

The bryostatins are a family of natural products of marine origin that display both intriguing structural complexity and a fascinating profile of biological activity.[1] These materials were isolated (from Bugula neritina) and their structures determined through the pioneering work of Pettit and coworkers.[2] Subsequently, a monumental large scale collection and isolation effort managed to yield some 18 grams of bryostatin 1, the most abundant and now most thoroughly investigated member of this family, from some 13,000 kg of the source organism.[3] This world’s supply of material has supported numerous biological investigations and roughly 80 clinical trials against various cancers.[4] Recently, a clinical trial against Alzheimer’s disease has also commenced.[5]
Despite this intense interest in the bryostatins as potential therapeutics, the mechanisms by which bryostatin 1 elicits its biological responses are only partially understood. It has been established that bryostatin 1 binds with high affinity to the regulatory C1 domains of protein kinase C (PKC) isozymes and thereby activates these enzymes.[6] It likewise binds to the homologous regulatory C1 domains of six other families of signaling proteins, e.g. the chimaerins and RasGRPs, to modulate their activities.[7] Physiologically, all of these proteins function, through their C1 domains, as sensors for the lipophilic second messenger sn-1,2-diacylglycerols. Paradoxically, however, whereas bryostatin 1 binds to the same binding site as do the diacylglycerols or their high affinity analogs, the phorbol esters, bryostatin 1 induces only a subset of the responses observed with these other ligands.[8] Moreover, bryostatin 1 blocks those responses that it does not itself induce and, in particular, is not tumor promoting, in contrast to most phorbol esters (e.g. phorbol 12-myristate-13-acetate, PMA). Despite the intense synthetic attention this family of compounds has attracted, bryostatin 1 has not as yet been synthesized, although the structurally similar and high affinity bryostatins 2, 3, and 7 have been prepared.[9] In addition, bryostatin 16, which has markedly diminished affinity for PKC (Ki = 118 nM) relative to bryostatin 1 (Ki = 1.35 nM), has also been prepared.[10] Several analogues of bryostatin have also been prepared, primarily by the Wender group and by us.[1,11]
Recently, our group has been focused on elucidating the structural features of bryostatin that are responsible for its function as a phorbol ester antagonist, as distinct from its activity simply as a ligand for PKC. We have previously reported on the synthesis of the bryopyran core structure [12] and of bryopyran analogues with greatly simplified A- and B-rings that function as phorbol ester mimics,[13] and have shown that functionality on the A-ring of bryostatin 1 is critical in preserving bryostatin-like biological effects.[14] In the present report, we describe the results of studies designed to reveal the influence of the C9 hydroxyl substituent of the A ring on the biological responses elicited by bryostatin 1.
Among the models that have been put forward previously for the binding of bryostatin 1 to the C1 domain of PKC, the computationally derived model of Itai and co-workers proposed an explicit H-bonding interaction (one of four) between the C9-OH and the C1 domain of the protein.[15] We began by independently examining the docking of bryostatin 1 and its C9-deoxy analogue to the C1 domain. Before beginning the docking we first performed a conformational search of bryostatin 1 in implicit water and octanol solvents. The global energy minimum conformation found in both solvents was essentially identical to the crystal[2] and NMR[16]conformations, and is characterized by an intramolecular H-bonding network in which the proton of the C3-OH makes a bifurcated H-bond between with the pyran oxygens of the A- and B-rings, and a second H-bond is established between the proton of the C19-OH and the oxygen of the C3-OH. The four lowest-energy conformations from the search were saved, and corresponding structures of C9-deoxy bryostatin 1 for each of these conformations were built by simply replacing the C9 hydroxyl with a hydrogen atom.
These conformations of bryostatin 1 and C9-deoxy bryostatin were then docked into the crystal structure of the C1b domain of PKC-δ.[17] We included a similarity constraint to the crystallized phorbol-13-acetate ligand to bias the optimization toward solutions where the acceptor atoms in bryostatin 1 and phorbol are close in space. The highest-scoring pose for both compounds was the global minimum conformation in solution, suggesting that bryostatin 1 does not undergo a conformational change upon binding to the C1 domain. The docked structures are shown in Figure 2.
Figure 2.

Bryostatin 1 (A) and C9-Deoxy Bryostatin 1 (B) docked into the C1 domain of PKCδ
The overall binding mode does not change with the presence or absence of the C9 hydroxyl, and is very similar to the model proposed by Itai et al.[15] The A- and B-rings lie above the binding site in the plane of the bilayer. In both compounds, the C26 hydroxyl is buried in the binding site, and forms hydrogen bonds with the backbone carbonyl of Leu 251 and the backbone NH of Thr 242. The ester oxygen adjacent to C1 appears to make a weak hydrogen bond to the NH of Gln 257. The methoxycarbonyl group at C34 extends over the edge of the binding site and hydrogen bonds to the backbone NH of Gly 253. Bryostatin also forms an additional hydrogen bond between the C9 hydroxyl and the backbone carbonyl of Met 239. This hydrogen bond is not formed by the phorbol ester compounds, lending support to the hypothesis that the C9 hydroxyl might be an important structural determinant for bryostatin’s biological activity.
To evaluate experimentally the contribution of the C9-OH substituent to the observed biological profile associated with bryostatin 1, we targeted C9-deoxy bryostatin 1 (Merle 30) for synthesis.[18] Here we were able to utilize an advanced intermediate (1) that we had prepared previously in our laboratories enroute to another “almost bryostatin” analogue, Merle 28.[14] The synthesis of the new C9-deoxy analogue Merle 30 is outlined in Scheme 1.
Scheme 1.

Synthesisof C9-Deoxy Bryostatin 1. (a) TMSOTf, Et3SiH, CH2Cl2, −78 °C, 82%; (b) O3,CH2Cl2,−78 °C, then DMS, 89%; (c) DDQ, pH7 buffer, CH2Cl2, 0 °C, 91%; (d) mCPBA, THF/H2O, 83%; (e) 2,4,6-Cl3PhCOCl, Et3N, THF, then DMAP, tol, 40 °C, 79%; ( f) 8, NaHMDS, THF, −78 °C, then diketone, 0 °C, Z:E =4:1, 85%; (g) K2CO3, MeOH, MeO(OH)CCO2Me; (h) Ac2O, DMAP, Py, 60% 2 steps; (i) NaBH4, CeCl3, MeOH, −40 °C; (j) (C8H11O)2O, DMAP,Py, dr =5:1, 75% 2 steps; (k)HF·Py, THF/MeOH/Py, (l) LiBF4, MeCN/H2O(20:1), 80 °C, 68%2 steps
The regio- and stereoselective deoxygenation of the C9 methyl ketal in the presence of the C19 methyl ketal was carried out by treating the bisketal compound 1 with TMSOTf and triethylsilane which afforded the reduced product 2 in excellent yield. In order to install the enoate on the B ring, a cross metathesis with methyl acrylate was first attempted.[19] However, this reaction provided an inseparable mixture of E/Z isomeric products in low yield. Alternatively, installation of this enoate could be approached by an asymmetric Horner-Wadsworth-Emmons (HWE) reaction with the corresponding ketone.[20] The oxidative cleavage of the C13-C33 olefin in the presence of the C16-C17 olefin proved to be unselective utilizing OsO4/NaIO4; however, when the bisolefin 2 was subjected to stoichiometric ozonolysis conditions using an ozone solution, the reaction was selective for the exocyclic olefin and after reductive workup provided the bisketone 3. When the asymmetric Horner-Wadsworth-Emmons reaction was conducted on bisketone 3 using the Fuji phosphonate reagent 8 derived from R-BINOL, once again, an inseparable 1:1.4 mixture of olefin isomers resulted. This result prompted us to attempt the HWE reaction later on a macrocyclic compound.
To prepare the seco acid 4 required for macrolactonization, the PMB group was removed under standard oxidative conditions and the thiolester was hydrolyzed using mCPBA/H2O in THF.[21] Yamaguchi macrolactonization of the seco acid then afforded the macrolactone in good yield.[22] With the macrolactone in hand, an asymmetric Horner-Wadsworth-Emmons reaction was carried out using the reagent 8[20] to give a 4:1 mixture of Z/E olefin isomers, which were separated by preparative thin layer chromatography.
In order to install the enoate functionality on the C ring, an aldol reaction was attempted between the C20 ketone and methyl glyoxylate using 1.1 equivalent of LDA. Surprisingly, and in contrast to previous experience with similar compounds, the aldol reaction took place exclusively on the C7 acetate of the C9-deoxy compound 5. Gratifyingly, when the ketone 5 was subjected to reaction with the methyl acetal of methyl glyoxylate,[23] using K2CO3/MeOH, a mixture of aldol adduct and the condensed product were obtained in which the C7 acetate had been hydrolyzed in both cases. A simple acetylation of the crude product both completed conversion of the aldol product into the desired enoate and reinstalled the C7 acetate, yielding the desired enoate 6 as a single isomer. Finally, Luche reduction of the C20 ketone followed by esterification with octadienoic anhydride provided the protected form of C9-deoxy bryostatin 1. The removal of the BPS (tert-butyldiphenylsilyl) group using HF·Py followed by global deprotection using LiBF4 in aqueous CH3CN completed the synthesis of C9-deoxy bryostatin 1 (Merle 30).[24]
Biological evaluation of this new analogue began with an assay of the binding affinity with PKCα, which gave Ki = 0.38 nM. Thus, this analogue binds comparably to bryostatin 1, despite the absence of the proposed interaction between the C9-OH and the Met 239 carbonyl.[15] Hence the computational result that the structure of the docked complex does not change if the H-bonding contact between the C9-OH and the protein is deleted is apparently mirrored experimentally in the binding affinity of Merle 30.
Functional activity of Merle 30 in living cells was assessed using the U937 leukemia cell system that we have employed previously. These cells give differential responses to PMA and bryostatin 1.[25] PMA is strongly anti-proliferative while bryostatin 1 causes only a minor, biphasic decrease in cell proliferation. When used in combination, bryostatin 1 is observed to block the anti-proliferative effect of PMA in a dose dependent manner. In addition, PMA induces attachment of the U937 cells while bryostatin 1 does not; again, bryostatin 1 antagonizes the PMA effect. When the results for these experiments are displayed as a function of dose, a characteristic visual “fingerprint” for each agent results, which can then be easily compared to those for PMA and for bryostatin 1. The results with Merle 30 in these assays, as well as the results for the previous bryostatin-like analogue Merle 28, are shown in Figure 3.
Figure 3.

Results of Proliferation and Attachment Assays in U937 Leukemia Cells for C9-Deoxy Bryostatin 1 (Merle 30)
It can be seen that analogue Merle 30 is largely bryostatin–like in its biological response in these assays, although with a little more PMA-like character (larger decrease in cell proliferation and greater attachment). Both analogues Merle 28 and Merle 30, which correspond in structure to deletion of just one polar group from the A- or B- ring of bryostatin 1, are only slightly less potent than bryostatin 1 itself in blocking the effects induced by PMA. Presumably, deletion of both polar groups simultaneously would provide an analogue that is much less bryostatin-like. Although that precise experiment has not yet been accomplished, it is of interest to note that deletion of both of these polar groups as well as deletion of the gem dimethyl group provides analogue Merle 27, which we have previously characterized as exhibiting PMA-like biology in the U937 cell system.[26]
The biological activity of Merle 30 was also examined in another system, the androgen-dependent human prostate cancer cell line LNCaP, for two different endpoints: proliferation and secretion of tumor necrosis factor α (TNFα), a key mediator of inflammation. In this system PMA inhibits cell proliferation and induces apoptosis whereas bryostatin 1 does not.[27] For proliferation, Merle 30 behaves almost identically to bryostatin 1; it does not inhibit LNCaP proliferation but does antagonize the inhibition by PMA. For induction of TNFα secretion, the three agents behave differently: PMA induces a potent response, bryostatin 1 induces no response, and Merle 30 induces a weak biphasic response. (See supporting information.) Once again, those compounds which themselves do not induce secretion block the response to PMA. We conclude that the C9 hydroxyl of bryostatin 1, at first hypothesized to be a key substituent, makes only a minor specific contribution to both the binding affinity for bryostatin 1 and the unique patterns of biological response to bryostatin 1.
Our current understanding of C1 domain function is that ligand binding drives interaction of the top face of the C1 domain with the lipid bilayer. Modeling indicates that the bryostatin overlays this top face, providing a new surface arrayed with both hydrophobic and polar groups. The complexity of the bryostatin–C1 domain surface provides the opportunity to differentially drive its interactions, whether with different membrane environments within the cell, with protein partners, or with other domains of the PKC itself. Both specific substituents on the bryopyran as well as its overall physico-chemical properties might be expected to make contributions. The diversity of these interactions could well underlie the complex structure activity relations of the bryostatins and their bryopyran analogues.
Figure 1.
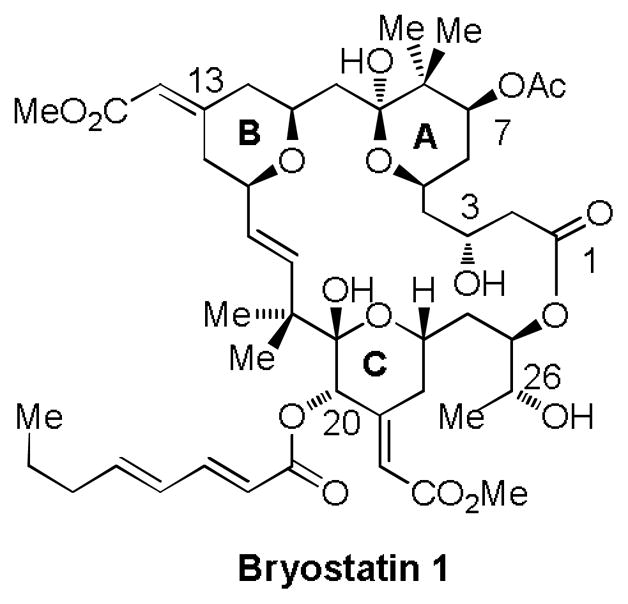
Structure of Bryostatin 1
Figure 4.

Structures of Merle 27, Merle 28 and PMA
Footnotes
Financial support for this work was provided by the National Institutes of Health through grant GM28961, in part by the intramural research program of the NIH, NCI, CCR, and in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E.
Supporting information for this article is available on the WWW at http://www.angewandte.org.
Contributor Information
Prof. Gary E. Keck, Email: keck@chem.utah.edu, Department of Chemistry, University of Utah, 315 South 1400 East, Rm 2020, Salt Lake City, UT 84112 (USA), Fax: (+1) 801-585-0024
Yam B. Poudel, Department of Chemistry, University of Utah, 315 South 1400 East, Rm 2020, Salt Lake City, UT 84112 (USA), Fax: (+1) 801-585-0024
Arnab Rudra, Department of Chemistry, University of Utah, 315 South 1400 East, Rm 2020, Salt Lake City, UT 84112 (USA), Fax: (+1) 801-585-0024.
Jeffrey C. Stephens, Department of Chemistry, University of Utah, 315 South 1400 East, Rm 2020, Salt Lake City, UT 84112 (USA), Fax: (+1) 801-585-0024
Noemi Kedei, Laboratory for Cancer Biology and Genetics, CCR, NCI, NIH, Bethesda, MD 20892 (USA).
Nancy E. Lewin, Laboratory for Cancer Biology and Genetics, CCR, NCI, NIH, Bethesda, MD 20892 (USA)
Megan L. Peach, Email: mpeach@helix.nih.gov, Basic Research Program SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD 21702 (USA)
Dr. Peter M. Blumberg, Email: blumberp@dc37a.nci.nih.gov, Laboratory for Cancer Biology and Genetics, CCR, NCI, NIH, Bethesda, MD 20892 (USA)
References
- 1.For reviews of the bryostatins and bryostatin analogues, see: Hale KJ, Hummersome MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413–453. doi: 10.1039/b009211h.Hale KJ, Manaviazar S. Chem Asian J. 2010;5:704–754. doi: 10.1002/asia.200900634.
- 2.Pettit GR, Herald CL, Doubek DL, Herald DL. J Am Chem Soc. 1982;104:6846–6848. [Google Scholar]
- 3.Schaufelberger DE, Koleck MP, Beutler JA, Vatakis AM, Alvarado AB, Andrews P, Marzo LV, Muschik GM, Roach J, Ross JT, Lebherz WB, Reeves MP, Eberwein RM, Rodgers LI, Testerman RP, Snader KM, Forenza S. J Nat Prod. 1991;54:1265–1270. doi: 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- 4.For current information, see http://clinicaltrials.gov.
- 5.For further information see: http://www.brni.org/news/PressRelease42209.htm
- 6.Reyland ME, Insel PA, Messing RO, Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP. Am J Physiol Lung Cell Mol Physiol. 2000;279:429–438. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 7.Griner EM, Kazanietz MG. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 8.Blumberg PM, Pettit GR. In: New Leads and Targets in Drug Research. Krosgaard–Larsen P, Christensen CB, Kodof H, editors. Munksgaard; Copenhagen: 1992. pp. 273–285. [Google Scholar]
- 9.(a) Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J Am Chem Soc. 1999;121:7540–7552. [Google Scholar]; (b) Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J Am Chem Soc. 1990;112:7407–7408. [Google Scholar]; (c) Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew Chem, Int Ed. 2000;39:2290–2294. [PubMed] [Google Scholar]; (d) Manaviazar S, Frigerio M, Bhatia GS, Hummersone MG, Aliev AE, Hale KJ. Org Lett. 2006;8:4477–4480. doi: 10.1021/ol061626i. [DOI] [PubMed] [Google Scholar]
- 10.Trost BM, Dong G. Nature. 2008;456:485–488. doi: 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For leading references, see: Wender PA, DeChristopher BA, Schreier AJ. J Am Chem Soc. 2008;130:6658–6659. doi: 10.1021/ja8015632.Wender PA, Baryza JL, Bennett CE, Bi FC, Brenner SE, Clarke MO, Horan JC, Kan C, Lacote E, Lippa BS, Nell PG, Turner TM. J Am Chem Soc. 2002;124:13648–13649. doi: 10.1021/ja027509+.Wender PA, De Brabander J, Harran PG, Jimenez J, Koehler MFT, Lippa B, Park C, Shiozaki M. J Am Chem Soc. 1998;120:4534–4535.Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. In: Drug Discovery Research: New Frontiers in the Post-Genomic Era. Huang Z, editor. Wiley-VCH; Hoboken, NJ: 2007. pp. 127–162.
- 12.(a) Keck GE, Truong AP. Org Lett. 2005;7:2149–2152. doi: 10.1021/ol050511w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keck GE, Truong AP. Org Lett. 2005;7:2153–2156. doi: 10.1021/ol050512o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Keck GE, Welch DS, Vivian PK. Org Lett. 2006;8:3667–3670. doi: 10.1021/ol061173h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Keck GE, Welch DS, Poudel YB. Tetrahedron Lett. 2006;47:8267–8270. doi: 10.1016/j.tetlet.2006.09.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin NE, Blumberg PM. J Am Chem Soc. 2008;130:6660–6661. doi: 10.1021/ja8022169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keck GE, Poudel YB, Welch DS, Kraft MB, Truong AP, Stephens JC, Kedei N, Lewin NE, Blumberg PM. Org Lett. 2009;11:593–596. doi: 10.1021/ol8027253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura K, Mizutani MY, Tomioka N, Endo Y, Shudo K, Itai A. Chem Pharm Bull. 1999;47:1134–1137. [Google Scholar]
- 16.Kamano Y, Zhang H, Morita H, Itokawa H, Shirota O, Pettit GR, Herald DL, Herald CL. Tetrahedron. 1996;52:2369–2376. [Google Scholar]
- 17.Zhang GG, Kazanietz MG, Blumberg PM, Hurley JH. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 18.The “Merle numbers” used herein are permanent identifiers for the analogue structures.
- 19.Choi T, Lee CW, Chatterjee AK, Grubbs RH. J Am Chem Soc. 2001;123:10417–10418. doi: 10.1021/ja016386a. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K, Ohta Y, Fuji K. Tetrahedron Lett. 1993;34:4071–4074. [Google Scholar]
- 21.Kraft MB. unpublished results. University of Utah; [Google Scholar]
- 22.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 23.The use of this hemiacetal in a similar aldol condensation has been previously described. See: Baryza JL. PhD Thesis. Stanford University; 2005.
- 24.Lipschutz BH, Harvey DF. Synth Commun. 1982;14:267–277. [Google Scholar]
- 25.Vrana JA, Saunders AM, Srikumar PC, Grant S. Differentiation. 1998;63:33–42. doi: 10.1046/j.1432-0436.1998.6310033.x. [DOI] [PubMed] [Google Scholar]
- 26.Keck GE, Li W, Kraft MB, Kedei N, Lewin NE, Blumberg PM. Org Lett. 2009;11:2277–2280. doi: 10.1021/ol900585t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gschwend JE, Fair WR, Powell CT. Mol Pharmacol. 2000;57:1224–1234. [PubMed] [Google Scholar]