

Abstract
Ranolazine (Ran), an anti-anginal drug, is a late Na+ channel current blocker that is also believed to attenuate fatty acid oxidation and mitochondrial respiratory complex I activity, especially during ischemia. In this study, we investigated if Ran's protective effect against cardiac ischemia/reperfusion (IR) injury is mediated at the mitochondrial level and specifically if respiratory complex I (NADH oxidoreductase) function is protected. We treated isolated and perfused guinea pig hearts with Ran just before 30 min ischemia and then isolated cardiac mitochondria at the end of 30 min ischemia and/or 30 min ischemia followed by 10 min reperfusion. We utilized spectrophotometric and histochemical techniques to assay complex I activity, western blot analysis for complex I subunit NDUFA9, electron paramagnetic resonance for activity of complex I Fe-S clusters, ELISA for determination of protein acetylation, native gel histochemical staining for respiratory supercomplex assemblies, and high pressure liquid chromatography for cardiolipin integrity; cardiac function was measured during IR. Ran treated hearts showed higher complex I activity and greater detectable complex I protein levels compared to untreated IR hearts. Ran treatment also led to more normalized electron transfer via Fe-S centers, supercomplex assembly and cardiolipin integrity. These improvements in complex I structure and function with Ran were associated with improved cardiac function after IR. However, these protective effects of Ran are not mediated by a direct action on mitochondria, but rather indirectly via cytosolic mechanisms that lead to less oxidation and better structural integrity of complex I.
Keywords: Complex I, mitochondria, IR injury, ranolazine, EPR, heart
1. Introduction
It is now evident that mitochondria play an important role in mediating both protection and damage during cardiac ischemia reperfusion (IR) injury. An important target for protection is mitochondrial complex I (NADH-Ubiquinone oxidoreductase) [1, 2]. Complex I is a large, multi-subunit, integral membrane protein highly susceptible to functional and structural damage during IR injury [3, 4]. Complex I is bound by cardiolipin, a highly unsaturated fatty acid lipid in the inner mitochondrial membrane (IMM). Cardiolipin is essential for maintaining functional and structural integrity of the respiratory complexes and to assure efficient transfer of electrons (e-) within subunits of the complexes and between the complexes [5]. The transfer of e- via sequential oxidation-reduction of Fe in the seven Fe-S clusters of complex I exemplifies this critical role of cardiolipin [6, 7]. Because cardiolipin is susceptible to oxidative attack by reactive O2 species (ROS) [8, 9] leading to peroxidation and carbon chain breakdown [10], the assemblies of respiratory complexes are also dependent on integrity of cardiolipin.
Ranolazine (Ran) is a clinically used drug known to reduce cardiac dysrhythmias [11-15] and tissue damage after IR [16]. During IR injury, Na+ can slowly enter myocytes during phase 3 of the action potential to initiate dysrhythmias [11]; thus as a late Na+ channel current blocker [13], Ran is believed to protect hearts by reducing the incidence of dysrhythmias during IR injury. However, since Ran prevents intracellular Na+ loading, particularly during IR, it could also decrease cytosolic Ca2+ overload via Na+/Ca2+ exchange (NCE) and consequently decrease mitochondrial Ca2+ (m[Ca2+]) overload [17]; m[Ca2+] overload is thought to cause increased production of ROS and to trigger cell apoptosis by release of cytochrome c. These events may underlie, at least in part, the protection afforded by Ran against cardiac tissue damage during IR.
Indeed, in a recent isolated heart study of IR injury [18] we found that Ran treatment just before ischemia reduced cytosolic and m[Ca2+] loading and ROS emission and, in isolated mitochondria, reduced cytochrome c release and slightly delayed opening of the mitochondrial permeability transition pore (mPTP) during cardiac IR injury. Other reports suggest that Ran exerts a cardioprotective effect by switching substrate utilization from fatty acids to glucose metabolism [16, 19, 20], which reduces mitochondrial O2 demand and that Ran partially blocks respiratory complex I activity [21]. Although these prior studies give insight into Ran's mode of cardioprotection, there is no direct evidence that Ran actually attenuates mitochondrial dysfunction during IR injury
Given the role of complex I in generating ROS during IR [4, 22] and the crucial role of cardiolipin [4, 10, 22, 23] in promoting efficient e- transfer, we sought first to assess changes in many markers of complex I function during IR injury and second to determine if there were any direct beneficial biophysical and biochemical effects of Ran on complex I during IR. Because it was unclear if Ran directly targets mitochondria to protect against IR injury, we hypothesized that Ran, when present during ischemia, preserves cardiolipin and supercomplex assembly integrity, and especially complex I activity, by its direct effect to reduce ROS emission and oxidation of mitochondrial components as a primary mechanism to protect against cardiac IR injury; alternatively, Ran has indirect effects to restore complex I function.
2. Materials and Methods
2.1. Isolated heart preparation and functional assessment
The Institutional Animal Care and Usage Committee of the Medical College of Wisconsin approved all animal studies. Guinea pig hearts (n=43) were isolated and prepared for constant pressure perfusion studies as previously published [2, 24-26]. Briefly, animals were injected i.p. with 10 mg/kg ketamine to induce anesthesia and with 5000 units heparin to prevent coagulation. Following decapitation and thoracotomy, hearts were removed and perfused with Krebs-Ringer's buffer (KR) (in mM 138 Na+, 4.5 K+, 1.2 Mg2+, 2.5 Ca2+, 134 Cl-, 15 HCO3-, 1.2 H2PO4-, 11.5 glucose, 2 pyruvate, 16 mannitol, 0.05 EDTA and 5 U/L insulin) gassed with 3% CO2, 97% O2 at pH 7.4 and 37°C. A saline filled balloon catheter attached to a transducer was used to measure left ventricular pressure (LVP). Diastolic LVP was set initially to zero in order to monitor any increase and an indicator of diastolic contracture, a marker of ischemic injury. Coronary flow was measured using an ultrasonic flowmeter (model T106X, Transonic Systems) placed directly into the aortic inflow line. Spontaneous heart rate was monitored using bipolar electrodes placed on the right atrial and ventricular walls.
2.2. Protocols for isolated, perfused heart
Given that IR injury is time dependent [27-29], we used an experimental protocol of 30 min of ischemia followed by 10 min reperfusion to assess changes in complex I. Once heart rate and LVP had stabilized each heart was subjected to one of the following six protocols: 60 min of control perfusion (TC) (n=8); 30 min global ischemia (I30) (n=8); 30 min global ischemia followed by 10 min of reperfusion (IR) (n=8); 10 μM Ran given 1 min prior to 30 min global ischemia (RanI30) (n=8); 10 μM Ran given 1 min before 30 min global ischemia followed by 10 min reperfusion (RanIR) (n=8), and 10 μM Ran given for 1 min followed by 40 min of KR perfusion (RanTC) (n=3). At the end of each experiment, some hearts were immediately immersed into liquid N2 for later assessment of changes in electron paramagnetic resonance (EPR) signals, or for later determination of cardiolipin integrity by high pressure liquid chromatography (HPLC); alternatively mitochondria were isolated, as described below, for western blotting, analysis of supercomplex assemblies, and assessment of complex I activity (NADH oxidation).
2.3. Isolation of mitochondria
At the conclusion of each protocol, some hearts were immediately removed from the perfusion apparatus and mitochondria were isolated as previously described [30, 31]. In brief, ventricles were excised, placed in isolation buffer (in mM: 200 mannitol, 5 KH2PO4, 5 MOPS, 1 EGTA, 0.1% BSA and 0.5 mg/ml butylated hydroxy toluene as an anti-oxidant (pH adjusted to 7.15 with KOH)), and minced into fine pieces. The suspension was homogenized for 30 s in 2.5 ml isolation buffer containing 5 U/ml protease (Bacillus licheniformis, Sigma) and for another 1 min after adding 17 ml isolation buffer. The suspension was centrifuged at 8,000 g for 10 min. The pellet was re-suspended in 25 ml isolation buffer and centrifuged at 750 g for 10 min, the supernatant was centrifuged again at 8,000 g for 10 min, and the final pellet, enriched in mitochondria, was re-suspended in 0.5 ml isolation buffer and stored at -80°C until further use.
2.4. Assessment of mitochondrial complex I activity
Mitochondria isolated as described above were centrifuged at 10,000 g and the pellet was re-suspended and washed in hypotonic buffer containing 25 mM potassium phosphate buffer with 5 mM MgCl2 at pH 7.2 by centrifugation at 10,000 g. After dilution to the appropriate concentration, mitochondria were subjected to three rounds of freeze-thaw cycles in the same hypotonic buffer. The fractured mitochondria were used for assay of dynamic respiratory complex I activity using a cuvette-based spectrophotometer (PTI- model QM-8, Photon Technology International). The spectrophotometer was configured for measuring transmittance and used specifically to derive the kinetic profile of complex I activity over time.
Complex I activity was measured by monitoring the dynamic change in transmittance from oxidation of NADH to NAD+ at 340 nm in a pH 7.2 buffer containing 25 mM potassium phosphate buffer with 5 mM MgCl2, 2 mM KCN, 2.5 mg/ml BSA, 0.13 mM NADH, and 65 μM ubiquinone (UQ10) in a total volume of 1 ml [32]. Briefly, after a 3 min baseline measurement, 5 μg of the mitochondrial lysate was added and transmittance was recorded continuously for another 5 min; this was followed by transmittance recorded for 3 min after the addition of 2 μg rotenone to completely block complex I activity. The rate of transmittance after adding rotenone was subtracted from the initial rate of change in transmittance, due to addition of mitochondria, to obtain rotenone sensitive activity. The rates of enzyme activity of all groups were normalized to citrate synthase activity to account for difference in active enzyme at the time of isolation. Rate of activity was calculated using the extinction coefficient of NADH, 6.81 mM-1 cm-1 with slopes derived over the period between 200 and 260 s (first order rate kinetics) [32].
2.5.Histochemical staining of complex I
The amount of total complex I and activity was also determined using histochemical staining. First, freshly isolated mitochondria were disrupted using the method of Schägger et al. [35]. Briefly, 400 μg of pelleted mitochondria was re-suspended in pH 7.0 buffer containing in mM: 50 NaCl, 50 imidazole/HCl, 2 6-aminohexanoic acid, and 1 EDTA. Next mitochondria were solubilized by adding dodecylmaltoside (2.5 g/g) and digitonin (6.0 g/g), followed by incubation on ice for 10 min and centrifugation at 16,000 g for 30 min. Then the supernatant was retained and 5 μl of 50% glycerol was added. Gradient gels with a 4-13% polyacrylamide gradient were used for separation of respiratory complexes using 25 mM imidazole anode buffer and 50 mM tricine, 7.5 mM imidazole cathode buffer. Gels were then used for histochemical staining to assess mitochondrial complex I actvity, by incubating the gel in 5 mM Tris-HCl, pH 7.4, containing 0.5 mg/ml Nitro Blue Tetrazolium chloride (NBT) and 2.5 mg/ml NADH. Bands were visible after 5 min incubation. The gels were transferred into Tris-HCl at pH 7.4 and scanned for densitometry. Individual band densities were normalized to their total respective lane Coomassie stain intensities.
2.6. Determination of supercomplex assemblies
Mitochondrial respiratory complexes are integrated into multi-complex assemblies that can be detected using native gel electrophoresis [33, 34] with the use of mild detergents to solubilize the mitochondrial membranes while preserving both the functional and structural integrity of the complexes. Mitochondria were prepared as described for histochemical staining for complex I activity, except that 6.0g/g digitonin, and no DDM, was used for solubilization of mitochondria and retaining supercomplex assemblies [36]. Proteins were then separated as described above in a 3-13% polyacrylamide gradient gel, and used for either Coomassie staining or histochemical staining of respiratory complexes.
For Coomassie staining, gels were stained for 20 min in 0.2% Brilliant Blue G in methanol:acetic acid:water (MAW) (40:7:53 v/v), and later destained with MAW. After destaining, the gels were scanned using a desktop scanner (CanoScan 8400F) and densitometry was performed using the ImageJ program (National Institutes of Health, Bethesda, MD). Respiratory complex I function was detected as in Section 2.5. Complex III and IV functions were determined by incubating the gels in 50 mM sodium phosphate buffer at pH 7.2 containing 5 mM cytochrome c and 0.5 mg/ml diaminobenzidine [36]. Histochemical staining of complexes III and IV was performed to determine supercomplex components. The observed band intensities were normalized against total lane Coomassie stain intensities. The components of supercomplexes were further determined by 2nd dimension SDS-PAGE. Briefly, supercomplex bands positive for complexes I, III and IV were excised from the gel and incubated in 1% SDS and 1% β- mercaptoethanol for 1 hr at room temperature. The bands were then placed in lanes of a 10% polyacrylamide gel and electrophoresed in Tris-Glycine-SDS buffer for 1.5 hrs at 125V. The proteins were then transferred onto PVDF membranes and used for Western blots using a cocktail of antibodies (Invitrogen, CA) against the following proteins: complex I subunit ND6, complex II FeS subunit, complex III core 2,complex IV subunit I, and complex V subunit ∝.
2.7.Western blot detection of complex I
Complex I subunit NDUFA9 is an integral membrane subunit reported to be more prone to post translational modifications (PTM) than other subunits [37, 38]. Hence any change in levels of this subunit indicates compromised membrane integrity and possibly PTM of the subunit. Mitochondrial protein (50 μg) was solubilized in Laemmli buffer and resolved using sodium dodecyl sulphate- polyacrylamide gel electrophoresis (SDS-PAGE) as described by Laemmli [39] and transferred onto poly vinilidine difluoride membranes using Transblot System (Bio-Rad, Carlsbad, CA) in 50 mM tricine and 7.5 mM imidazole transfer buffer. Membranes were blocked with 10% non fat dry milk in Tris Buffered Saline- TBSt (25 mM Tris-HCl at pH 7.5, 50 mM NaCl and 0.1% Tween 20) by incubating for 1 h followed by incubation in anti- NDUFA9 antibody (Invitrogen, CA) solution overnight at 4°C. After three washes with TBSt the membrane was incubated with an appropriate secondary antibody conjugated to horse-radish peroxidase for 3 h. After five washes with TBSt the membrane was incubated in enhanced chemiluminescence detection solution (ECL-Plus, GE-Amersham) and exposed to X-ray film for autoradiography. Equivalent protein loading was confirmed using an antibody against cytochrome c oxidase (complex IV) subunit 1 (Invitrogen, CA) since complex IV is relatively more resistant to IR injury than is complex I at 10 min reperfusion [27-29].
2.8. Acetylation of mitochondrial protein
A common PTM is acetylation. Total mitochondrial protein acetylation was analyzed using Enzyme Linked Immuno Sorbent Assay (ELISA). Mitochondria, isolated as described above, were solubilized by suspension in phosphate buffered saline, pH 7.4, by the addition of 0.5 % Tween-20. 20 μg of the lysed protein was suspended in 50 μl of sodium carbonate-bicarbonate buffer, pH 7.4 and coated overnight in a 96 well plate. The wells were washed with PBS and blocked with 1% BSA. After three washes, anti-acetyl lysine antibody was added to each well. Following a three-hour incubation, the wells were washed again in PBS, and incubated with appropriate secondary antibody. Color development was achieved by the use of a solution containing o-phenyline diamine, citric acid, and H2O2. The plate was scanned for changes in optical density at 490 nm [40].
2.9. Electron paramagnetic resonance
An EPR system (Elexsys 580 Pulse EPR spectrometer, Bruker, Billerica, MA) was used to detect the transfer of e- through respiratory complexes I and II (succinate dehydrogenase) resulting from the successive oxidation/reduction of Fe in Fe-S clusters. Some treated or untreated isolated hearts (n=4 per group) were immediately transferred to liquid N2 at the end of the protocol and ground to a fine powder. The powder was transferred into quartz glass tubes which were stored at -80°C until analysis. EPR signals were obtained by scanning the samples in a high intensity magnetic field (3000-4000 G) at 10 Kelvin, 9.6 GHz, 5 mW microwave power and 5 G amplitude modulation. Each sample was scanned 9 times and spectra were averaged [41, 42]. Signal intensities were measured for g’s of 2.023 (3Fe-4S attributed to cluster S3 of complex II, or to mitochondrial aconitase), 2.006 (attributed to semi-ubiquinone radical, UQ•), 1.94 (2Fe-2S attributed to N1b of complex I, or to S1 of complex II), and 1.89 (4Fe-4S attributed to N4 of complex I, or to the Rieske center of complex III [43].
2.10. Isolation and analysis of cardiolipin by HPLC
The integrity of cardiolipin isolated from minced heart tissue was assessed using HPLC (System Gold, Beckman Coulter, Fullerton, CA). HPLC detects difference in molecular weight, and thus different species, by the change in mobility of individual molecules through the HPLC column. Lipids were extracted from hearts using a modified method of Hara et al. [44]. Briefly, hearts were removed from the perfusion system after treatments and quickly frozen in liquid N2 and ground to a fine powder. Approximately 1 g of powdered tissue was added to 18 ml hexane:isopropanol (3:2) and mixed for 1 min. The suspension was filtered through Whatman no. 5 filter paper. The powdered tissue was washed twice with 2 ml hexane:isopropanol. Isolated lipids were then analyzed and estimated for cardiolipin content using thin layer chromatography (TLC). Briefly, 200 μl of each sample was loaded onto a Whatman silica gel-60 TLC plate and allowed to dry; 50 μg of purified bovine heart cardiolipin was used as the standard. Lipids were separated using chloroform:methanol:acetic acid:water (55:37.5:3:2) [45]. Bands were visualized by exposure to iodine vapors for 5 min. The ratio of band intensity for each group's cardiolipin was estimated and total lipids in each fraction were calculated. Appropriate amount of lipids equivalent to 200 μg of cardiolipin was dried under N2 gas and resuspended in 20 μl hexane:isopropanol (3:2) and then injected into the HPLC. HPLC analysis was performed using the method of Barcelo-Coblijn et al. [45] at 208 nm.
2.11. Statistical evaluation of data
For the intact heart studies, measurements for each group were compared at baseline, during the brief treatment with or without Ran before ischemia, at 30 min ischemia, and at 10 min reperfusion. For all other studies, measurements for each group were measured at the end of the protocol. All data are expressed as mean ± SEM. Between groups and within group individual values were subject to two-way analysis of variance to determine overall significance. If F values were significant (P < 0.05), post hoc comparisons of means tests (Student-Newman-Keuls) were used to compare the groups within each subset. In mitochondrial studies, statistical analysis was performed similarly. Differences between means were considered significant when P < 0.05 (two-tailed).
3. Results
3.1. Spectrophotometric determination of complex I activity
Representative kinetic traces of complex I activity, monitored in a spectrophotometer as the rate of oxidation of NADH to NAD+ in the presence of UQ10, illustrate differences dependent on the treatment (Fig. 1A). In summarized data from 4 hearts from each of the 5 experimental groups (Fig. 1B) controls (TC) exhibited a NADH oxidation rate of 122±5 nmol/min/mg protein with a large decrease in oxidation rate after I30 to 49±8 nmol/min/mg, but partial restoration to 106±1 nmol/min/mg in the RanI30 group. Similarly, there was a large decrease in complex I activity after IR to 70±4 nmol/min/mg, which was completely restored to 123±7 nmol/min/mg in the RanIR group. No changes in complex I activity were observed in the RanTC group (data not shown). To determine if Ran directly alters mitochondrial complex I activity in fractured mitochondria, in preliminary experiments Ran was added to freeze-thaw fractured mitochondria from the TC, I30 and IR groups, followed by measurement of complex I activity; the result was a slight but insignificant decrease in activity (data not shown).
Fig. 1.
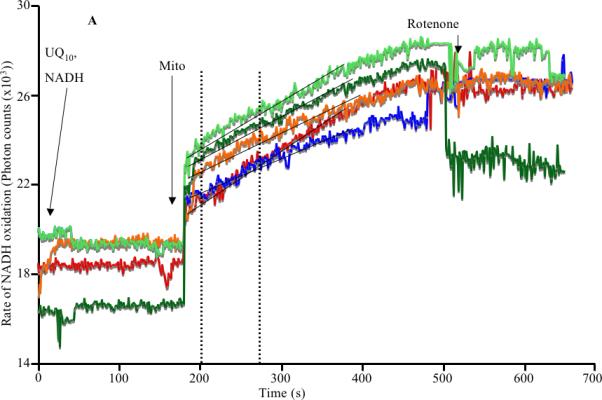
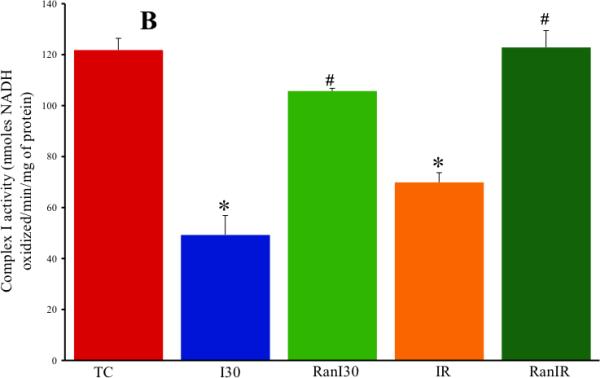
A: Representative spectrophotometric assay of mitochondrial complex I activity during cardiac ischemia reperfusion depicting the time points of addition of substrate, enzyme and the inhibitor. B: Summary data shows ischemia alone reduced the activity of the enzyme, which was restored by treatment with ranolazine. Reperfusion itself corrected the decrease in activity, but this was not as pronounced as with ranolazine (Ran) on reperfusion. Note that the activities depicted in B have been corrected for rotenone sensitivity and normalized to citrate synthase levels. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.2. Histochemical staining for complex I function in gel
A representative gel (Fig. 2, upper panel) for NBT-oxidoreductase activity, reflecting complex I function with or without IR and Ran, illustrates decreased band density after I30 and IR but a higher band density after RanI30. Summary data (Fig. 2, lower panel) showed that band density decreased after I30 (87±3%) and IR (87±5%) compared to the normalized TC group (100%). Ran treatment before ischemia resulted in a significantly higher band density for RanI30 (95±4%) vs. I30 alone, whereas band density for RanIR (91±4%) was not significantly greater than that for IR alone. The observed band densities were normalized to the total lane intensities.
Fig. 2.
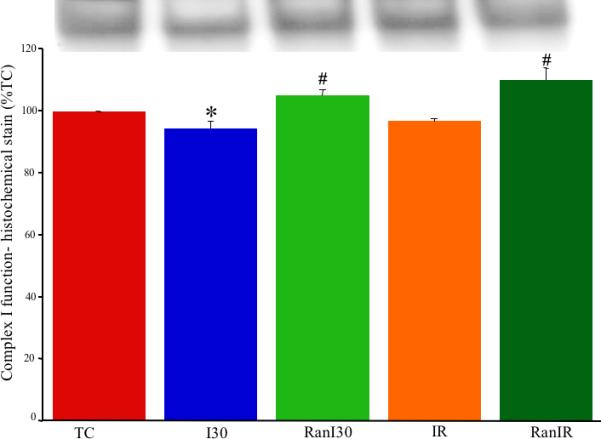
Upper panel: Representative histochemical gel staining of complex I activity, measured as NBT-oxidoreductase, during cardiac ischemia reperfusion. Lower panel: Summary data shows ischemia alone resulted in lower staining than in time controls and reperfusion. Ran treatment increased staining, indicating improved complex I function. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.3. Supercomplex assemblies detected by native gels
A representative image of a native gel (Fig. 3, panel A) stained by Coomassie Brilliant Blue illustrates supercomplexes- SC1 and SC2 comprised of complexes I, III and IV as identified by histochemical staining for complex I (panel B) and III and IV (panel C), and respiratory complexes after 2D electrophoresis detected by Western blot using antibodies against the indicated subunits (panel D). The two supercomplexes presumably are composed of multiple copies of respiratory complexes. Summary data of supercomplex assemblies from Coomassie stained gels (Fig. 3E) showed that band intensity for the supercomplex SC1 (TC =100%) was lower in I30 (86±1 %), IR (88±1%) and RanI30 (87±2%). Ranolazine treatment improved the band intensity of SC1 following IR (96±1%). Band intensity for supercomplex SC2 showed similar results. Compared to TC (100%), there was a significant decrease in band intensity in I30 (77±5%) and IR (85±2%). Ranolazine treatment improved band intensity following I30 (RanI30=93±1%) and IR (RanIR=95±5%). Complex I was not found to associate further with complexes II and V to form other supercomplexes (data not shown). The observed band densities were normalized to the total lane intensities.
Fig. 3.
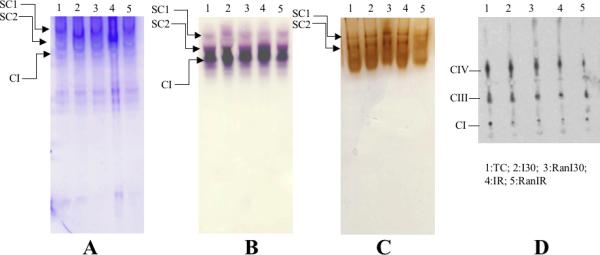
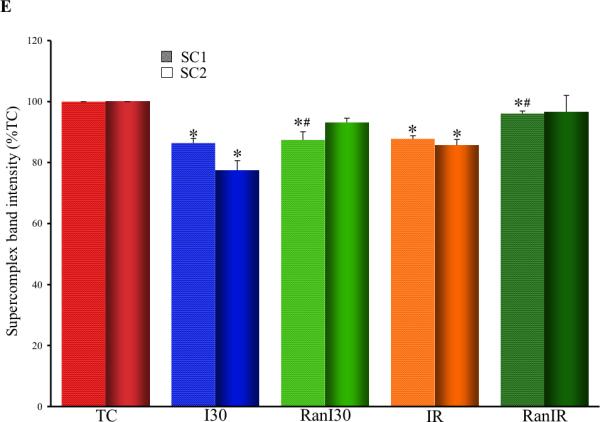
Determination of supercomplex assemblies using native PAGE. Panel A: Coomassie stained gel after native PAGE. SC1 and SC2 indicate supercomplexes composed of complexes I, III and IV, with varying copies of each complex. Components of supercomplex determined by histochemical staining for complex I (panel B), complexes III and IV (panel C) and by Western blot analysis of SC1 and SC2 against respiratory complex subunits (panel D). Summary data of supercomplex assemblies from Coomassie stained gels (panel E) shows reduction in supercomplex assemblies following ischemia and reperfusion, and restoration by ranolazine treatment. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.4. Determination of integrity of complex I subunit NDUFA9 by Western blots
A representative blot for the complex I subunit NDUFA9 (Fig. 4, upper panel) from hearts subjected to IR ± Ran illustrates decreased intensity after I30 and restoration after RanI30 and RanIR. Complex IV subunit I was used as the loading control. Summary data (Fig. 4, lower panel) showed that compared to TC (100%) the anti-NDUFA9 immune reactive band intensity was significantly lower after I30 (84±9%) but was restored after RanI30 (109±7%). Band densities for IR (101±4%) and RanIR (112±7%) groups were not different from the TC group.
Fig. 4.
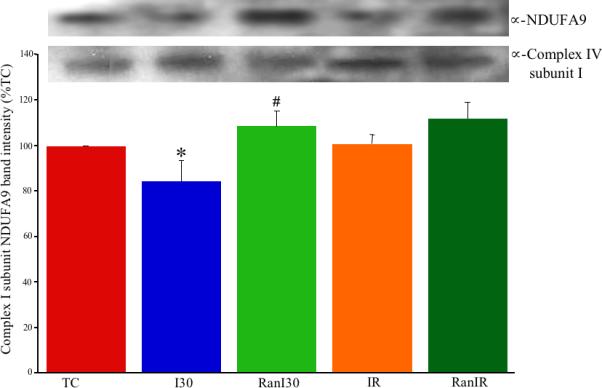
Upper panel: Representative western blot detection of complex I subunit NDUFA9 during cardiac ischemia reperfusion. Lower panel: Summary data shows that ischemia reduced the amount of detectable subunit, indicating a loss of protein or protein damage. Both reperfusion and Ran treatment during ischemia restored protein levels to time control levels. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.5. Analysis of acetylation of mitochondrial proteins using ELISA
The optical density for acetylated mitochondrial proteins, as detected by ELISA, showed an overall decrease in all groups compared to TC (76±4.9 arb. units) (data not represented graphically). I30 showed the least acetylation (41.7±3.1 arb. units), whereas IR partially restored the acetylation (57.8±4.6 arb. units). Treatment with Ran partially restored acetylation in both groups (Ran I30 = 56.5±2.7 arb. units; RanIR = 59.8±2.3 arb. units). Values for each treatment group were significantly lower than the TC group, and there was a statistically significant difference (p<0.05) between I30 and RanI30, but not between IR and RanIR groups.
3.6. Electron transfer in Fe-S clusters by electron paramagnetic resonance
Averaged peak intensities (4 hearts per group) of Fe-S resonance signals (in arb. units) showed differences in peak signals dependent on the treatments (Fig 5A). The g=2.023 signal was assigned to S3, the 3Fe-4S cluster of complex II, or to mitochondrial aconitase, g=2.006 to the semi-ubiquinone radical (UQ•), g=1.94 to cluster N1b of complex I, or to cluster SI of complex II, and g=1.89 to cluster N4 of complex I, or to the Rieske center of complex III. Summary data (Fig. 5B) showed that compared to TC (3.1±1.4 arb. units), neither I30 (2.7±0.8) nor IR (4.2±0.6) significantly altered the g=2.023 signal. However, Ran treatment reduced the g=2.023 signal further after RanI30 (1.6±0.4), but not after RanIR (4.2±1.0). There was a significant increase in the g=2.006 radical after I30 (1.49±0.07), which was lower in TC (0.87±0.09) and after IR (1.06±0.03). Ran treatment also reduced the g=2.006 signal (RanI30 = 1.29±0.09; RanIR = 1.02±0.04). There was also a significant increase in signals pertaining to g=1.94 and 1.89 during ischemia, which returned to TC levels after reperfusion. The contribution at g=1.94 for N4 was verified by the changes noted at g=1.89; i.e. the contribution of N1b and S1 can be estimated after evaluating the contribution of N4. The signal at g=1.94, contributed by N3, was small as estimated by the weak signal at g=1.86, the low field g-value for the signal from N3. Similarly the contribution to the signal at g=1.92 from N2 was weak as determined by the absence of signal from the low field g-value for N2 at 2.05.
Fig. 5.
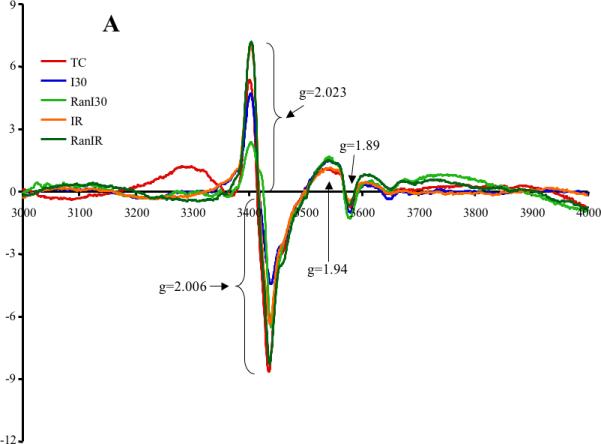
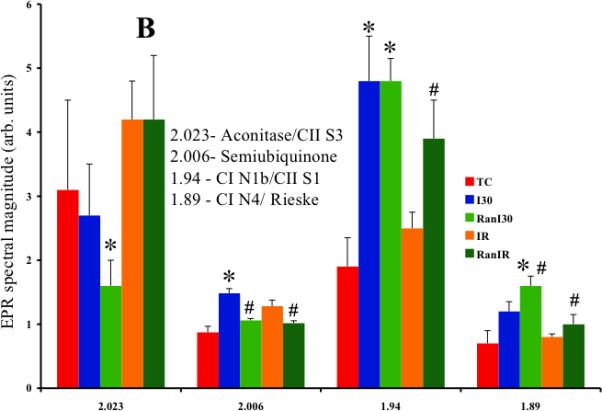
A: Representative traces of EPR signals during cardiac ischemia reperfusion denoting changes in three mitochondrial Fe-S clusters and semiubiquinone. B: Summary data shows the changes in spectral magnitudes of the Fe-S clusters and semiubiquinone. Ischemia increased electron transfer within complex I and semiubiquinone; this was reversed on reperfusion. Ran had a small effect to decrease electron transfer during reperfusion. I, II, III = respiratory complexes; N and Rieske = FeS clusters. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.7. Determination of cardiolipin integrity by HPLC
Representative HPLC traces of cardiolipin integrity were different depending on treatment (Fig. 6A). Compared to the cardiolipin standard TC (93±2 arb. units), there were significant decreases in the areas under the curves in I30 (56±21 arb. units) and more so in IR (32±12 arb. units) groups, reflective of damaged cardiolipin. In summary data (Fig. 6B) the RanI30 group (49±15 arb. units) showed no improvement over the I30 group alone, but the RanIR (69±8 arb. units) group showed a larger area under the peak that is reflective of less fragmented cardiolipin. The number of peaks as detected by HPLC was also higher (reflecting more fragmented species) in I30 and IR groups than in RanI30 and RanIR groups, respectively (Fig. 6A).
Fig. 6.
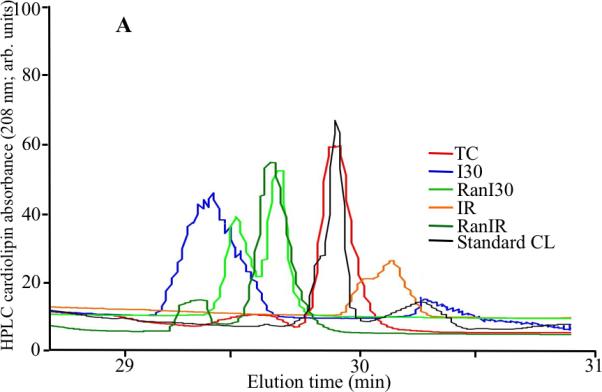
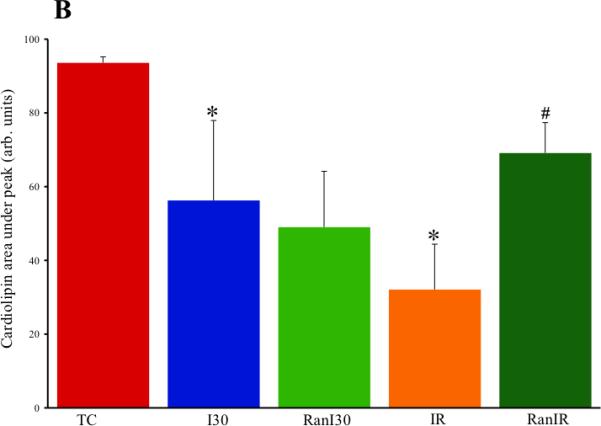
A: Representative traces of cardiolipin integrity by HPLC analysis during cardiac ischemia reperfusion. B: Summary data shows that ischemia alone reduced the area under the major peak of cardiolipin, and reperfusion decreased it further. The additional peaks seen in the spectra could be modified/breakdown products of cardiolipin. Treatment with ranolazine restored peak heights and also decreased the number of peaks, indicating that ranolazine treatment preserves cardiolipin. * indicates p<0.05 for I30/IR vs. TC; # indicates p<0.05 for RanI30/RanIR vs. I30/IR
3.8. Improved cardiac function after IR injury with ranolazine
Heart rate, coronary flow, diastolic LVP, and developed (systolic-diastolic) LVP remained unchanged during continuous perfusion of hearts for 60 min (TC, data not displayed) after which time hearts were harvested for examination of complex I function using the methods described above. At the end of 30 min no flow global ischemia, there was no heart beat and diastolic LVP was elevated (Table 1); after 10 min reperfusion heart rate and coronary flow were similar to those of the TC in all IR groups. On the other hand, diastolic LVP was more elevated in the IR group than in the RanIR group and developed LVP was more depressed in the IR group than in the RanIR group. These data indicate that Ran had a protective effect on reducing contracture during ischemia and on increasing contractile function on reperfusion. These protective effects were associated with improvements in several assays of complex I function and the integrity of its support structure, cardiolipin.
Table 1.
Cardiac function before, during and after IR injury.
| Baseline | Ischemia (30 min) | Reperfusion (10 min) | |
|---|---|---|---|
| dLVP/dtmax (mmHg/s) | |||
| IR10 | 2342±228 | 1342±136 | |
| RanIR | 2449±133 | 2406±151# | |
| dLVP/dtmin (mmHg/s) | |||
| IR10 | 2052±129 | 955±106 | |
| RanIR | 1923±185 | 1971±166# | |
| Systolic-diastolic LVP (mmHg) | |||
| IR10 | 88±4 | 38±5 | |
| RanIR | 90±4 | 57±7# | |
| Diastolic LVP (mmHg) | |||
| I30 | 0±0 | 3±1 | |
| RanI30 | 0±0 | 1±1# | |
| IR10 | 0±0 | 5±2 | |
| RanIR | 0±0 | 0±0# | |
| Coronary flow (ml/min) | |||
| IR10 | 15.9±0.7 | 14.8±0.6 | |
| RanIR | 15.2±1.1 | 15.3±1.1 | |
| Heart rate (beats/min) | |||
| IR10 | 227±4 | 228±2 | |
| RanIR | 233±5 | 226±2 |
LVP = Left ventricular pressure
I30 = 30 min of global ischemia
IR10 = 30 min of global ischemia + 10 min reperfusion
RanI30 = 10 μM Ran given 1 min before I30
RanIR = 10 μM Ran given 1 min before IR10
indicates p<0.05 for RanI30 or RanIR vs. I30 or IR10
4. Discussion
4.1. Summary of findings
Complex I is a major source and target of ROS during IR [4, 28] and excess ROS is a major determinant of poor return of cardiac function after IR injury [2, 25]. For that reason we used several experimental tools to search for deleterious biochemical and biophysical changes in respiratory complex I function and structural integrity in hearts subjected to 30 min ischemia alone, or to ischemia plus 10 min reperfusion. Ran, which is reported to have cardio-protective effects [11, 12, 14, 15] by switching substrate preference, among other proposed effects [16], was examined for its potential to attenuate damage to complex I and to improve cardiac function when present during ischemia.
We found that ischemia alone a) reduced the rate of NADH oxidation, b) reduced protein acetylation, c) decreased complex I function, d) reduced supercomplex assembly, e) decreased the detection of a complex I subunit, f) increased the reduced state several fold in three distinct Fe-S clusters in complex I and/or complex II [2Fe-2S in N1b and/or S1 and the 4Fe-4S cluster N4, and in complex I N4 and/or Rieske center in complex III], increased the signal at 2.006 (UQ•) but did not alter the signal at 2.023 [S3 in complex II and/or aconitase] and, g) reduced the structural integrity of cardiolipin, a supporting structure for mitochondrial complexes. Compared to ischemia alone, treatment with Ran during ischemia resulted in full or partial restoration of: a) the rate of NADH oxidation, b) the acetylation of mitochondrial proteins, c) the detection of complex I subunit, and d) the supercomplex assembly.
During the short 10 min reperfusion period, complex I dysfunction persisted, acetylation levels remained lower, and cardiolipin showed greater disintegration. Because the expression levels of respiratory complex I subunits are not likely to change given the short time period of our protocols, any reduction in complex I subunit band intensity probably reflects compromised integrity of the subunit's structure. The reduction in NADH oxidation rate with reperfusion alone was reversed and cardiolipin integrity was improved by Ran treatment. Electron transfer through N1b of complex I and/or SI of complex II was improved by Ran on reperfusion and through N4 of complex I or the Rieske center of complex III. Each of these changes observed after Ran treatment was associated with improved diastolic LVP during ischemia and improved contractile function during reperfusion. However, Ran did not alter complex I activity when applied directly to fragmented mitochondria in either the TC, I30, or IR groups.
Our study supports our alternative hypothesis that Ran treatment indirectly leads to partial protection of mitochondria, as shown in part, by improved complex I activity, restored e- transfer through some Fe-S clusters, and retained supercomplex assembly and cardiolipin integrity. This range of protection implies that Ran is not likely to have a direct molecular interaction with complex I, which is substantiated by our preliminary finding that Ran had no effect on complex I activity when added directly to fractured mitochondria. Hence we conclude that Ran acts indirectly at another site to reduce damage to mitochondrial structure. We propose that by maintaining complex I e- flow and preserving the supercomplex assemblies, the tendency for electron leak and subsequent generation of O2•- is reduced. This notion is supported by our recent observation [18] that by lowering ROS emission with reduced cytosolic and m[Ca2+] loading, Ran indiscriminately reduces oxidative damage to complex I and to its supporting structures (among other mitochondrial and cytosolic molecules). Our comprehensive examination of complex I function during IR injury demonstrates the extent of structural and functional damage to complex I and the impact of a non-mitochondrial-targeted drug to indirectly protect mitochondrial function, likely by reducing ROS emission.
4.2. ROS and damage to respiratory complexes during cardiac injury
Superoxide radicals (O2•-) are produced in small amounts in mitochondria under O2 and substrate replete conditions, but the balance between O2•- generation and free radical scavenging, (ROS emission), is altered in high oxidative conditions such as IR injury [1, 22, 46, 47]. These O2•- radicals and their downstream products attack all types of cellular macro molecules, i.e. proteins, lipids, carbohydrates, and D(R)NA, thereby altering their structure and function [48, 49]. Although all major biomolecules are susceptible to oxidative damage, the extent of this damage is dependent on the molecular structure, the length of exposure to ROS, the concentration and kind of ROS, and the capacity of ROS scavengers available.
Respiratory complexes differ in their susceptibility to IR injury. Complexes I and III are believed to be more susceptible to IR injury than complexes II, IV, and V [22]. A useful kinetic test for an effect of Ran to protect respiratory complex I against IR injury is the measure of catalytic conversion rate of NADH to NAD+ in complex I during IR. Although respiratory complexes I and III are reported to be a major source of O2•- generation [1, 3, 50, 51], many studies have implicated complex I to also be the most susceptible of the respiratory complexes to ischemic damage [4, 52]. Complex I is also a major site of e- leak during IR; this is likely to drive e- transfer backward through complex I toward the NADH binding site [2, 53, 54]. Indeed, in our collateral histochemical and kinetic experiments we found that complex I function was attenuated; this is consistent with the increased (backward) e- transfer in complex I and the decreased (forward) e- transfer in complex II [22, 55].
Interventions to limit backward e- transfer from complex II into complex I may be helpful in attenuating ROS release [2, 53, 56]. Indeed, we [56] showed previously that amobarbital, a complex I blocker at the rotenone site, itself slightly enhanced generation of O2•- before ischemia but attenuated O2•- emission during IR [2]. Other modulators of complex I are also known to have protective effects against IR injury [52]. The decreased rate of NADH oxidation in ischemic hearts, compared to reperfused hearts, might also be due to a lack of the e- acceptor O2. When reperfusion begins there is an abrupt availability of O2, and hence a higher rate of respiratory chain activity. In support of this, we found faster respiratory complex I activity in Ran -treated hearts than in the untreated ischemia and IR groups
4.3. Mechanism of mitochondrial protection by ranolazine during cardiac IR injury
Ranolazine is a clinically useful anti-anginal drug [15, 57] that was found originally to block the late Na+ current that occurs with ischemia [11, 58-60]. But others have reported that Ran induces a switch from the usual substrate preference from fatty acids to glucose, and that it partially blocks complex I at an unconfirmed site, particularly in uncoupled mitochondria [21]. If Ran were indeed a partial complex I blocker in vivo as Wyatt et al. [21] reported in in vitro isolated mitochondrial experiments, this might contribute to preserving its structure and function during IR injury as we found. But it is unlikely that Ran penetrates membranes effectively due to its hydrophilic structure. It is conceivable that a fat soluble metabolite of Ran is a direct mitochondrial protective drug, but this will require direct evidence of a metabolite with actions on complexes and other mitochondrial targets. Because we found that Ran had no direct effects on mitochondrial function when applied directly to isolated, energized mitochondria [18], or to fractured mitochondria, we believe it is unlikely to directly target complex I or any other mitochondrial protein and its supporting structures.
During ischemia there is an increase in cytosolic [Na+] due to failure of the Na+/K+ ATPase pump and an increase in toxic intermediates [60]. This triggers NCE, which results in increased cell Ca2+ loading, and consequently, m[Ca2+] loading which may lead to mitochondrial oxidative damage by “Ca2+-induced ROS release” [46, 61]. Increased m[Ca2+] contributes to an increase in ROS emission under certain circumstances, either by increasing the rate of O2•- generation or by reducing the rate of ROS scavenging [17, 61, 62]. Therefore we postulated that Ran induces its protective effect by reducing NCE in the cytosolic compartment to reduce Ca2+ loading, and by extension, to mitigate m[Ca2+] loading. Our conclusion derives from the principal earlier finding that Ran is a late Na+ channel current blocker [11, 58-60], and our report that Ran reduces m[Ca2+] overload and ROS emission during late ischemia and reperfusion [18]. This decrease in ROS may or may not be a direct consequence of an improved electron transport chain activity or reduced m[Ca2+] overload. However, Song et al. [63] reported that Ran attenuated H2O2 -induced cytosolic Ca2+ overload and cardiac contractile dysfunction suggesting that “ROS induced Ca2+ loading” can be attenuated by Ran.
The oxidative stress that occurs in ischemia is known to activate deacetylases [64, 65], which cause a decrease in lysine acetylation. Deacetylation can disturb the secondary and tertiary associations among proteins and/or their subunits. Hence it is possible that by preserving acetylation, a reversible PTM, this might help preserve the integrity of mitochondrial proteins. Ran treatment attenuated the lysine deacetylation of mitochondrial proteins during ischemia. If Ran could access the mitochondrial matrix, it might interfere with the action of deacetylases to protect the proteins but there is no direct evidence for this.
4.3. Electron transfer in complex I and protective effect of ranolazine during cardiac IR injury
Changes in EPR spectra can occur due to many factors, including but not limited to: a loss of one or more Fe-S clusters, a saturated oxidation or reduction potential, and proximity of neighboring clusters. Complex I subunits with their Fe-S clusters mediate single e- transfer so any damage to these subunits or Fe-S clusters will cause disrupted e- transfer. Once an e- enters the Fe-S cluster chain, its further movement is affected by the redox states of the following Fe-S clusters [6, 66]. From FMN one e- enters a transport chain consisting of one binuclear and the six tetranuclear Fe-S clusters leading (during forward e- transfer) to the Q10 binding site (FMN → N3→N1b–N4–N5–N6a–N6b→N2→UQ•→Q10) [73] and ultimately to complex III. In our study, we observed no significant change in the g=2.023 signal representing the 3Fe-4S cluster, S3, of complex II with IR. In contrast, results of Myers et al. [42] reported a decrease in the aconitase signal (with the same g value) following oxidative stress with chromium radicals. There was a significant increase in the amount of the EPR detectable UQ• radical during ischemia, and this was reduced by reperfusion as well as by Ran treatment. It should be noted however, that UQ• is one of the several free radicals generated during ischemia, so this is not representative of the total oxidative stress in the tissue.
In contrast to S3 of complex II, there were significant increases in the signal intensities for g=1.94 and 1.89 during ischemia that decreased again on reperfusion; this could represent reverse e- transfer during ischemia within complex I. It is difficult to ascertain the exact source of this signal because it can emanate from cluster N1b of complex I or SI of complex II. Although other clusters like the 4Fe-4S and N3 have a g-value close to 1.94 (1.93 for N3) the complementary peak (2.04 for N3) is too weak to detect, implying there is little contribution of signal from N3 at 1.93. Thus we attribute the 1.94 signal to N1b and/or S1.
Reverse e- transfer is believed to occur in the respiratory chain during ischemia, particularly within complex I [54]. Our results support this assumption. It is plausible from the EPR spectra of N1b (g=1.94) and N4 (g=1.89) clusters that ischemia can cause a severe back up of e- to cause reverse e- transfer back in complex I. Reperfusion appeared to largely correct this e- back up, whereas Ran did not affect signal intensity during ischemia, but increased it on reperfusion. The reason for this is unclear. Similarly, we observed an increase in signal intensity for g=1.89 during ischemia. Again, this signal can be attributed to either cluster N4 of complex I or the Rieske center of complex III; however, since most of this signal was generated by the N4 cluster (as noticed after spectral splitting at higher resolution), we conclude that the observed changes were due to cluster N4 and not to the Rieske center.
The signal at g=2.023, assigned to the oxidized state of the 3Fe-4S, S3 cluster in complex II decreased with ischemia and fell even more during 30 min ischemia with Ran, but recovered during reperfusion with or without Ran. We attribute the decrease in the 2.023 signal to greater e- flow through complex II, thus reducing the S3 center. This complements the suggestion that there is a reverse e- transfer during ischemia.
4.4. Cardiolipin integrity and protective effect of ranolazine during cardiac injury
Cardiolipin, a highly unsaturated fatty acid molecule, is prone to ROS attack with the most commonly encountered changes being lipid peroxidation and carbon chain breakdown [49]. The qualitative mobility of cardiolipin was assessed using HPLC by a change in area under the peak, compared to the cardiolipin standard, and by the appearance of secondary peaks with different retention times; these changes represent distortion of cardiolipin. Moreover, cardiolipin has a high content of oxidatively sensitive linoleic acid, a phospholipid unique to mitochondria. Its location in the IMM makes it extremely susceptible to oxidative damage by ROS, whether acutely as in IR injury or chronically as in aging [8, 9]. Cardiolipin is necessary not only for the assembly of each respiratory complex and the supercomplexes, but also for the proper functioning of respiratory complexes and for that matter an efficient e- transfer. Thus any fragmentation or damage to cardiolipin structure reflects on the performance of the respiratory complexes [4, 5, 10]. Indeed, in a pilot study, analysis of the complex I subunit NDUFS3, which is in the peripheral arm of complex I [67] showed no changes in Western blot band intensity with and without IR and with and without treatment with Ran (data not shown), compared to the decrease in band intensity of NDUFA9, which is a membrane integral subunit. This shows the importance of the integrity of cardiolipin on maintaining complex I structure and function.
Ran is also reported to partially inhibit fatty acid oxidation [21] and to stimulate glucose oxidation in hearts during normoxia, ischemia, and IR [20, 68]. Thus Ran may also act as a metabolic modulator [19, 20] that promotes more efficient use of O2 and substrates. A reduction in ROS emission would likely reduce oxidative damage to cardiolipin, as suggested by our HPLC experiments, in which Ran -treated hearts exhibited a partial restoration toward the control cardiolipin HPLC spectra. Our data showing restoration of cardiolipin integrity, along with improvements in complex I structure and activity in the Ran treated hearts, are consistent with another study showing that restoration of cardiolipin content in mitochondria can improve complex I activity [4].
4.5.Conclusions
Protection of hearts against IR injury by Ran is associated with mitochondrial protection. Although Ran exerts a protective effect on complex I and its supporting structures, it is doubtful from our study that Ran mediates its protection by directly interacting with complex I, given our finding that Ran had no direct effect on complex I activity, or on any other mitochondrial function we measured in intact or fragmented mitochondria. The reversal or attenuation of complex I dysfunction, albeit indirect, by Ran points out the importance of complex I integrity and function as important factors in restoring cardiac function. Moreover, given the effects we and others observed, Ran appears to protect hearts in various ways, including but not limited to blocking the late Na+ current. Concomitant decreases in m[Ca2+] overload and ROS emission, induced by Ran, are the probable factors that lead to preservation of cardiolipin, maintaining supercomplex structure and less backward e- transfer through the Fe-S clusters of complex I, thereby improving its efficiency in function and NADH oxidizing capability. Nevertheless, our study emphasizes the extent of injury to complex I and other mitochondrial structures, and expresses the importance of understanding the mechanisms of compounds to protect mitochondria during cardiac IR injury.
Highlights.
Mitochondrial complex I is a major target of cardiac ischemia/reperfusion (IR) injury.
IR injury caused specific biophysical, biochemical and molecular changes in complex I.
A cardio-protective drug, ranolazine, was found to indirectly reduce complex I damage.
Cardiac function after IR injury can be improved by indirectly reducing complex I dysfunction.
Acknowledgments
This work was supported in part by grants from the American Heart Association (0855940G, D.F. Stowe), the National Institutes of Health (R01 HL095122, A.K.S. Camara, and R01 HL089514, D.F. Stowe), and the Veterans Administration (VA Merit 8204-05P, D.F. Stowe).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no disclosures to make.
References
- 1.Stowe DF, Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antiox. Redox. Signal. 2009;11:1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 2008;77:406–415. doi: 10.1016/j.cardiores.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, Moraes CT, Enriquez JA. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 5.Fry M, Green DE. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 1981;256:1874–1880. [PubMed] [Google Scholar]
- 6.Euro L, Bloch DA, Wikstrom M, Verkhovsky MI, Verkhovskaya M. Electrostatic interactions between FeS clusters in NADH:ubiquinone oxidoreductase (Complex I) from Escherichia coli. Biochemistry. 2008;47:3185–3193. doi: 10.1021/bi702063t. [DOI] [PubMed] [Google Scholar]
- 7.Hinchliffe P, Sazanov LA. Organization of iron-sulfur clusters in respiratory complex I. Science. 2005;309:771–774. doi: 10.1126/science.1113988. [DOI] [PubMed] [Google Scholar]
- 8.Lesnefsky EJ, Hoppel CL. Cardiolipin as an oxidative target in cardiac mitochondria in the aged rat. Biochim. Biophys. Acta. Bioenerg. 2008;1777:1020–1027. doi: 10.1016/j.bbabio.2008.05.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesnefsky EJ, Minkler P, Hoppel CL. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009;46:1008–1015. doi: 10.1016/j.yjmcc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Rad. Bio. Med. 1999;27:42–50. doi: 10.1016/s0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 11.Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiological basis for the antiarrhythmic actions of ranolazine. Heart rhythm. 2011;8:1281–1290. doi: 10.1016/j.hrthm.2011.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boden WE. Ranolazine and its anti-ischemic effects: revisiting an old mechanistic paradigm anew? J. Am. Coll. Cardiol. 2010;56:943–945. doi: 10.1016/j.jacc.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Hale SL, Kloner RA. Ranolazine, an inhibitor of the late sodium channel current, reduces postischemic myocardial dysfunction in the rabbit. J. Cardiovasc. Pharmacol. 2006;11:249–255. doi: 10.1177/1074248406294607. [DOI] [PubMed] [Google Scholar]
- 14.Stone PH. Ranolazine: new paradigm for management of myocardial ischemia, myocardial dysfunction, and arrhythmias. Cardiol. Clin. 2008;26:603–614. doi: 10.1016/j.ccl.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Stone PH, Chaitman BR, Stocke K, Sano J, DeVault A, Koch GG. The anti-ischemic mechanism of action of ranolazine in stable ischemic heart disease. J. Am. Coll. Cardiol. 2010;56:934–942. doi: 10.1016/j.jacc.2010.04.042. [DOI] [PubMed] [Google Scholar]
- 16.Zacharowski K, Blackburn B, Thiemermann C. Ranolazine, a partial fatty acid oxidation inhibitor, reduces myocardial infarct size and cardiac troponin T release in the rat. Eur. J. Pharmacol. 2001;418:105–110. doi: 10.1016/s0014-2999(01)00920-7. [DOI] [PubMed] [Google Scholar]
- 17.An J, Varadarajan SG, Camara A, Chen Q, Novalija E, Gross GJ, Stowe DF. Blocking Na+/H+ exchange reduces [Na+]i and [Ca2+]i load after ischemia and improves function in intact hearts. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H2398–2409. doi: 10.1152/ajpheart.2001.281.6.H2398. [DOI] [PubMed] [Google Scholar]
- 18.Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol. Res. 2011;64:381–390. doi: 10.1016/j.phrs.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCormack JG, Baracos VE, Barr R, Lopaschuk GD. Effects of ranolazine on oxidative substrate preference in epitrochlearis muscle. J. Appl. Physiol. 1996;81:905–910. doi: 10.1152/jappl.1996.81.2.905. [DOI] [PubMed] [Google Scholar]
- 20.McCormack JG, Barr RL, Wolff AA, Lopaschuk GD. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135–142. doi: 10.1161/01.cir.93.1.135. [DOI] [PubMed] [Google Scholar]
- 21.Wyatt KM, Skene C, Veitch K, Hue L, McCormack JG. The antianginal agent ranolazine is a weak inhibitor of the respiratory complex I, but with greater potency in broken or uncoupled than in coupled mitochondria. Biochem. Pharmacol. 1995;50:1599–1606. doi: 10.1016/0006-2952(95)02042-x. [DOI] [PubMed] [Google Scholar]
- 22.Han F, Da T, Riobo NA, Becker LB. Early mitochondrial dysfunction in electron transfer activity and reactive oxygen species generation after cardiac arrest. Crit. Care Med. 2008;36:S447–453. doi: 10.1097/ccm.0b013e31818a8a51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pope S, Land JM, Heales SJ. Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim. Biophys. Acta Bioenerg. 2008;1777:794–799. doi: 10.1016/j.bbabio.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Camara AK, An J, Riess ML, Novalija E, Stowe DF. Cardiac preconditioning with 4-h, 17 degrees C ischemia reduces [Ca2+]i load and damage in part via KATP channel opening. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H1961–1969. doi: 10.1152/ajpheart.01032.2001. [DOI] [PubMed] [Google Scholar]
- 25.Heinen A, Aldakkak M, Stowe DF, Rhodes SS, Riess ML, Varadarajan SG, Camara AK. Reverse electron flow-induced ROS production is attenuated by activation of mitochondrial Ca2+-sensitive K+ channels. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H1400–1407. doi: 10.1152/ajpheart.00198.2007. [DOI] [PubMed] [Google Scholar]
- 26.Riess ML, Camara AK, Chen Q, Novalija E, Rhodes SS, Stowe DF. Altered NADH and improved function by anesthetic and ischemic preconditioning in guinea pig intact hearts. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H53–60. doi: 10.1152/ajpheart.01057.2001. [DOI] [PubMed] [Google Scholar]
- 27.Ambrosio G, Zweier JL, Flaherty JT. The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J. Mol. Cell. Cardiol. 1991;23:1359–1374. doi: 10.1016/0022-2828(91)90183-m. [DOI] [PubMed] [Google Scholar]
- 28.Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am. J. Physiol. 1983;244:H743–748. doi: 10.1152/ajpheart.1983.244.6.H743. [DOI] [PubMed] [Google Scholar]
- 29.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aldakkak M, Stowe DF, Cheng Q, Kwok WM, Camara AK. Mitochondrial matrix K+ flux independent of large-conductance Ca2+-activated K+ channel opening. Am. J. Physiol. Cell. Physiol. 2010;298:C530–541. doi: 10.1152/ajpcell.00468.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinen A, Camara AK, Aldakkak M, Rhodes SS, Riess ML, Stowe DF. Mitochondrial Ca2+-induced K+ influx increases respiration and enhances ROS production while maintaining membrane potential. Am. J. Physiol. Cell. Physiol. 2007;292:C148–156. doi: 10.1152/ajpcell.00215.2006. [DOI] [PubMed] [Google Scholar]
- 32.Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Meth. Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- 33.Schägger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta Bioenerg. 2002;1555:154–159. doi: 10.1016/s0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- 34.Schagger H, Pfeiffer K. The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001;276:37861–37867. doi: 10.1074/jbc.M106474200. [DOI] [PubMed] [Google Scholar]
- 35.Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 36.Wittig I, Carrozzo R, Santorelli FM, Schägger H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis. 2007;28:3811–3820. doi: 10.1002/elps.200700367. [DOI] [PubMed] [Google Scholar]
- 37.Fearnley IM, Walker JE. Conservation of sequences of subunits of mitochondrial complex I and their relationships with other proteins. Biochim. Biophys. Acta Bioenerg. 1992;1140:105–134. doi: 10.1016/0005-2728(92)90001-i. [DOI] [PubMed] [Google Scholar]
- 38.Carroll J, Shannon RJ, Fearnley IM, Walker JE, Hirst J. Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I. Identification of two new subunits. J. Biol. Chem. 2002;277:50311–50317. doi: 10.1074/jbc.M209166200. [DOI] [PubMed] [Google Scholar]
- 39.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Alamdari DH, Kostidou E, Paletas K, Sarigianni M, Konstas AG, Karapiperidou A, Koliakos G. High sensitivity enzyme-linked immunosorbent assay (ELISA) method for measuring protein carbonyl in samples with low amounts of protein. Free Radic. Biol. Med. 2005;39:1362–1367. doi: 10.1016/j.freeradbiomed.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, Antholine WE, Zielonka J, Srinivasan S, Avadhani NG, Kalyanaraman B. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys. J. 2009;96:1388–1398. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myers CR, Antholine WE, Myers JM. The pro-oxidant chromium(VI) inhibits mitochondrial complex I, complex II, and aconitase in the bronchial epithelium: EPR markers for Fe-S proteins. Free Radic. biol. Med. 2010;49:1903–1915. doi: 10.1016/j.freeradbiomed.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohnishi T, Nakamaru-Ogiso E. Were there any “misassignments” among iron-sulfur clusters N4, N5 and N6b in NADH-quinone oxidoreductase (complex I)? Biochim. Biophys. Acta Bioenerg. 2008;1777:703–710. doi: 10.1016/j.bbabio.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hara A, Radin NS. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978;90:420–426. doi: 10.1016/0003-2697(78)90046-5. [DOI] [PubMed] [Google Scholar]
- 45.Barcelo-Coblijn G, Murphy EJ. An improved method for separating cardiolipin by HPLC. Lipids. 2008;43:971–976. doi: 10.1007/s11745-008-3212-3. [DOI] [PubMed] [Google Scholar]
- 46.Camara AK, Aldakkak M, Heisner JS, Rhodes SS, Riess ML, An J, Heinen A, Stowe DF. ROS scavenging before 27° C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am. J. Physiol. Cell. Physiol. 2007;292:C2021–2031. doi: 10.1152/ajpcell.00231.2006. [DOI] [PubMed] [Google Scholar]
- 47.Camara AK, Lesnefsky EJ, Stowe DF. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 2010;13:279–347. doi: 10.1089/ars.2009.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies KJ, Goldberg AL. Proteins damaged by oxygen radicals are rapidly degraded in extracts of red blood cells. J. Biol. Chem. 1987;262:8227–8234. [PubMed] [Google Scholar]
- 49.Davies KJ, Goldberg AL. Oxygen radicals stimulate intracellular proteolysis and lipid peroxidation by independent mechanisms in erythrocytes. J. Biol. Chem. 1987;262:8220–8226. [PubMed] [Google Scholar]
- 50.Malinska D, Kulawiak B, Kudin AP, Kovacs R, Huchzermeyer C, Kann O, Szewczyk A, Kunz WS. Complex III-dependent superoxide production of brain mitochondria contributes to seizure-related ROS formation. Biochim. Biophys. Acta Bioenerg. 2010;1797:1163–1170. doi: 10.1016/j.bbabio.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 52.Fato R, Bergamini C, Bortolus M, Maniero AL, Leoni S, Ohnishi T, Lenaz G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2009;1787:384–392. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell. Physiol. 2008;294:C460–466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 54.Grivennikova VG, Vinogradov AD. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta Bioenerg. 2006;1757:553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 55.Sadek HA, Humphries KM, Szweda PA, Szweda LI. Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch. Biochem. Biophys. 2002;406:222–228. doi: 10.1016/s0003-9861(02)00446-0. [DOI] [PubMed] [Google Scholar]
- 56.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 57.Szel T, Koncz I, Jost N, Baczko I, Husti Z, Virag L, Bussek A, Wettwer E, Ravens U, Papp JG, Varro A. Class I/B antiarrhythmic property of ranolazine, a novel antianginal agent, in dog and human cardiac preparations. Eur. J. Pharmacol. 2011;662:31–39. doi: 10.1016/j.ejphar.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 58.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the proarrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J. Cardiovasc. Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 60.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ. Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- 61.Camara AK, An J, Chen Q, Novalija E, Varadarajan SG, Schelling P, Stowe DF. Na+/H+ exchange inhibition with cardioplegia reduces cytosolic [Ca2+] and myocardial damage after cold ischemia. J. Cardiovasc. Pharmacol. 2003;41:686–698. doi: 10.1097/00005344-200305000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Miyamae M, Camacho SA, Weiner MW, Figueredo VM. Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 1996;271:H2145–2153. doi: 10.1152/ajpheart.1996.271.5.H2145. [DOI] [PubMed] [Google Scholar]
- 63.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 2006;318:214–222. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 64.Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc. Res. 2007;76:473–481. doi: 10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 66.Ingledew WJ, Ohnishi T. An analysis of some thermodynamic properties of iron-sulphur centres in site I of mitochondria. Biochem. J. 1980;186:111–117. doi: 10.1042/bj1860111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim. Biophys. Acta Bioenerg. 2007;1767:1215–1227. doi: 10.1016/j.bbabio.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 68.Clarke B, Wyatt KM, McCormack JG. Ranolazine increases active pyruvate dehydrogenase in perfused normoxic rat hearts: evidence for an indirect mechanism. J. Mol. Cell. Cardiol. 1996;28:341–350. doi: 10.1006/jmcc.1996.0032. [DOI] [PubMed] [Google Scholar]