

Abstract
Development of novel prevention and treatment strategies for herpes simplex virus (HSV) mediated diseases is dependent upon an accurate understanding of the central molecular events underlying the regulation of latency and reactivation. We have recently shown that the transactivation function of the virion protein VP16 is a critical determinant in the exit from latency in vivo. HSV-1 strain SJO2 carries a single serine to alanine substitution at position 375 in VP16 which disrupts its interaction with its essential co-activator Oct-1. Here we report that SJO2 is severely impaired in its ability to exit latency in vivo. This result reinforces our prior observations with VP16 transactivation mutant, in1814, in which VP16 interaction with Oct-1 is also disrupted and solidifies the importance of the VP16-Oct-1 interaction in the early steps in HSV-1 reactivation.
Introduction
Over 80% of the human population is infected with herpes simplex virus type 1 (HSV-1) and/or type 2 (HSV-2). Two key features of infection by HSV, its permanence and its ability to periodically switch from a latent to a lytic infection, underlie the pandemic levels of HSV infection ongoing worldwide. Recurrent HSV disease is a major contributor to blindness (Pepose, Keadle et al. 2006) and encephalitis (Stone and Hawkins 2007), to devastating neonatal disease (Roberts 2009), and to increased risk of type II diabetes (Sun, Pei et al. 2005), cardiovascular (Visser and Vercellotti 1993; Mukamal, Kronmal et al. 2004) and Alzheimer's (Itzhaki and Wozniak 2008) diseases, and sexually acquired HIV infections (Glynn, Biraro et al. 2009). Despite the importance of HSV reactivation to human health, the regulation of this process remains poorly understood.
In contrast, activation of the viral lytic cycle in a newly infected cell is understood in some detail, at least in so far as it occurs in cultured cell monolayers (Roizman, Gu et al. 2005; Knipe 2007). Under biologically relevant infection conditions, that is, at a multiplicity of infection less than 1, the viral tegument protein VP16 augments entry into the lytic cycle 10-100 fold depending upon cell type (McFarlane, Daksis et al. 1992). Upon infection, VP16 is released into the cell and through interactions with host cell proteins HCF and Oct-1 is recruited specifically to TAATGARAT motifs in the viral immediate early (IE) gene promoters (Wysocka and Herr 2003). Although VP16 is a multifunctional protein, its transactivation function resides within a carboxyterminal acidic activation domain and a region upstream, termed the core domain, which is required for coactivator interactions (Wysocka and Herr 2003). Mutations in VP16 that disrupt either coactivator interactions or the acidic activation domain result in viral mutants that exhibit similar phenotypes of diminished IE gene expression and infectivity in the context of low multiplicity viral infection in vitro (McFarlane, Daksis et al. 1992; Lam, Smibert et al. 1996; O'Reilly, Hanscombe et al. 1997; Smiley and Duncan 1997; Wysocka and Herr 2003; Ottosen, Herrera et al. 2006). However, the replication phenotypes in vivo are dramatically different. Strain in1814 bears a 4 amino acid insertion following residue 379 (sometimes erroneously cited as 397) that disrupts the interaction of VP16 with Oct-1 on IE gene promoters (Ace, McKee et al. 1989; Steiner, Spivack et al. 1990). Two activation domain (AD) truncation mutants, RP5 and A422, have also been described (Tal-Singer, Pichyangkura et al. 1999; Thompson, Preston et al. 2009). The core domain mutant in1814 shows a 20-fold reduction in viral replication in trigeminal ganglia (TG), whereas the activation domain deletion mutant Δ422 shows a 10,000-fold reduction (Thompson, Preston et al. 2009). Since the late functions of VP16 remain intact in these mutants (Smiley and Duncan 1997; Tal-Singer, Pichyangkura et al. 1999; Mossman, Sherburne et al. 2000), these findings reveal that initiation of the lytic cycle in vivo depends almost completely upon transactivation by VP16. Further, these studies suggest that whereas the AD is essential for activity, weak residual interaction of VP16 on IE promoters occurs in the core domain mutant in1814 (Smiley and Duncan 1997), confirming the importance of the VP16-Oct-1-DNA interaction in the mouse model. The disparity in the phenotypes displayed in vitro and in vivo reveals that despite extensive biochemical detail, understanding of the biological significance of these interactions in the context of the complete viral life cycle is extremely limited.
Interestingly, activation of the lytic cycle from the latent viral genome (reactivation) has been considered to occur via a mechanism independent of VP16 transactivation (Steiner, Spivack et al. 1990; Sears, Hukkanen et al. 1991; Tal-Singer, Pichyangkura et al. 1999). This conclusion was based on ex vivo reactivation studies with VP16 transactivation mutants and attempts to express VP16 from an inducible promoter in vivo (Sears, Hukkanen et al. 1991). In addition, the expectation was that the VP16 tegument protein, which in cultured cells is expressed late in the lytic cycle (Honess and Roizman 1974) would not be present to activate the lytic cycle from the latent genome. Contrary to this expectation, we recently demonstrated that VP16 transactivation function is required for the very early stages in reactivation in vivo (in the context of the intact animal). We observed that neurons latently infected with the in1814 mutant virus failed to exit latency and generate viral proteins. Other reports eliminate other candidate viral proteins as essential for the exit from latency (Sawtell, Thompson et al. 2006; Thompson and Sawtell 2006; Thompson, Preston et al. 2009). Collectively these observations support the hypothesis that VP16 activates expression of IE genes from the latent viral genome to initiate reactivation from latency in vivo. The importance of defining the regulatory mechanisms controlling in vivo reactivation warrants further examination of this hypothesis. To this end, we now report on the ability of mutant SJO2, an HSV-1 mutant that contains a single amino acid substitution in VP16 which disrupts Oct-1 interaction (Ottosen, Herrera et al. 2006) and transactivation of IE genes by the VP16 induced complex (VIC), to exit latency in vivo. We propose that expression and potentially post-translational modification of VP16 are essential and early events in the reactivation of HSV-1 from latency in vivo.
Results
The purpose of this study was to further test the hypothesis that in vivo, initiation of the viral lytic cycle from the latent viral genome is dependent upon VP16 transactivation function. Our previous study utilized the well-characterized mutant in1814, which contains an insertion of 4 amino acids after residue 379 of VP16 (Ace, McKee et al. 1989) (Steiner, Spivack et al. 1990). Viral mutant SJO2, previously generated and characterized in vitro (Ottosen, Herrera et al. 2006), contains a single serine to alanine substitution in VP16 at position 375 (fig.1A). Each of these mutations results in a VP16 protein that in biochemical assays fails to interact with Oct-1, fails to form the VIC, and transactivates a TAATGARAT promoter many fold less efficiently than wt protein in transient transfection assays (O'Reilly, Hanscombe et al. 1997; Wysocka and Herr 2003). Thus our hypothesis predicts that SJO2, like in1814, should be deficient in exiting latency in vivo.
Figure 1. VP16 transactivation mutant SJO2.
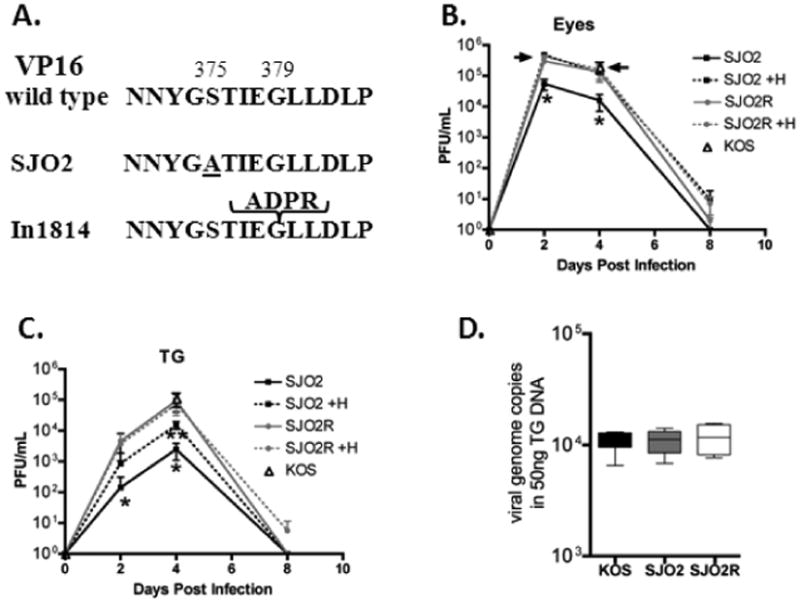
(A). SJO2 contains a single Ser to Ala substitution at amino acid 375 within the VP16 core region. For reference, the 4 amino acid insertion in in1814 is shown. (B,C). Mice were infected on scarified corneas with ×106 SJO2 or ×106 of either SJO2R or KOS. Replication of SJO2 and its rescuant, SJO2R or parental strain KOS (d 4 only) in the eyes (B) and TG (C) of infected mice (n=3) per group. Single asterisk (*) indicates significant differences (p= 0.002, unpaired, t-test) between viral titers in SJO2 homogenates when plated in the presence or absence of HMBA. Double asterisk (**) indicates significant difference between SJO2 titers (plated with HMBA) and SJO2R titers (p=0.004, unpaired, t-test). D. The number of viral genome copies per 50 ng total DNA in TG pairs latently infected (40 dpi) with KOS (n=4), SJO2 (n=4), or SJO2R (n=4). The box extends from the 25th to the 75th percentiles. The horizontal line indicates the median and the whiskers show the range.
Prior to testing SJO2 in vivo, a rescue of this mutant virus was generated by recombining a DNA fragment restricted to the VP16 gene (bp 103, 441 to bp 105,107) into SJO2. Three independent isolates were plaque-purified. The restoration of serine at position 375 and the absence of any adventitious mutations in the VP16 gene were confirmed by DNA sequencing (not shown). Because mutants lacking the transactivation function of VP16 enter the lytic cycle inefficiently at low multiplicity, the standard plaque assay (a low multiplicity assay which is used to quantify infectious viral particles) will underestimate the amount of virus present (Ace, McKee et al. 1989; Smiley and Duncan 1997; Preston and McFarlane 1998). The addition of the cell differentiating agent hexamethylbisacetamide (HMBA) increases the plaquing efficiency of in1814 and other VP16 transactivation deficient mutants with minimal effect on wild type virus (Preston and McFarlane 1998). A comparison of the plaquing efficiency of SJO2, SJO2R, and parental strain KOS in rabbit skin cells (RSC) demonstrated that as reported for in1814 (McFarlane, Daksis et al. 1992; Smiley and Duncan 1997; Preston and McFarlane 1998), treatment with 5mm HMBA resulted in a ∼10 fold increase in plaques of SJO2 (for example, a typical SJO2 stock yields 1.×107pfu/ml in the absence of HMBA and 1.7×108 pfu/ml in the presence of HMBA). In contrast, SJO2R and parental strain KOS exhibited < 1.8 fold differential, confirming that by this criterion, the rescued virus was restored to wild type. In addition, infection of wells of 24–well plates (∼×105 RSC per well) at an moi of 10 revealed that like in1814, mutant SJO2 generated viral titers equivalent to parental strain KOS (6.×106 pfu ±1.×106 and 6.×106 pfu ±1.×106, respectively) when measured by plaque assay in the presence of HMBA.
The in vivo replication of SJO2, SJO2R, and parental strain KOS was evaluated in groups of male, 8-10 wk old DBA2J mice (Jackson) which were inoculated on scarified corneas with either ×106 pfu (SJO2) or ×106pfu (SJO2R or KOS). The upward adjustment of inoculum titer of SJO2 was used to achieve similar levels of latency between the groups (Sawtell 1997; Sawtell and Thompson et al. 2006; Thompson and Sawtell 2006; Thompson, Preston et al. 2009). Relative to HSV-1 strain 17syn+, strain KOS reactivates at a lower frequency in our standard model of in vivo reactivation in Swiss Webster mice (∼70% vs 35%). In order to ensure that the reactivation and exit from latency assays were sufficiently robust, a preliminary screen of the relative efficiency of reactivation by strain KOS in the more susceptible mouse strain DBA2J compared to Swiss Webster mice was performed. KOS remained largely avirulent in DBA2J mice but the frequency of reactivation was similar to that of strain 17syn+ in Swiss Webster mice (data not shown). On days 2, 4, and 8 days pi, eyes and TGs were harvested from 3 mice/group and viral titers were evaluated in individual tissues by plaque assay in the presence and absence of HMBA. The rise and fall of infectious virus production during acute infection in vivo in the eyes and TG of SJO2 and SJO2R infected mice is shown in fig.1B,C. These data reveal first, that the number of plaque forming units (pfu) measured in tissue homogenates from SJO2 but not SJO2R infected mice exhibit an ∼10 fold increase (p=0.022, unpaired t-test) when plated in the presence of HMBA (McFarlane, Daksis et al. 1992). This confirms the VP16 transactivation deficit in virus replicated in mice infected with SJO2 and also confirms that VP16 transactivation function has been fully restored in SJO2R. Secondly, the maximum titers achieved in the eyes when assayed in the presence of HMBA were not different among the groups (fig.1.B). However, the defect in VP16 transactivation in SJO2 resulted in an ∼8 fold reduction in viral titers in TG when assayed in the presence of HMBA on day 4 pi (p=0.03, unpaired t-test) (fig.1.C). The VP16 S375A mutation resulted in a less severe replication defect in both the eyes and TG than previously reported for mutant in1814 in Swiss Webster mice indicating that SJO2 retains some VP16 transactivation function in vivo. To assess the degree to which latency is established in neurons infected with the mutant viruses, we assayed the viral DNA present in TG at day 40 pi using quantitative real time PCR as previously detailed (Sawtell, Thompson et al. 2006). Importantly, the levels of viral genomes were not different among the groups (n=4 TG pairs/group) (fig.1.D). This result is consistent with the finding that the transactivation impaired mutant in1814 establishes latency efficiently (Steiner, Spivack et al. 1990; Ecob-Prince, Preston et al. 1993) and our more detailed quantitative analysis confirming the ability of in1814 to establish latency with a similar number of neurons and copy number profile as wild type and in1814R (Thompson, Preston et al. 2009). These experiments demonstrate that in the TG, SJO2 mutant virus has diminished replicative capacity but can effectively establish latency. We conclude that VP16 Ser375 is important for lytic replication in TG in vivo.
In order to test the ability of mutant SJO2 to reactivate from latency, groups of mice (n=8) latently infected with either SJO2, SJO2R, or KOS were subjected to stress using hyperthermia and at 22hr post-treatment, ganglia were harvested and processed for infectious virus (n=8) using methods previously reported (Sawtell and Thompson 1992; Sawtell 2003; Thompson, Preston et al. 2009). When assayed in the presence of HMBA, no infectious virus was detected in TG harvested from SJO2 infected mice (0/8), whereas infectious virus was recovered from 5/8 mice infected with SJO2R and 6/8 mice infected with KOS (p=0.04 and p=0.009, unpaired t-test). While this finding is consistent with a requirement for VP16 transactivation in reactivation, this assay reflects any deficit that impairs infectious virus production and does not distinguish between a function required very early from one required late in the process.
To test the hypothesis that VP16 transactivation is required at a very early stage in the reactivation process (that is to initiate IE gene expression from the latent viral genome), we employed a sensitive assay to detect the expression of viral proteins in single neurons in the intact ganglia (avoiding tissue loss during processing and sectioning) (Sawtell 2003). We have validated this assay extensively in previous studies and have demonstrated that viral mutants that cannot fully reactivate, (as measured by infectious virus production), including those lacking the viral thymidine kinase or ICP0 (Sawtell 2003; Sawtell, Thompson et al. 2006; Thompson and Sawtell 2006; Thompson, Preston et al. 2009), do exit latency and express viral protein in neurons following stress in vivo (see fig. 2B). Ganglia were harvested from all three groups of latently infected mice prior to heat stress and examined for the presence of neurons expressing viral proteins. In the absence of heat stress, no positive neurons in any of 8 TG per group were detected. In contrast, at 22hr following heat stress, neurons expressing viral protein were detected in TG from mice infected with either SJO2R or KOS (fig.2A). Specifically, 9/10 TG infected with KOS and 12/16 TG SJO2R contained a mean of ∼3 (range of 1-9) neurons positive for HSV proteins, indicating that these neurons had entered the viral lytic cycle (fig.2B). In contrast, only 1 neuron from a total of 15 TG infected with SJO2 was found to have exited latency. These results are similar to those reported for in1814, and provide further evidence that the VP16 transactivation function is a function critical for the very early stages in the in vivo reactivation process.
Figure 2. Mutant SJO2 exits latency 50 fold less efficiently than SJO2R.
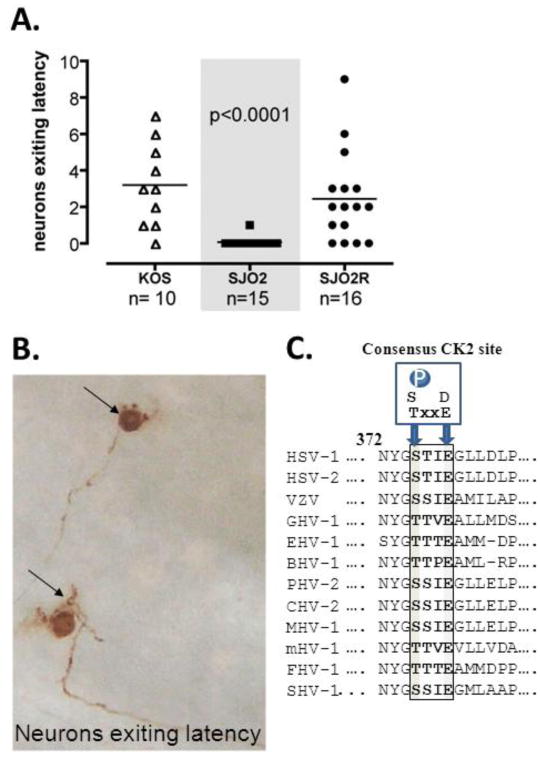
(A). Mice latently infected with KOS, SJO2 or SJO2R were subjected to hyperthermic stress and 22hr post-treatment, TG were removed and processed to detect HSV proteins in the whole ganglia using immunohistochemistry. Rabbit anti-HSV-1antibody (AXL237, Accurate) diluted 1:1000 was utilized as detailed (Sawtell 2003). Neurons exiting latency are then visualized and counted. Each symbol represents the number of neurons that exited latency in a single TG. n indicates the number of TG examined. (B). Photomicrograph of unsectioned ganglion (KOS infected) containing neurons expressing viral proteins indicating that resident viral genomes have exited latency. (C). The region of VP16 critical for Oct-1 interaction, including the presence of a Serine or Threonine at position 375 (dark bar) and a consensus CK2 site (S/T xxE/D) is conserved among VP16 homologues from diverse α-herpes viruses.
In contrast to the in vivo setting, several previous studies have demonstrated that ganglia latently infected with in1814 can exit latency and can produce infectious virus (reactivate) when explanted and maintained in culture (Steiner, Spivack et al. 1990; Harris and Preston 1991; Valyi-Nagy, Deshmane et al. 1991; Ecob-Prince, Preston et al. 1993; Ecob-Prince, Rixon et al. 1993; Thompson, Preston et al. 2009). The expectation, then, is that TG latently infected with SJO2 would also exit latency and reactivate following explant. To test this prediction, TG were harvested from mice latently infected with SJO2 or SJO2R and cultured as described (Thompson, Preston et al. 2009). At 5 days post-explant, TG were homogenized and the presence of virus determined by plaque assay in the presence of HMBA. Infectious virus was detected in 100% of TG explanted from SJO2 (4/4) and SJO2R (3/3) infected mice. Thus, as shown for in1814, SJO2 can exit latency and reactivate in a VP16- independent manner upon explant whereas upon heat stress in vivo, the exit from latency is more than 50 fold reduced.
These findings solidify the hypothesis that in vivo the very early stages in reactivation from latency occur through a VP16-dependent mechanism. Of interest, VP16 S375 resides in a consensus CK2 site (S/TxxE/D) and can be phosphorylated in transfected cells and by CK2 in a kinase assay (O'Reilly, Hanscombe et al. 1997). Both the CK2 site and a Ser/Thr at position 375 are highly conserved among the α-herpesviruses (fig.2C). Replacing S375 with Thr (phosphorylatable by CK2) but not Ala retains the transactivation function of VP16 in a transactivation assay (O'Reilly, Hanscombe et al. 1997). Thus, a phosphorylatable amino acid at 375 appears important for regulating the interaction of VP16 with Oct-1-DNA (O'Reilly, Hanscombe et al. 1997). Although not reported here, additional ongoing studies strongly support that phosphorylation of VP16 at Ser375 is an important posttranslational modification that regulates both entry into the lytic cycle and the earliest stages of reactivation from latency in vivo (Sawtell, unpublished). A role for VP16 at the earliest stages of reactivation requires that its expression be independent of an ongoing lytic cycle. We previously published evidence that VP16 is expressed de novo from the latent viral genome (Thompson, Preston et al. 2009) and have since identified a region of the VP16 promoter that regulates this de novo expression in neurons (manuscript in preparation). We propose that reactivation (infectious virus production) is preceded by multiple events which occur stochastically in response to stress (Thompson, Preston et al. 2009). From analyses at the cell level, identification of neurons exiting latency as defined by viral protein expression reveal the low probability (∼1in 2-3,000) of a given latently infected neuron proceeding to this stage (Sawtell 1998). The probability of this event is greatly reduced in the absence of VP16 transactivation (<1 in 100-150,000). While valuable information can be gained by global analyses in the TG, capturing this process fully will ultimately require the development of tools providing single cell level information on events upstream of viral protein expression. In a recent report, utilizing a novel in vivo strategy to record “promoter activation” in neurons through time in the context of HSV infection, the number of neurons evidencing activity from the VP16 promoter was very low but measurable (Proenca, Coleman et al. 2011), consistent with a multi-stage model in which VP16 expression, translation, and posttranslational modification are stochastic events required for the initiation of the reactivation process. Thus activation of the VP16 promoter may not be universally linked to activation of the lytic cycle and loss of neurons. We propose that alignment of both VP16 expression and activity mediated by post translational modifications of VP16 that influence cofactor partnering, such as phosphorylation at S375, modulate the exit from latency.
Acknowledgments
This work was supported by Public Health Service NIH grants AI32121 to NMS and EY13168 to RLT.
Contributor Information
Nancy M. Sawtell, Department of Pediatrics, Division of Infectious Diseases, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio 45229-3039, USA
Steven J. Triezenberg, Van Andel Research Institute, Grand Rapids, MI 49503
Richard L. Thompson, Department of Molecular Genetics, Biochemistry, and Microbiology, University of Cincinnati College of Medicine, Cincinnati, Ohio, 45267-0524, USA
References
- Ace CI, McKee TA, et al. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J Virol. 1989;63(5):2260–9. doi: 10.1128/jvi.63.5.2260-2269.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecob-Prince MS, Preston CM, et al. Neurons containing latency-associated transcripts are numerous and widespread in dorsal root ganglia following footpad inoculation of mice with herpes simplex virus type 1 mutant in1814. J Gen Virol. 1993;74(Pt 6):985–94. doi: 10.1099/0022-1317-74-6-985. [DOI] [PubMed] [Google Scholar]
- Ecob-Prince MS, Rixon FJ, et al. Reactivation in vivo and in vitro of herpes simplex virus from mouse dorsal root ganglia which contain different levels of latency- associated transcripts. J Gen Virol. 1993;74(Pt 6):995–1002. doi: 10.1099/0022-1317-74-6-995. [DOI] [PubMed] [Google Scholar]
- Glynn JR, Biraro S, et al. Herpes simplex virus type 2: a key role in HIV incidence. Aids. 2009;23(12):1595–8. doi: 10.1097/QAD.0b013e32832e15e8. [DOI] [PubMed] [Google Scholar]
- Harris RA, Preston CM. Establishment of latency in vitro by the herpes simplex virus type 1 mutant in1814. J Gen Virol. 1991;72(Pt 4):907–13. doi: 10.1099/0022-1317-72-4-907. [DOI] [PubMed] [Google Scholar]
- Honess RW, Roizman B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol. 1974;14(1):8–19. doi: 10.1128/jvi.14.1.8-19.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki RF, Wozniak MA. Herpes simplex virus type 1 in Alzheimer's disease: the enemy within. J Alzheimers Dis. 2008;13(4):393–405. doi: 10.3233/jad-2008-13405. [DOI] [PubMed] [Google Scholar]
- Knipe D. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- Lam Q, Smibert CA, et al. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. Embo J. 1996;15(10):2575–81. [PMC free article] [PubMed] [Google Scholar]
- McFarlane M, Daksis JI, et al. Hexamethylene bisacetamide stimulates herpes simplex virus immediate early gene expression in the absence of trans-induction by Vmw65. J Gen Virol. 1992;73(Pt 2):285–92. doi: 10.1099/0022-1317-73-2-285. [DOI] [PubMed] [Google Scholar]
- Miller CS, Danaher RJ, et al. ICP0 is not required for efficient stress-induced reactivation of herpes simplex virus type 1 from cultured quiescently infected neuronal cells. J Virol. 2006;80(7):3360–8. doi: 10.1128/JVI.80.7.3360-3368.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossman KL, Sherburne R, et al. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J Virol. 2000;74(14):6287–99. doi: 10.1128/jvi.74.14.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamal KJ, Kronmal RA, et al. Traditional and novel risk factors in older adults: cardiovascular risk assessment late in life. Am J Geriatr Cardiol. 2004;13(2):69–80. doi: 10.1111/j.1076-7460.2004.02123.x. [DOI] [PubMed] [Google Scholar]
- O'Reilly D, Hanscombe O, et al. A single serine residue at position 375 of VP16 is critical for complex assembly with Oct-1 and HCF and is a target of phosphorylation by casein kinase II. Embo J. 1997;16(9):2420–30. doi: 10.1093/emboj/16.9.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosen S, Herrera FJ, et al. Phosphorylation of the VP16 transcriptional activator protein during herpes simplex virus infection and mutational analysis of putative phosphorylation sites. Virology. 2006;345(2):468–81. doi: 10.1016/j.virol.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepose JS, Keadle TL, et al. Ocular herpes simplex: changing epidemiology, emerging disease patterns, and the potential of vaccine prevention and therapy. Am J Ophthalmol. 2006;141(3):547–557. doi: 10.1016/j.ajo.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Preston CM, McFarlane M. Cytodifferentiating agents affect the replication of herpes simplex virus type 1 in the absence of functional VP16. Virology. 1998;249(2):418–26. doi: 10.1006/viro.1998.9314. [DOI] [PubMed] [Google Scholar]
- Proenca JT, Coleman HM, et al. An investigation of HSV promoter activity compatible with latency establishment reveals VP16 independent activation of HSV immediate early promoters in sensory neurones. J Gen Virol. 2011 doi: 10.1099/vir.0.034728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S. Herpes simplex virus: incidence of neonatal herpes simplex virus, maternal screening, management during pregnancy, and HIV. Curr Opin Obstet Gynecol. 2009;21(2):124–30. doi: 10.1097/GCO.0b013e3283294840. [DOI] [PubMed] [Google Scholar]
- Roizman B, Gu H, et al. The first 30 minutes in the life of a virus: unREST in the nucleus. Cell Cycle. 2005;4(8):1019–21. doi: 10.4161/cc.4.8.1902. [DOI] [PubMed] [Google Scholar]
- Sawtell NM. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997;71(7):5423–31. doi: 10.1128/jvi.71.7.5423-5431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM. The probability of in vivo reactivation of herpes simplex virus type 1 increases with the number of latently infected neurons in the ganglia. J Virol. 1998;72(8):6888–92. doi: 10.1128/jvi.72.8.6888-6892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM. Detection and quantification of the rare latently infected cell undergoing herpes simplex virus transcriptional activation in the nervous system in vivo. Methods Mol Biol. 2005;292:57–72. doi: 10.1385/1-59259-848-x:057. [DOI] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J Virol. 1992;66(4):2157–69. doi: 10.1128/jvi.66.4.2157-2169.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, et al. Herpes simplex virus DNA synthesis is not a decisive regulatory event in the initiation of lytic viral protein expression in neurons in vivo during primary infection or reactivation from latency. J Virol. 2006;80(1):38–50. doi: 10.1128/JVI.80.1.38-50.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears AE, Hukkanen V. Expression of the herpes simplex virus 1 alpha transinducing factor (VP16) does not induce reactivation of latent virus or prevent the establishment of latency in mice. J Virol. 1991;65(6):2929–35. doi: 10.1128/jvi.65.6.2929-2935.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley JR, Duncan J. Truncation of the C-terminal acidic transcriptional activation domain of herpes simplex virus VP16 produces a phenotype similar to that of the in1814 linker insertion mutation. J Virol. 1997;71(8):6191–3. doi: 10.1128/jvi.71.8.6191-6193.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner I, Spivack JG, et al. A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990;64(4):1630–8. doi: 10.1128/jvi.64.4.1630-1638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MJ, Hawkins CP. A medical overview of encephalitis. Neuropsychol Rehabil. 2007;17(4-5):429–49. doi: 10.1080/09602010601069430. [DOI] [PubMed] [Google Scholar]
- Sun Y, Pei W, et al. An association of herpes simplex virus type 1 infection with type 2 diabetes. Diabetes Care. 2005;28(2):435–6. doi: 10.2337/diacare.28.2.435. [DOI] [PubMed] [Google Scholar]
- Tal-Singer R, Pichyangkura R, et al. The transcriptional activation domain of VP16 is required for efficient infection and establishment of latency by HSV-1 in the murine peripheral and central nervous systems. Virology. 1999;259(1):20–33. doi: 10.1006/viro.1999.9756. [DOI] [PubMed] [Google Scholar]
- Thompson RL, Preston CM, et al. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009;5(3):e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J Virol. 2006;80(22):10919–30. doi: 10.1128/JVI.01253-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyi-Nagy T, Deshmane SL, et al. Investigation of herpes simplex virus type 1 (HSV-1) gene expression and DNA synthesis during the establishment of latent infection by an HSV-1 mutant, in1814, that does not replicate in mouse trigeminal ganglia. J Gen Virol. 1991;72(Pt 3):641–9. doi: 10.1099/0022-1317-72-3-641. [DOI] [PubMed] [Google Scholar]
- Visser MR, Vercellotti GM. Herpes simplex virus and atherosclerosis. Eur Heart J. 1993;14 K:39–42. [PubMed] [Google Scholar]
- Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003;28(6):294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]