Bhlhb5;Cdh11−/− double mutant mice. The loss of Cdh11 alone has no effect on the formation of the corticospinal tract. H) Bhlhb5 mutant mice develop skin lesions by ~ 6 weeks of age. The onset of skin lesions is significantly delayed to ~13 weeks in Bhlhb5−/−; Cdh11−/− double mutant mice. n = 12 littermate pars; * indicates significant difference (p < 0.05, t-test.)
From this list of putative Bhlhb5 target genes, we focused initially on Prdm8, a protein belonging to the PRDI-BF1 and RIZ homology domain containing family that have recently emerged as key mediators of development (Baudat et al., 2010; Berg et al., 2010; Ohinata et al., 2005; Parvanov et al., 2010; Seale et al., 2008). Members of this family are transcriptional regulators that are characterized by the presence of a SET domain, a signature motif found in members of the histone methyltransferase superfamily. Consistent with this, several Prdm proteins, including Prdm8, have been reported to have intrinsic histone methyltransferase activity (Eom et al., 2009; Hayashi et al., 2005; Kim et al., 2003; Wu et al., 2010), while others are known to function as repressors by recruiting histone modifying enzymes (Ancelin et al., 2006; Davis et al., 2006; Duan et al., 2005; Gyory et al., 2004). Since Prdm8 is significantly over-expressed upon the loss of Bhlhb5 (Figures 1A, 1B and 1C), we reasoned that Prdm8 might function as part of a linear repressor cascade in which Bhlhb5 represses Prdm8 and Prdm8 represses other targets. The other possibility that we considered was that Bhlhb5 and Prdm8 function together, and that Prdm8 is up-regulated in the absence of Bhlhb5 due to a mis-regulated negative feedback loop.
To begin to investigate these possibilities, we investigated whether mice lacking Bhlhb5 or Prdm8 share any common phenotypes. As reported previously, we observe that the axons from corticospinal motor neurons of Bhlhb5 mutant mice terminate prematurely and fail to enter the spinal cord (Figures 2A, S2A, S2B and (Joshi et al., 2008)). In addition, we noted that loss of Bhlhb5 in the dorsal telencephalon resulted in the almost complete absence of the three fiber tracts that connect the cerebral hemispheres: the corpus callosum, hippocampal commissure and the anterior commissure (Figures 2B and S2C). Strikingly, we found that these major axon tracts are also mistargeted in Prdm8 mutant mice, which showed a similar absence of the corticospinal tract (Figure 2A) as well as agenesis of the corpus callossum and hippocampal commissure (Figure 2B). In contrast, cortical layering is unaffected in both Bhlhb5 and Prdm8 knockout mice (Figure S3). Thus, both Bhlhb5 and Prdm8 are required for the correct targeting of projection neurons of the dorsal telencephalon.
Figure 2. Mice lacking either Bhlhb5 or Prdm8 have similar phenotypes.

A) Absence of the corticospinal tract in the spinal cord of mice lacking either Bhlhb5 or Prdm8. In wild type mice (WT), PKCγ immunostaining labels the axons from corticospinal motor neurons as they descend in the dorsal funiculus of the spinal cord (white arrow). In both Bhlhb5−/− and Prdm8−/− mice, this axon tract is absent at all levels of the spinal cord. Similar results are observed when this fiber tract is labeled genetically (data not shown). Note that PKCγ also labels a subset of lamina II neurons in the dorsal horn, which are unaffected by the loss of Bhlhb5 or Prdm8. Images are of representative cervical spinal cords from adult mice (8 wks). B) Agenesis of the corpus callosum and hippocampal commissure in mice lacking either Bhlhb5 or Prdm8. In wild type mice (WT), dense bundles of axons connect the two hemispheres of the dorsal telencephalon (black arrow). In Bhlhb5−/− and Prdm8−/− mice, there is severe agenesis of these colossal fiber tracts. This axon targeting phenotype was observed in 20/20 Bhlhb5−/− mice, 5/7 Prdm8−/− mice, and none of the corresponding wild type littermates. Images are of representative coronal brain sections from adult mice stained with luxol fast blue and cresyl violet. C) Mice lacking either Bhlhb5−/− or Prdm8−/− show elevated scratching behavior that results in the development of skin lesions. Photos illustrating characteristic lesions are shown. All Bhlhb5−/− mice develop skin lesions at approximately 6 wks of age; ~75% of Prdm8−/− mice eventually develop skin lesions. D) Both Bhlhb5−/− and Prdm8−/− mice show an unusual movement in which they walk on their forepaws. Note that this ‘handstand’ phenotype, which appears to be secondary to abnormal contraction of the hindpaws, is only observed in a small fraction (~1– 5%) of Bhlhb5 or Prdm8 mutants. Also see Figure S2 for a more detailed analysis of axon targeting defects and Figure S3 for analysis of cortical lamination, which is unaffected by the loss of either Bhlhb5 or Prdm8.
In addition to showing common axonal targeting defects, we found that Bhlhb5 and Prdm8 mutant mice have similar behavioral abnormalities. For example, both mutants show abnormal itching behavior that results in the formation of skin lesions, which is observed in 100% of Bhlhb5 mutant mice and ~75% of Prdm8 mutant mice (Figure 2C). Furthermore, ~5% of mice lacking either Bhlhb5 or Prdm8 occasionally display an unusual movement in which they walk on their forepaws (Figure 2D). The remarkable similarity of the cellular and behavioral phenotypes observed in mice lacking either Bhlhb5 or Prdm8 strongly suggests that these factors function together, possibly as obligate partners of the same transcriptional repressor complex.
Bhlhb5 and Prdm8 form a transcriptional repressor complex
If Bhlhb5 and Prdm8 form a protein complex that represses transcription, we would expect Bhlhb5 and Prdm8 to i) be expressed in the same subsets of neurons, ii) inhibit the same target genes, and iii) bind together to the same regulatory elements in DNA. Thus, we set out to test each of these possibilities.
We began by generating antibodies to Prdm8 (Figure S3) to characterize its expression pattern. At the subcellular level, both Bhlhb5 and Prdm8 show a similar distribution in the nucleus (Figures 3A and 3B). Moreover, analysis of sections from wild type mice at a variety of embryonic and early post-natal ages revealed that Bhlhb5 and Prdm8 show a high degree of co-localization in select subpopulations of differentiating neurons. For instance, Bhlhb5 and Prdm8 are both expressed in the intermediate zone and the cortical plate of dorsal telencephalon from E13.5 – E17.5 (Figure S5A). By P0, both factors are highly expressed in superficial layers of the cortex (Figure 3B). In other regions of the nervous system (where Bhlhb5 and Prdm8 are expressed more sparsely) the co-expression of these two proteins is even more apparent; Bhlhb5 and Prdm8 clearly mark a shared subset of neurons in the diencephalon (Fig. 3B, inset ii), the brainstem (Figure S5B) and the spinal cord (Ross et al., 2010). This co-expression in specific populations of neurons suggests that Bhlhb5 and Prdm8 might work together to regulate common aspects of neuronal differentiation.
Figure 3. Bhlhb5 and Prdm8 are colocalized in subsets of neurons.
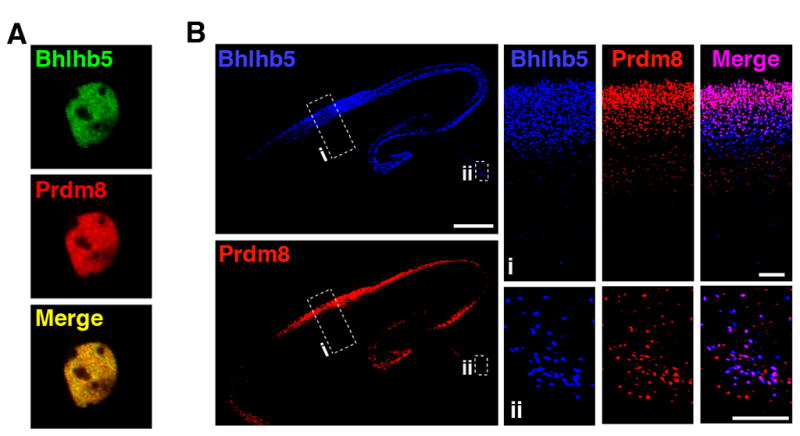
A) Double-labeling of neurons with antibodies to Bhlhb5 (green) and Prdm8 (red) reveals that Bhlhb5 and Prdm8 are localized (merge, yellow) to the nucleus where they show a diffuse pattern of expression, suggesting that they may modulate the expression level of transcribed genes found within euchromatic DNA. B) Immunohistochemistry of sagittal sections from wild type mice at P0 using antibodies to Bhlhb5 (blue) and Prdm8 (red) reveals that both proteins are co-expressed to a large extent in superficial layers of the cortical plate and in the developing hippocampus. Scale bar = 500 μm. Insets are enlarged on the right, top (i), showing co-localization (purple) of Bhlhb5 and Prdm8 in the superficial cortex, and bottom (ii), showing co-localization in sparse subsets of neurons in the diencephalon. Scale bar = 100 μm. Also see Figure S5 for a more extensive analysis of Bhlhb5 and Prdm8 co-localization at other times during development and in other regions of the nervous system.
Next we investigated whether the same genes are misexpressed in Bhlhb5−/− and Prdm8−/− mice. To obtain an unbiased view, we independently determined the gene expression profiles of each mutant, analyzing mRNA expression in the dorsal telencephalon of mice. This approach allowed us to investigate whether the genes that are misregulated in Bhlhb5 mutant mice are also misregulated in Prdm8 mutant mice, and vice versa, rather than simply testing previously discovered Bhlhb5 targets in the Prdm8 mutant mice. For these new gene profiling experiments, we chose P0 as the time point, both for practical reasons and with the hope of discovering new target genes.
Interestingly, we found that just as Prdm8 mRNA is upregulated in Bhlhb5 mutant mice, so Bhlhb5 mRNA is upregulated in Prdm8 mutant mice (Figures 4A and 4B). But what about other Bhlhb5 target genes—are they likewise upregulated in Prdm8 mutant mice? We found that loss of either Bhlhb5 or Prdm8 resulted in changes in gene expression in a small number of genes and, remarkably, all of the genes that were significantly upregulated in one mutant were also upregulated in the other. These genes include Antxr2, Connexin36, NMDA3A and Paqr3 (Figures 4C, 4D, 4E and 4F) as well as Fgf5 and Netrin1 (data not shown). We therefore conclude that Bhlhb5 and Prdm8 inhibit the expression of a common set of genes, consistent with the possibility that they function together as part of the same repressor complex.
Figure 4. Mice lacking either Bhlhb5 or Prdm8 have a common molecular profile.
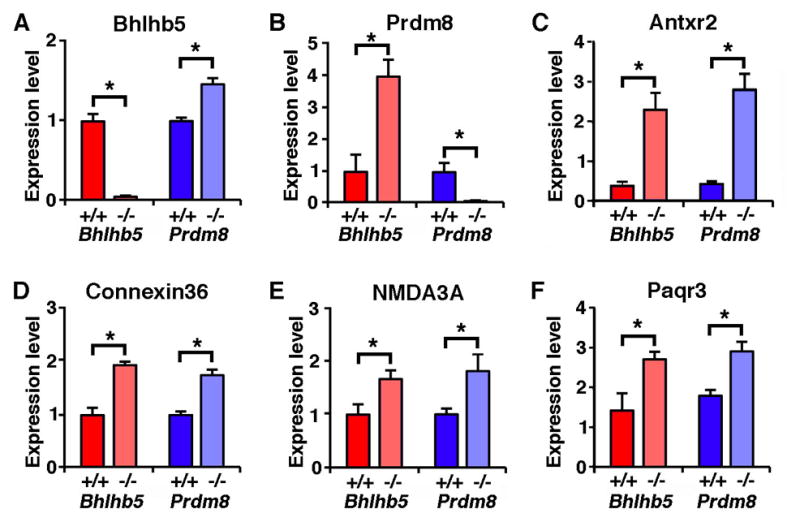
Affymetrix microarray-based gene profiling was performed to identify genes that are misexpressed in the dorsal telencephalon of either Bhlhb5−/− or Prdm8−/− mice at P0. Each of the mutant strains was compared to its respective littermate control, using four independent biological replicates per genotype, and observations were validated by quantitative qPCR. A) Bhlhb5 mRNA is significantly upregulated in Prdm8−/− mice. B) Prdm8 mRNA is significantly upregulated in Bhlhb5−/− mice. C – F) Common genes are similarly misregulated in both Bhlhb5−/− and Prdm8−/− mice, including Antxr2 (C), Connexin36 (D), NMDA3A (E) and Paqr3 (F). Data are presented as mean +/− SEM of biological replicates and * indicates significant difference relative to controls (p < 0.05, t-test). Of note, most of the genes that are misregulated in Bhlhb5 mutant mice at P0 (i.e., as illustrated here) are different that those that are misregulated from E13.5 – E17.5 (Figure S1), suggesting that Bhlhb5 may regulate different genes at different times during development. Also see Figure S6 for analysis of possible cross-regulation of Bhlhb5 and Prdm8 at the protein level.
We next investigated more directly the possibility that Bhlhb5 and Prdm8 form a repressor complex by characterizing Bhlhb5 occupancy throughout the genome and testing whether Prdm8 is bound to the same genomic loci as Bhlhb5. The genomic binding sites of Bhlhb5 in the dorsal telencephalon were mapped by chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq). As a negative control, we also performed ChIP-seq using tissue from Bhlhb5−/− mice, thereby confirming the specificity of the Bhlhb5 antibody. In these ChIP-seq experiments, we identified ~ 2300 specific Bhlhb5 binding sites, representing approximately one binding site per million bases (see Ross-et-al-2011.hms.harvard.edu to visualize genomic data online). In addition to describing Bhlhb5 binding sites in the brain across the genome, the identification of these sequences allowed us to uncover an eight nucleotide consensus binding motif for Bhlhb5: CATATGNTNT (Figure 5A). Thus, Bhlhb5 binds to a sequence element consisting of a canonical E-box (underlined), a motif common to many members of the basic helix-loop-helix family, together with several other key nucleotides that likely confer additional sequence specificity.
Figure 5. Bhlhb5 and Prdm8 bind to common genomic loci.

DNA sequences to which Bhlhb5 is bound in the dorsal telencephalon at E17.5 were identified by chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq). A) Bhlhb5 consensus binding motif identified by the analysis of genomic Bhlhb5 binding sites. E-box is underlined. B – D) ChIP-seq data shows Bhlhb5 binding at its own proximal promoter (B), the proximal promoter of RP58 (C) and the first intron of Cdh11 (D). The height of the black bars indicates the number of input-normalized ChIP-Seq reads, representing the amount of Bhlhb5 binding. Consensus binding motifs for Bhlhb5 are found within the Bhlhb5 binding site at each gene, shown in red. Vertical gray bars indicate the genomic regions amplified in subsequent ChIP-qPCR experiments, either at the Bhlhb5 binding site (BS) or at a negative control region located −2 or −3 kb away, as indicated. The binding site for Bhlhb5 within the first intron of Cdh11 is indicated by the red bar. E – G) ChIP-qPCR using antibodies to Bhlhb5 confirms that Bhlhb5 is bound to its own proximal promoter (E), the proximal promoter of RP58 (F) and the first intron of Cdh11 (G). Experiments show significantly enriched Bhlhb5 binding at the identified Bhlhb5 binding site (BS) relative to the negative control region. H – J) ChIP-qPCR using antibodies to Prdm8 reveals that Prdm8 is also bound to the Bhlhb5 binding site at the proximal promoter of Bhlhb5 (H), the proximal promoter of RP58 (I) and the first intron of Cdh11 (J). Experiments show significantly enriched Prdm8 binding at the identified Bhlhb5 binding site (BS) relative to the negative control region. Also see Figure S7, in which 12 additional genomic loci are tested for the co-occupancy of Bhlhb5 and Prdm8. For E – J, chromatin immunoprecipitates were prepared from the dorsal telencephalon of P0 mice and y-axis (Binding) represents enrichment of gDNA over input (×10−3). For each ChIP experiment, antibody and/or knockout controls were performed in parallel, and in each case, these showed extremely low apparent binding, indicating that the ChIP experiments were specific (e.g., Figures S4C, S4D, 6A, 6B and S8). Data are representative of at least three independent experiments. * indicates significant (p < 0.05, t-test). K) Bhlhb5 and Prdm8 co-associate in neurons. Co-immunoprecipitation experiments (using ChIP conditions to preserve protein-DNA complexes) were performed to address whether Bhlhb5 and Prdm8 exist in a common complex. Bhlhb5 was immunoprecipited from the dorsal telencephalon of WT or Bhlhb5−/− mice and subjected to western blotting using antibodies to Prdm8 or Bhlhb5, as indicated. Data are representative of four independent experiments. This interaction appears to be specific since the transcriptional activator CREB was not observed in Bhlhb5-associated complexes (data not shown). Note that we were unable to co-immunoprecipitate Bhlhb5 and Prdm8 under native conditions, possibly because the interaction between Bhlhb5 and Prdm8 is indirect, or because they interact selectively in the presence of DNA.
Having identified genomic Bhlhb5 binding sites in the brain, we were in a position to ask whether Prdm8 binds to the same DNA sequence elements. To address this question, we chose several genes from the ChIP-seq data to test, including two genes that showed Bhlhb5 binding in the proximal promoter, Bhlhb5 itself and Repressor Protein 58 (RP58) (Figures 5B and 5C). In addition, we selected one of the putative Bhlhb5 target genes identified by expression profiling, Cdh11, which showed Bhlhb5 binding within its first intron (Figure 5D). First, we confirmed that Bhlhb5 bound to these genomic locations by ChIP-qPCR, comparing the binding of Bhlhb5 at its putative binding site to that observed at a negative control region 2 – 3 kilobases away (Figures 5E, 5F and 5G). Next, we performed ChIP-qPCR using specific antibodies to Prdm8 and examined whether the Bhlhb5 binding sites are likewise occupied by Prdm8. Notably, these experiments revealed that each of the loci tested that are bound by Bhlhb5 are also bound by Prdm8 (Figures 5H, 5I, and 5J). To ensure that the binding of Bhlhb5 and Prdm8 at these loci is specific we performed a number of negative controls. We observed no binding at these sites when preimmune antisera is used instead of immune antisera (e.g., Figures S4C and S4D) and none of these sites is bound by the transcription factors Npas4, CREB or SRF (Kim et al., 2010), thereby confirming specificity. To address whether the precise correspondence in binding sites for Bhlhb5 and Prdm8 is a widespread phenomenon, we tested twelve other genomic loci, including all of the putative Bhlhb5 binding sites that are found within 200 kb of genes that are misregulated in the Bhlhb5 knockout mouse. In general, a very good correspondence in binding between Bhlhb5 and Prdm8 was observed, suggesting that the vast majority of Bhlhb5 binding sites are also occupied by Prdm8 (Figure S7). Consistent with this idea, we found that Bhlhb5 and Prdm8 are associated with one another under the conditions used for ChIP, as revealed by co-immunoprecipitation and western blotting (Figure 5K). Taken together, these data strongly indicate that Bhlhb5 and Prdm8 are bound concurrently to common DNA elements throughout the genome where they repress transcription.
A Bhlhb5 homodimer binds specifically to DNA and then recruits Prdm8 to mediate repression
Our experiments provided several lines of evidence in support of the idea that Bhlhb5 and Prdm8 form a neural repressor complex: these factors are co-localized in neurons where they bind to the same genomic loci, and loss of either factor results in highly similar cellular and behavioral phenotypes, as well as the upregulation of a common set of genes. However, the discovery of this neural repressor complex left open a key remaining question—how does each component of the Bhlhb5/Prdm8 repressor complex function at a molecular level to repress gene expression?
As a first step to gain molecular insight into the nature of the Bhlhb5 repressor complex, we investigated whether Bhlhb5 forms a homo or heterodimer. Many members of the basic helix-loop-helix family of transcription factors bind DNA as a heterodimer with E-proteins (E2A, E2-2 and/or HEB) and of these, only E2-2 (also known as Tcf4) is expressed in post-mitotic neurons of the dorsal telencephalon (see www.stjudebgem.org). We therefore considered the possibility that Bhlhb5 might dimerize with E2-2. Alternatively, given that the Olig2, which is closely related to Bhlhb5, forms avid homodimers, we also tested whether Bhlhb5 might likewise partner with itself (Lee et al., 2005b; Li et al., 2011). To distinguish between these possibilities, we performed co-immunoprecipitation of tagged constructs expressed in heterologous cells. These experiments revealed that myc-tagged Bhlhb5 is able to pull down HA-tagged Bhlhb5 but not HA-tagged E2-2, suggesting that Bhlhb5 more readily forms a homodimer than a heterodimer with E-proteins (Figure 6A).
Figure 6. A Bhlhb5 dimer recruits Prdm8 to form a repressor complex at specific DNA targets.

A) Bhlhb5 forms a homodimer. HEK293T cells were transfected with the indicated constructs, immunoprecipitated with antibodies to myc, and blotted using antibodies to both myc and HA, revealing that myc-tagged Bhlhb5 can co-immunoprecipitate HA-tagged Bhlhb5 but not HA-tagged E2-2. Top blot shows the immunoprecipitated protein (IP); bottom blot shows 2.5% of input. Note that myc-Bhlhb5 (with 6 myc tags) and the IgG heavy chain have the same apparent molecular weight (~ 50 kDa), whereas HA-Bhlhb5 (with 3 HA tags) is ~ 40 kDA. The isoform of E2-2 used in these experiments was the longer form, E2-2B. B) ChIP experiments performed using tissue from knockout mice reveal that Prdm8 cannot target to Bhlhb5 binding sites in the absence of Bhlhb5. In WT mice, Bhlhb5 (red) and Prdm8 (blue) bind to the proximal promoter of RP58, as revealed by ChIP-qPCR for Bhlhb5 and Prdm8 (i). In Prdm8−/− mice, Bhlhb5 still binds to the proximal promoter RP58 (ii). In Bhlhb5−/− mice, Prdm8 can no longer bind to Bhlhb5 binding sites, indicating that Bhlhb5 is required for Prdm8 targeting to this loci (iii). Similar results were seen for the Bhlhb5 binding sites at the Cdh11 and Bhlhb5 genes (see Figure S4). Data are representative of three independent experiments. The y-axis (Binding) represents enrichment of gDNA over input (×10−3).
Next we addressed the molecular role of Prdm8 within this repressor complex. Some members of the Prdm family have been shown to function as sequence specific transcription factors, while others are known to function as co-factors to mediate transcriptional repression (Davis et al., 2006; Duan et al., 2005; Gyory et al., 2004; Hayashi et al., 2005; Kim et al., 2003). Given the phenotypic similarity between Bhlhb5 and Prdm8 mutant mice, we first considered the hypothesis that a Bhlhb5 dimer is recruited to specific DNA elements through its consensus binding motif and then recruits Prdm8 to mediate transcriptional repression. If so, we reasoned that Bhlhb5 would bind normally to its DNA targets in the absence of Prdm8, but that Prdm8 would not associate with these sites in the absence of Bhlhb5.
To address this hypothesis, we performed ChIP-qPCR from the dorsal telencephalon of wild type or mutant mice and analyzed the binding of Bhlhb5 and Prdm8 at the RP58 promoter. As we had shown above (Figures 5F and 5I), we again found that both Bhlhb5 and Prdm8 display robust binding to the proximal promoter of RP58 in wild type mice (Figure 6Bi). Furthermore, Bhlhb5 shows similar binding to the RP58 promoter when ChIP-qPCR was performed in Prdm8 mutant mice, indicating that the binding of Bhlhb5 at this promoter is not dependent on Prdm8 (Figure 6Bii). In sharp contrast, however, we found that Prdm8 was not bound to the RP58 promoter in Bhlhb5 mutant mice (Figure 6Biii). Note that the observed absence of Prdm8 binding at this site is not due to a general absence of Prdm8 protein in Bhlhb5−/− mice (e.g., see Figure 1C). Thus, the inability of Prdm8 to bind to the RP58 promoter in the absence of Bhlhb5 suggests that Prdm8 requires Bhlhb5 for targeting to this genetic locus. Furthermore, the dependence of Prdm8 on Bhlhb5 for sequence-specific targeting to DNA appears to be a general phenomenon since we observed similar results when we tested several other genomic loci including the Bhlhb5 promoter (Figure S8A) and the Bhlhb5 binding site in the first Cdh11 intron (Figure S8B).
Based on these findings, we suggest a model in which Bhlhb5 functions by binding to specific DNA elements possibly as a homodimer and then recruiting Prdm8 to mediate the repression of target genes (Figure 8A). When the Bhlhb5 alone is present, it can bind to target genes, but it cannot repress them. Likewise, when Prdm8 alone is present, target genes are also not repressed, in this case because Prdm8 does not bind to DNA in the absence of Bhlhb5. Thus, both factors are required to mediate transcriptional repression of a specific set of target genes so that, when either Bhlhb5 or Prdm8 is knocked out, common genes are upregulated and highly similar phenotypes result.
Figure 8. Model: Mechanism of repression.

A) A Bhlhb5 homodimer binds to its consensus binding element (1) and then recruits Prdm8 to mediate repression (2). Bhlhb5 requires Prdm8 to mediate repression, whereas Prdm8 requires Bhlhb5 for proper targeting to Bhlhb5 target genes. B) A schematic illustrating phylogenetic relationships among murine Bhlhb5-related proteins and Prdm8-related proteins, including Zfp488, a zinc finger protein that shows a very high degree of similarity with Prdm8 over the C-terminus (including the C2H2 zinc fingers) but lacks the SET domain characteristic of other Prdm family members. Our study reveals that Bhlhb5 and Prdm8 are obligate partners and others have shown that Olig2 interacts physically and functionally with Zfp488 (Wang et al., 2006), suggesting that the interaction between bHLH transcription factors and Prdm-related proteins may be a general mechanism of repression during neuronal development. Note that branch lengths are not scaled to distances. C) Prdm8 and Zfp488 show a very high degree of similarity across their C-termini. Amino acids 609 – 687 (for murine Prdm8) and 258 – 337 (for murine Zfp488) are shown. Identity = 53/80 (66%); Positives = 66/80 (82%); Gaps = 1/80 (1%). E-value, 3-e28. NLS, nuclear localization sequence.
Cdh11 is a critical target of the Bhlhb5/Prdm8 repressor complex
With this new insight into Bhlhb5 function, we next turned our attention to identifying a mechanistic link between the misexpressed target genes and the neural phenotypes observed upon the loss of the Bhlhb5/Prdm8 repressor complex. We focused on the genes that are misregulated in Bhlhb5 mutant mice at the time of the axon targeting defects (i.e. from E13.5 – E17.5). One of these genes is Cdh11, a classic type II cadherin that mediates homophilic cell-cell adhesion (Kimura et al., 1995). Cdh11 mRNA is expressed in differentiating neurons of the cortical plate, including layer V projection neurons that form the corticospinal tract (Kimura et al., 1996). Furthermore, we found that Cdh11 protein is highly expressed in the axons of corticofugal neurons at E16.5, when these projection neurons are extending their projections through the internal capsule (Figure 7C, white arrows), consistent with the idea that Cdh11 may play a role in their guidance. In addition, the subcortical expression pattern of Cdh11 is suggestive of a possible role in regulating the connectivity of corticospinal motor neurons. In particular, Cdh11 is specifically expressed in a number of intermediate subcortical targets where corticospinal motor neurons form collaterals, namely the red nucleus, the basilar pons, and the inferior olive (Kimura et al., 1996). Importantly, Bhlhb5 and Prdm8 bind to an intron within the Cdh11 gene (Figures 5D, 5G and 5J) and Cdh11 mRNA is up-regulated in both Bhlhb5 and Prdm8 mutant mice during embryonic development (Figures 7A and 7B). Upon loss of Bhlhb5, the overall level of Cdh11 protein appears elevated, whereas the pattern of Cdh11 expression is unaffected (Figures 7C and D), suggesting that a Bhlhb5/Prmd8 repressor complex may function to restrain the level of Cdh11 rather than its distribution.
Based on these observations, we hypothesized that Cdh11 might be a target of the Bhlhb5/Prdm8 repressor complex whose upregulation in the absence of Bhlhb5 or Prdm8 leads to a disruption of the formation of the corticospinal tract. Specifically, over-expression of Cdh11 in axons of corticospinal motor neurons might impede their progress due to enhanced adhesion to Cdh11-expressing intermediate targets. This might then prevent these Cdh11-overexpressing axons from extending past Cdh11-expressing intermediate targets and into the spinal cord. If so, we reasoned that reducing the level of Cdh11 in Bhlhb5 mutant mice might at least partially rescue the axon extension defects in corticospinal motor neurons. To test this idea we obtained Cdh11 mutant mice (Cdh11−/−), which lack functional Cdh11 due to a targeted disruption of the extracellular domain and most of the transmembrane domain (Horikawa et al., 1999). Importantly, mice lacking Cdh11 show normal targeting of corticospinal axons (Figure 7G and data not shown). We compared the size of the corticospinal tract in mice lacking both Bhlhb5 and Cdh11 (Bhlhb5−/−; Cdh11−/−) to littermates in which Bhlhb5 alone is disrupted (Bhlhb5−/−). For these experiments, we used anatomical landmarks to carefully identify matched coronal sections for each genotype, and then quantified the area marked by PKCγ, a protein that labels axons of the corticospinal tract. This analysis revealed that, relative to wild type mice, both Bhlhb5−/− single mutants and Bhlhb5−/−; Cdh11−/− double mutants show a significant reduction in the size of corticospinal tract as it passes through the caudal brain. However, the severity of this defect was significantly reduced in the Bhlhb5−/−; Cdh11−/− double mutants, which showed a significantly larger area of PKCγ staining than that observed in mice lacking Bhlhb5 alone, both in the ventral medulla and in the dorsal funiculus of the cervical spinal cord (Figures 7E, 7F and 7G). These findings indicate that reducing the level of expression of Cdh11 in Bhlhb5 mutant mice results in a partial rescue of the area of the corticospinal tract, suggesting that Cdh11 is one of the Bhlhb5/Prdm8 target genes that must be repressed for proper axon targeting of corticospinal motor neurons to occur.
We next considered the possibility that upregulation of Cdh11 expression in the absence of the Bhlhb5/Prdm8 complex might contribute to other phenotypes observed in Bhlhb5 and Prdm8 mutant mice. Consistent with this idea, we observed that, whereas virtually all Bhlhb5−/− mice developed skin lesions by eight weeks of age, almost none of the Bhlhb5−/−; Cdh11−/− double mutants had skin lesions at this time. To confirm this observation, we carefully documented the age of onset of skin lesions in littermates that were either single (Bhlhb5−/−) or double (Bhlhb5−/−; Cdh11−/−) mutants. Although mice of both genotypes eventually developed skin lesions, the appearance of lesions occurred significantly later in Bhlhb5−/−; Cdh11−/− mice compared to Bhlhb5−/− mice (13 vs. 6 weeks, respectively, n = 12 littermate pairs; Figure 7H), whereas mice lacking Cdh11 alone never developed skin lesions. Thus, loss of Cdh11 significantly delays the onset of skin lesions in Bhlhb5 mutant mice. The partial genetic rescue of two phenotypes (skin lesions and corticospinal tract axon mistargeting) observed in Bhlhb5−/− mice supports the idea that Cdh11 is a target of the Bhlhb5/Prdm8 repressor complex, and suggests that the precise regulation of this adhesion molecule through repression is critical for proper neural development.
DISCUSSION
We examined how Bhlhb5 functions at a molecular level and discovered that it couples specifically with Prdm8 to form a neuronal repressor complex. This conclusion is supported by our observation that a common set of genes is aberrantly over-expressed upon loss of either factor, likely accounting for the striking phenotypic similarities between Bhlhb5 and Prdm8 knockout mice. In addition, our studies reveal that Bhlhb5 and Prdm8 bind to common genetic loci and, importantly, each requires the other for its activity: Prdm8 can no longer target these loci in the absence of Bhlhb5, whereas Bhlhb5 can bind target genes but cannot repress them without Prdm8 (Figure 8A). Finally, genetic rescue experiments reveal that Cdh11 is one of the targets of the Bhlhb5/Prdm8 repressor complex whose upregulation in the absence of the Bhlhb5/Prdm8 repressor complex contributes to the abnormal phenotype observed in Bhlhb5 and Prdm8 mutant mice. Taken together, these findings suggest that Bhlhb5 and Prdm8 are obligate components of a transcriptional repressor complex. Bhlhb5 binds to specific DNA within the regulatory regions of its target genes and recruits Prdm8 to mediate repression of the transcription of these targets. In the absence of Bhlhb5 or Prdm8 the gene targets are upregulated resulting in abnormal development.
The coupling of bHLH factors and Prdm-related proteins as a common theme in neural development
Since Bhlhb5 and Prdm8 belong to conserved gene families, our finding that these two factors specifically interact raises the question as to whether the functional association between bHLH factors and Prdm-related proteins is a more general occurrence. Phylogenetic analysis of SET-domain containing proteins reveals that Prdm8 is most closely related to Prdm13, and recent studies show that Prdm8 and Prdm13 are expressed in non-overlapping patterns in the developing nervous system (Fumasoni et al., 2007; Kinameri et al., 2008). However, we unexpectedly discovered that the gene with which Prdm8 shares the highest degree of similarity (as revealed using a blastn algorithm comparing murine genes) is not a member of the Prdm family, but rather the zinc-finger protein, Zfp488. This gene shares 82% similarity with Prdm8 over 80 amino acids in the C-terminus (Figure 8C), suggesting that Prdm8 and Zfp488 may share a common ancestor. The idea that Prdm8 and Zfp488 are ancestrally related proteins is noteworthy because Zfp488 was recently shown to interact physically and functionally with one of Bhlhb5’s closest relatives, Olig2 (Wang et al., 2006). This intriguing connection suggests the possibility that the interaction between bHLH transcription factors and Prdm-related proteins is a general mechanism for the regulation of gene expression during neural development (Figure 8B). In this regard, it is notable that both Prdm13 and Olig3 are expressed in Class A progenitors in the dorsal spinal cord, consistent with the idea that these two factors may also couple selectively to mediate transcriptional repression in these cells (Kinameri et al., 2008; Muller et al., 2005).
We provide phenotypic and mechanistic evidence that Bhlhb5 and Prdm8 are obligate partners for certain aspects of development. However, it is likely that Prdm8 functions without Bhlhb5 in some contexts. This prediction is based on the observation that, while Prdm8 shows significant overlap with the Bhlhb5 expression domain, there are regions of the nervous system that express Prdm8 but not Bhlhb5. For instance, Prdm8 (but not Bhlhb5) is widely expressed in the developing diencephalon, throughout neurons of the dorsal root ganglia (DRG), and in rod bipolar cells of the retina (S.E. Ross, unpublished observation). This somewhat broader pattern of expression of Prdm8 relative to Bhlhb5 suggests that Prdm8 may have additional partners to which it can couple, and one attractive candidate in this regard is Bhlhb4: loss of function studies have revealed that Bhlhb4 is required for the survival of rod bipolar cells and, furthermore, that this factor is expressed, like Prdm8, in the embryonic diencephalon and DRG (Bramblett et al., 2002; Bramblett et al., 2004).
Molecular function of the Bhlhb5/Prdm8 repressor complex
Because Prdm8 contains a SET domain that is characteristic of histone methyltransferases, it is possible that it may directly mediate repression of target genes by methylating target gene-associated histones. Consistent with this idea, Prdm8 has been shown to methylate histone H3K9 in vitro (Eom et al., 2009), a modification associated with transcriptional repression. Likewise, the tumor suppressor Prdm2 and the meiotic recombination determinant Prdm9 also show intrinsic histone methyltransferase activity (Hayashi et al., 2005; Kim et al., 2003). However, several other Prdm family members, including Prdm1, Prdm5 and Prdm6, appear to mediate repression indirectly by recruiting the histone methyltransferase, G9A (Davis et al., 2006; Duan et al., 2005; Gyory et al., 2004). Thus, it is not yet clear whether Prdm8 functions directly or indirectly to mediate transcriptional repression. In either case, however, Prdm8 appears to be required for the repression of Bhlhb5 target genes.
A curious aspect of the Bhlhb5/Prdm8 repressor complex is that, while each requires the other to repress target gene expression, we do not observe a perfect coincidence the expression of Bhlhb5 and Prdm8. Indeed, in many cases, the expression of these two factors appears to be somewhat reciprocal—neurons with highest levels of Bhlhb5 tend to have low levels of Prdm8, and vice versa. This disparity in expression level implies that Bhlhb5 and Prdm8 do not always exist as part of a functional repressor complex, and furthermore suggests that the expression of these factors is very tightly controlled, possibly to limit the degree and/or the duration of gene repression mediated by Bhlhb5/Prdm8. In keeping with this idea, we find that the Bhlhb5/Prmd8 repressor appears to curb its own activity by restricting the expression of Prdm8, which is upregulated in Bhlhb5 knockout mice. These observations suggest that Bhlhb5 and Prdm8 are part of a complex regulatory network that needs to be precisely coordinated for proper development.
A possible role for Cdh11 in neural circuit formation
One of the consequences of disrupting the function of the Bhlhb5/Prdm8 repressor complex is that Cdh11 is aberrantly over-expressed, and our findings suggest that this misexpression has detrimental consequences for neuronal developement. Previous studies have revealed that cadherins form a large family of cell-cell adhesion molecules that have been proposed to serve as an adhesive code underlying neural assembly (Takeichi, 2007). Classic type II cadherins, in particular, are expressed in restricted groups of synaptically interconnected neurons in a manner that is highly suggestive of a role in neural circuit formation (Inoue et al., 1998; Suzuki et al., 1997). We speculate that the importance of Cdh11 for neural connectivity is masked in loss-of-function studies, possibly as a consequence of redundancy in the mechanisms that mediate neural circuit assembly. Here we have revealed a possible role for Cdh11 in circuit development by observing the effects of a Cdh11 gain-of-function—that is, overexpression of Cdh11 due to loss of the Bhlhb5/Prdm8 repressor complex. We suggest that Cdh11 may be involved in the interaction between axons from corticospinal motor neurons and intermediate subcortical targets, such the red nucleus, the basilar pons, and the inferior olive, which also express Cdh11. If so, overexpression of Cdh11 in axons of corticospinal motor neurons may impede their progress beyond these intermediate targets and into the spinal cord. In addition, our genetic rescue experiments implicate Cdh11 as one target of the Bhlhb5/Prdm8 repressor complex in the spinal cord, where Bhlhb5/Prdm8-mediated repression is required for the proper function of neural circuits that mediate itch sensation. Thus, Bhlhb5/Prdm8-mediated repression may be required to ensure that Cdh11 is expressed at the right time and place for proper connectivity and function of motor and sensory circuits. Formal proof that Cdh11 is a direct target of the Bhlhb5/Prdm8 repressor complex will require analysis of the axonal projection and itch phenotypes in a knock–in mouse in which the Bhlhb5 binding site within Cadh11 gene is disrupted.
Since the loss of Cdh11 in Bhlhb5 mutant mice results only in a partial rescue of axon extension in corticospinal neurons, it is likely that other misexpressed genes also contribute to this phenotype. In this regard, it is noteworthy that several additional putative Bhlhb5/Prdm8 target genes, including p75 NTR, Necdin, MageL2, and Netrin have been shown to play roles in axon extension and/or axon guidance (Lee et al., 2005a; Marthiens et al., 2005; Serafini et al., 1994; Yamashita et al., 1999). Furthermore, netrin signaling is required for the guidance of corticospinal tract axons (Finger et al., 2002). Thus, our work identifies several interesting candidates that warrant further study since they may contribute to the axon targeting defects observed when either Bhlhb5 or Prdm8 function is disrupted.
EXPERIMENTAL PROCEDURES
See Supplemental Experimental Procedures for details on animal husbandry and colony management, immunohistochemistry, gene expression analysis by qPCR, ChIP-Seq library construction, ChIP, read alignment, the identification of Bhlhb5 binding sites, the identification of Bhlhb5 consensus binding motif, co-immunoprecipitation, the generation of phylogenetic trees, and quantitative western blotting.
Quantification of corticospinal tract axon area
Five pairs of littermate adult mice (~ 8 wks) that were lacking either Bhlhb5 alone (Bhlhb5−/−) or both Bhlhb5 and Cdh11 (Bhlhb5−/−; Cdh11−/−) were perfused with 4% PFA and the brain was dissected out with the cervical spinal cord attached. For each sample, serial sections (20 μm) were collected from the cortex through to the cervical spinal cord. Every 5th section was co-stained with PKCγ (which marks the corticospinal tract), as well as NeuN and Hoescht to assist with matching levels between samples. Matched images corresponding to two regions were selected for analysis: I) caudal to the basilar pons and II) caudal to the pyramidal decussation. Images were analyzed in Metamorph. A constant threshold was applied to all images and the dorsal funiculus was masked. We then computed the area above threshold, which was normalized to the area observed in wild type mice. All measurements were conducted blind to genotype.
Generation of Phylogenetic trees
Phylogenetic trees of murine Bhlhb5- and Prdm8-related proteins were created using the amino acid sequences of each murine protein and the ClustalW algorithm, with MyoD and G9A as the outgroups, respectively. Apart from Zfp488, which we added based on our discovery of high similarity in protein sequences between Prdm8 and Zfp488 (Evalue 3e-28), the decision of which family members to include in the phylogenetic analysis was based on previous analyses for bHLH (Ledent et al., 2002; Ledent and Vervoort, 2001; Stevens et al., 2008) and Prdm families (Fumasoni et al., 2007).
Supplementary Material
Acknowledgments
We thank M. Takeichi for supplying the Cdh11 mutant mice; A. Cano for supplying the HA-tagged E2-2B expression vector; E. C. Griffith for critical readings of the manuscript; D. Harmin for help with statistical analysis; P. Zhang for assistance with mouse colony management; the Intellectual and Developmental Disabilities Research Center (IDDRC) Gene Manipulation Core (M. Thompson, Y. Zhou, and H. Ye); the Harvard Medical School Rodent Histopathology Core (R.T. Bronson), and the IDDRC Molecular Genetics Core. This work was supported by a Jane Coffin Childs Fellowship and a Dystonia Medical Research Foundation Fellowship to S.E.R., NIH grant NS028829 to M.E.G., and the Developmental Disabilities Mental Retardation Research Center grant NIH-P30-HD-18655.
Footnotes
Supplemental Information for this article includes eight figures, one table, and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.neuron.2010.02.025.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T, Surani MA. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol. 2006;8:623–630. doi: 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Lam KW, Sarbajna S, Odenthal-Hesse L, May CA, Jeffreys AJ. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Bramblett DE, Copeland NG, Jenkins NA, Tsai MJ. BHLHB4 is a bHLH transcriptional regulator in pancreas and brain that marks the dimesencephalic boundary. Genomics. 2002;79:402–412. doi: 10.1006/geno.2002.6708. [DOI] [PubMed] [Google Scholar]
- Bramblett DE, Pennesi ME, Wu SM, Tsai MJ. The transcription factor Bhlhb4 is required for rod bipolar cell maturation. Neuron. 2004;43:779–793. doi: 10.1016/j.neuron.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Davis CA, Haberland M, Arnold MA, Sutherland LB, McDonald OG, Richardson JA, Childs G, Harris S, Owens GK, Olson EN. PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol. 2006;26:2626–2636. doi: 10.1128/MCB.26.7.2626-2636.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol. 2005;25:10338–10351. doi: 10.1128/MCB.25.23.10338-10351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. Microarrays, Empirical Bayes and the Two-Groups Model. Statis Sci. 2008;23:1–22. [Google Scholar]
- Eom GH, Kim K, Kim SM, Kee HJ, Kim JY, Jin HM, Kim JR, Kim JH, Choe N, Kim KB, et al. Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun. 2009;388:131–136. doi: 10.1016/j.bbrc.2009.07.134. [DOI] [PubMed] [Google Scholar]
- Farah MH, Olson JM, Sucic HB, Hume RI, Tapscott SJ, Turner DL. Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development. 2000;127:693–702. doi: 10.1242/dev.127.4.693. [DOI] [PubMed] [Google Scholar]
- Feng L, Xie X, Joshi PS, Yang Z, Shibasaki K, Chow RL, Gan L. Requirement for Bhlhb5 in the specification of amacrine and cone bipolar subtypes in mouse retina. Development. 2006;133:4815–4825. doi: 10.1242/dev.02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger JH, Bronson RT, Harris B, Johnson K, Przyborski SA, Ackerman SL. The netrin 1 receptors Unc5h3 and Dcc are necessary at multiple choice points for the guidance of corticospinal tract axons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:10346–10356. doi: 10.1523/JNEUROSCI.22-23-10346.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyory I, Wu J, Fejer G, Seto E, Wright KL. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol. 2004;5:299–308. doi: 10.1038/ni1046. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature. 2005;438:374–378. doi: 10.1038/nature04112. [DOI] [PubMed] [Google Scholar]
- Horikawa K, Radice G, Takeichi M, Chisaka O. Adhesive subdivisions intrinsic to the epithelial somites. Dev Biol. 1999;215:182–189. doi: 10.1006/dbio.1999.9463. [DOI] [PubMed] [Google Scholar]
- Inoue T, Tanaka T, Suzuki SC, Takeichi M. Cadherin-6 in the developing mouse brain: expression along restricted connection systems and synaptic localization suggest a potential role in neuronal circuitry. Dev Dyn. 1998;211:338–351. doi: 10.1002/(SICI)1097-0177(199804)211:4<338::AID-AJA5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Joshi PS, Molyneaux BJ, Feng L, Xie X, Macklis JD, Gan L. Bhlhb5 regulates the postmitotic acquisition of area identities in layers II-V of the developing neocortex. Neuron. 2008;60:258–272. doi: 10.1016/j.neuron.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KC, Geng L, Huang S. Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res. 2003;63:7619–7623. [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Matsunami H, Inoue T, Shimamura K, Uchida N, Ueno T, Miyazaki T, Takeichi M. Cadherin-11 expressed in association with mesenchymal morphogenesis in the head, somite, and limb bud of early mouse embryos. Dev Biol. 1995;169:347–358. doi: 10.1006/dbio.1995.1149. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Matsunami H, Takeichi M. Expression of cadherin-11 delineates boundaries, neuromeres, and nuclei in the developing mouse brain. Dev Dyn. 1996;206:455–462. doi: 10.1002/(SICI)1097-0177(199608)206:4<455::AID-AJA11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kinameri E, Inoue T, Aruga J, Imayoshi I, Kageyama R, Shimogori T, Moore AW. Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS One. 2008;3:e3859. doi: 10.1371/journal.pone.0003859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent V, Paquet O, Vervoort M. Phylogenetic analysis of the human basic helix-loop-helix proteins. Genome Biol. 2002;3:RESEARCH0030. doi: 10.1186/gb-2002-3-6-research0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent V, Vervoort M. The basic helix-loop-helix protein family: comparative genomics and phylogenetic analysis. Genome Res. 2001;11:754–770. doi: 10.1101/gr.177001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O’Neill MA, Wevrick R. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum Mol Genet. 2005a;14:627–637. doi: 10.1093/hmg/ddi059. [DOI] [PubMed] [Google Scholar]
- Lee SK, Lee B, Ruiz EC, Pfaff SL. Olig2 and Ngn2 function in opposition to modulate gene expression in motor neuron progenitor cells. Genes & development. 2005b;19:282–294. doi: 10.1101/gad.1257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Kintner C, Anderson DJ. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Pfaff SL. Cracking the transcriptional code for cell specification in the neural tube. Cell. 2001;106:651–654. doi: 10.1016/s0092-8674(01)00499-8. [DOI] [PubMed] [Google Scholar]
- Marthiens V, Gavard J, Padilla F, Monnet C, Castellani V, Lambert M, Mege RM. A novel function for cadherin-11 in the regulation of motor axon elongation and fasciculation. Mol Cell Neurosci. 2005;28:715–726. doi: 10.1016/j.mcn.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–175. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, Barton SC, Obukhanych T, Nussenzweig M, Tarakhovsky A, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436:207–213. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi G, Mauri G, Pesole G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics. 2001;17(Suppl 1):S207–214. doi: 10.1093/bioinformatics/17.suppl_1.s207. [DOI] [PubMed] [Google Scholar]
- Peyton M, Stellrecht CM, Naya FJ, Huang HP, Samora PJ, Tsai MJ. BETA3, a novel helix-loop-helix protein, can act as a negative regulator of BETA2 and MyoD-responsive genes. Mol Cell Biol. 1996;16:626–633. doi: 10.1128/mcb.16.2.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SE, Greenberg ME, Stiles CD. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. doi: 10.1016/s0896-6273(03)00365-9. [DOI] [PubMed] [Google Scholar]
- Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, et al. Loss of inhibitory interneurons in the dorsal spinal cord and elevated itch in Bhlhb5 mutant mice. Neuron. 2010;65:886–898. doi: 10.1016/j.neuron.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurmans C, Guillemot F. Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol. 2002;12:26–34. doi: 10.1016/s0959-4388(02)00286-6. [DOI] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M. The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell. 1994;78:409–424. doi: 10.1016/0092-8674(94)90420-0. [DOI] [PubMed] [Google Scholar]
- Skellam JG. The frequency distribution of the difference between two Poisson variates belonging to different populations. J R Stat Soc Ser A. 1946;109:296. [PubMed] [Google Scholar]
- Sobrado VR, Moreno-Bueno G, Cubillo E, Holt LJ, Nieto MA, Portillo F, Cano A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J Cell Sci. 2009;122:1014–1024. doi: 10.1242/jcs.028241. [DOI] [PubMed] [Google Scholar]
- Stevens JD, Roalson EH, Skinner MK. Phylogenetic and expression analysis of the basic helix-loop-helix transcription factor gene family: genomic approach to cellular differentiation. Differentiation. 2008;76:1006–1022. doi: 10.1111/j.1432-0436.2008.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki SC, Inoue T, Kimura Y, Tanaka T, Takeichi M. Neuronal circuits are subdivided by differential expression of type-II classic cadherins in postnatal mouse brains. Mol Cell Neurosci. 1997;9:433–447. doi: 10.1006/mcne.1997.0626. [DOI] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol. 2002;12:1157–1163. doi: 10.1016/s0960-9822(02)00926-0. [DOI] [PubMed] [Google Scholar]
- Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8:11–20. doi: 10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- Wang SZ, Dulin J, Wu H, Hurlock E, Lee SE, Jansson K, Lu QR. An oligodendrocyte-specific zinc-finger transcription regulator cooperates with Olig2 to promote oligodendrocyte differentiation. Development. 2006;133:3389–3398. doi: 10.1242/dev.02522. [DOI] [PubMed] [Google Scholar]
- Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, et al. Structural biology of human H3K9 methyltransferases. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZP, Dutra A, Stellrecht CM, Wu C, Piatigorsky J, Saunders GF. Functional and structural characterization of the human gene BHLHB5, encoding a basic helix-loop-helix transcription factor. Genomics. 2002;80:311–318. doi: 10.1006/geno.2002.6833. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.