

Abstract
AIMS
To assess steady-state effects of therapeutic and supra-therapeutic doses of prucalopride on the QT interval using a novel design involving a parallel placebo group with nested crossover for positive control.
METHODS
A double-blind, double-dummy, placebo- and active-controlled study was conducted in 120 healthy male and female volunteers (NCT00903747). Volunteers were randomized to receive prucalopride 2–10 mg once daily (therapeutic and supratherapeutic doses, respectively) (group 1), placebo with 400 mg moxifloxacin on day 1 (group 2a), or placebo with moxifloxacin on day 15 (group 2b). Twelve-lead 24 h Holter ECGs recorded at various time-points were evaluated blind and centrally.
RESULTS
Estimated mean difference in study specific corrected QT interval (QTcSS) time-matched change from baseline between prucalopride (2 and 10 mg) and placebo was <5 ms at all time points (maximum mean difference: 3.83 ms at 3.5 h post dose on day 5 with 2 mg [90% Cl −0.33, 6.38 ms]). Upper limits of the two-sided 90% CI for QTcSS were all <10 ms. There were no outlying QTcSS values >450 ms and no subjects had an increase >60 ms following prucalopride. Moxifloxacin produced the expected significant changes in QTcSS (>5 ms, maximum of +12.7 ms at 5 h post dose) at all time-points except 1 h post dose. Prucalopride resulted in small increases in heart rate (maximum of 5.8 beats min–1), which were similar for 2 and 10 mg. Prucalopride was well tolerated after first day of treatment.
CONCLUSION
Prucalopride at both therapeutic and supra therapeutic doses has no clinically significant effects on cardiac repolarisation in healthy volunteers.
Keywords: 5-HT4, constipation, moxifloxacin, prucalopride, safety, study specific QT correction, thorough QT study
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Drugs interacting with the hERG potassium channel are associated with a prolongation of the QT interval, which can lead to life-threatening arrhythmias such as torsades de pointes.
Early prokinetic agents, such as cisapride, display poor selectivity for their target receptor, 5-HT4, also interacting with hERG channels and, as a consequence, have a poor safety and benefit/risk profile.
Prucalopride, a novel and selective 5-HT4 agonist with enterokinetic activity, is indicated for the treatment of chronic constipation in adults in whom laxatives fail to provide adequate relief.
WHAT THIS STUDY ADDS
This study describes the results of a novel thorough QT study design involving a parallel group with nested crossover for positive control.
The mean QTc interval was not prolonged in patients receiving therapeutic (2 mg) or supra-therapeutic (10 mg) doses of prucalopride, as assessed using QTcSS.
New generation prokinetic agents with improved selectivity for 5-HT4 receptors, such as prucalopride, should help improve the benefit/risk profile of these agents and, ultimately, their clinical usefulness.
Introduction
Prucalopride (Resolor®) is a novel enterokinetic agent. It is the first of a new generation of selective, high-affinity 5-HT4 receptor agonists [1–3]. This drug, which has been developed for the treatment of chronic constipation in adults in whom laxatives fail to provide adequate relief, stimulates gastrointestinal (GI) motility through its effects on 5-HT4 receptors [1, 3]. Clinical data, including those from three identical pivotal phase III studies, have demonstrated that 2 mg prucalopride once daily is effective in increasing stool frequency, reducing constipation-related symptoms and improving quality of life [4–6].
Concerns about possible cardiovascular effects of enterokinetic agents arose a few years ago, prompting market withdrawal and restrictions for the use of cisapride and tegaserod, in 2003 and 2006, respectively. It is now commonly accepted that there should be a 30-fold separation (or safety margin) between maximum unbound drug concentrations in humans and the human ether a-go-go related gene (hERG) potassium channel half maximal inhibitory concentration (IC50), to reduce the potential risk of cardiac pro-arrhythmia such as torsade de pointes [7]. In contrast with cisapride, which has a margin of less than 10-fold between its affinity for 5-HT4 and hERG channels (binding studies), prucalopride at its therapeutic dose is more than 200 times more selective for 5-HT4 than for the hERG channels [2]. With regard to functional tests measuring hERG currents, cisapride has no discernable safety margin between its hERG IC50 and its therapeutic concentration [7]. Conversely, prucalopride blocks hERG channels with an IC50 value ranging between 4.1 and 22 µm[8, 9], which is equivalent to a safety margin of at least 200 times the therapeutic plasma concentration and, as such, significantly exceeds the recommended threshold of 30-fold IC50[7].
Furthermore, there is no evidence of ECG irregularities or cardiac arrhythmias in healthy volunteer or clinical studies [1, 3–5, 10–15], including studies in elderly patients with previous cardiovascular history [16].
The main objective of this thorough QT study was to evaluate the cardiac safety of prucalopride in accordance with the ICH E14 guideline, by assessing its effect on the QT interval when administered to healthy volunteers at therapeutic (2 mg) and supratherapeutic (10 mg) doses [17, 18]. The supratherapeutic dose used in this study was assessed and found to be safe in a previous phase I cardiac safety study, and is likely to be the maximum dose tolerated in the majority of healthy volunteers (unpublished data). This dose, however, has to be titrated in order to be tolerated, which led to a novel thorough QT study design involving a parallel group with nested crossover for positive control, thereby reducing the number of subjects by half. In accordance with the E14 guideline, a positive control, moxifloxacin (400 mg), was included to establish the sensitivity of the study to detect relevant small changes in the QT interval [17].
Methods
Subjects
The study (NCT00903747) enrolled 120 healthy male and female non-smoking volunteers aged 18–50 years, with a body mass index (BMI) of 18–30 kg m−2 and body weight of ≥50 kg. Eligible volunteers were in good health (determined by physical examination, 12-lead ECG and clinical laboratory evaluations), had a normal ECG and no clinically significant medical history. Concomitant medications were not allowed, except for paracetamol up to 3 × 500 mg day−1 and no more than 3 g week−1. All volunteers gave written informed consent prior to any study related procedures.
The study was approved by an independent Ethics Committee (Welwyn Clinical Pharmacology Ethics Committee, Hatfield, Hertfordshire, UK) and was conducted in accordance with the principles of the Declaration of Helsinki (1996 version), Good Clinical Practice guidelines and other applicable regulatory requirements.
Study design
This was a phase I, double-blind, randomized, placebo- and positive-controlled parallel group/crossover study. A single oral dose of 400 mg moxifloxacin was included as a positive control, by means of a crossover design nested within the placebo arm of the parallel group design.
Volunteers were randomized to one of three treatment groups: group 1 (prucalopride, 2 mg tablets manufactured by Sanico NV, Turnhout, Belgium), group 2a or group 2b (placebo/moxifloxacin [Avelox®, Bayer HealthCare AG, Leverkusen, Germany]). Subjects in group 1 received a once daily dose of 2 mg prucalopride on days 1 to 5, after which dosing escalated by 2 mg day−1 to a maximum dose of 10 mg on day 9, and continued once daily dosing with 10 mg prucalopride on days 10 to 13. Subjects also received a single dose of placebo (in packaging identical to that of moxifloxacin) on days 1 and 15, to ensure blinding of moxifloxacin intake in groups 2a and 2b. Subjects in groups 2a and 2b received a single dose of 400 mg moxifloxacin on day 1 or day 15, respectively, and placebo tablets matching the group 1 prucalopride dosing on days 1 to 13, as well as a single dose of placebo (in packaging identical to that of moxifloxacin) on day 15 or day 1, for groups 2a and 2b, respectively. No dose was administered on day 14. Volunteers remained in the study centre from day −2 until day 16.
Randomization was performed using a randomly permuted 2:1:1 block allocation scheme for groups 1, 2a and 2b, stratified by gender.
Assessments
To ensure blinding, all assessments were performed in all groups at all time-points.
Twelve-lead ECGs were recorded using the Mortara Instrument (Milwaukee, WI, USA) H12+ (1000 Hz) continuous 12-lead ECG digital recorder and saved onto a flash card. ECGs were read centrally by an independent assessor, blinded to the type of treatment and the date and time of recording. Standard 12-lead ECGs were extracted in triplicate for each time point from the 12-lead Holter data recordings on day −1 (baseline, time-matched to ECGs on assessment days), days 5 and 13 (for prucalopride effect) at pre dose and 1, 2, 2.5, 3, 3.5, 4, 5, 6, 8, 12 and 23.5 h post dose, on day 13 at 36 and 48 h post dose and on days 1 and 15 (for assay sensitivity) at pre-dose, and 1, 2, 3, 4, 5 and 6 h post dose.
ECG intervals were measured digitally on a computer screen by trained cardiovascular technicians who were blinded to subject and treatment group using a proprietary validated electronic calliper system (WebHeart™). For each 12-lead ECG, three consecutive magnified PQRST complexes and their preceding RR intervals were manually measured. Measurements were performed from V3 (preferred) or lead II (back up), with the same lead used for each subject wherever possible. Cardiologists, each of whom read all of the ECGs for a given subject, verified the interval durations and performed the morphological analysis and diagnostic interpretation.
To assess plasma concentrations of prucalopride, blood samples (4 ml) were taken on days 5 and 13 at the same time point as the ECGs. A validated liquid chromatography with tandem mass spectrometry method was used with a lowest and highest limit of quantification of 0.2 and 100 ng ml−1, respectively. Accuracy ranged from 98.5 to 101.3% and the coefficient of variation ranged from 0.4 to 5.6%.
Safety assessments included physical examinations, vital signs, resting 12-lead ECGs, 24 h Holter monitoring, safety clinical laboratory tests (biochemistry, haematology and urinalysis) and adverse event (AE) monitoring.
Statistical methods
The primary endpoint was defined as the QTcSS time-matched change from baseline, where QTcSS = QT/RRλ, and the exponent λ was derived as the median value (within genders) of the regression coefficient of loge(QT) on loge(RR) using baseline data. The regression coefficients were estimated separately for each volunteer, and the median values were determined for males and females separately. Other corrections calculated were QTcF and QTcB using the Fridericia [19] and Bazett [20] corrections, respectively (data not reported here).
Analysis of covariance (ancova) was performed on the time-matched change from baseline in QTcSS, separately at each time point (mean of triplicate ECGs), with treatment (active and placebo) and gender as categorical variables and (time-matched) baseline QTcSS as a linear covariate. Groups 2a and 2b were pooled for this analysis. Absence of effects of prucalopride on cardiac repolarization (i.e. non-inferiority vs. placebo) was concluded if the upper limit of the 90% two-sided Cl for the difference of prucalopride from placebo for all time points was below the non-inferiority margin of 10 ms. All statistical analyses were conducted using SAS Version 9 or later.
In addition, a comparison between moxifloxacin (400 mg) and placebo, to demonstrate assay sensitivity, was performed in groups 2a and 2b, regarded as an AB/BA crossover design. The ancova model included treatment, period, sequence and subject. A two-sided 90% CI was derived for the QTcSS difference (moxifloxacin minus placebo) at all time points. Assay sensitivity was considered demonstrated if the lower limit of the CI for the QTcSS difference between moxifloxacin and placebo was ≥5 ms.
The sample size of 120 volunteers was based on a between and within subject standard deviation (SD) for the (baseline adjusted) gender dependent, study specific corrected QT interval (QTcSS) of 11 ms (unpublished data). For prucalopride, it was assumed that the true QTcSS effect relative to placebo was 3 ms. The moxifloxacin effect was expected to be at least 12 ms at its highest point [21].
Results
Subject disposition and demographics
A total of 352 volunteers were screened, and 120 of these were enrolled and randomized to either prucalopride (group 1, n = 60), moxifloxacin/placebo (Group 2a, n = 30) or placebo/moxifloxacin (group 2b, n = 30). There were four withdrawals from the study (all female): one from the prucalopride group (day 10, due to vomiting and nausea), and three from the moxifloxacin/placebo group (one on day 11 due to personal reasons, one on day 7 due to an AE [palpable lymph node] and one on day 10 due to a protocol deviation). All other subjects completed the study. The data from all randomized subjects were included in the analysis.
The majority of volunteers in the three treatment groups were Caucasian (group 1: 76.7%, group 2a: 66.7% and group 2b: 70.0%) and male (56.7% in all groups), with a BMI (mean ± SD) of 23.6 ± 2.6, 24.6 ± 3.0 and 23.9 ± 2.4 kg m−2 for group 1, 2a and 2b, respectively. Mean age (27.7–29.9 years), height (171.8–174.4 cm) and weight (70.5–73.0 kg) were comparable for all treatment groups.
Change in QTcSS from baseline
In order to derive the male and female study specific correction factor (λ), the relationship between QT and RR was assessed for each subject. The median values of the slopes of the regression analysis of log (QT) and log (RR) were 0.296 for men and 0.357 for women, both of which were close in value to the Fridericia correction factor (0.333). The QTcF values were therefore very similar to the QTcSS values and for that reason are not further discussed.
The mean changes in QTcSS for both 2 and 10 mg prucalopride were similar to placebo over the 24 h and 48 h period, respectively. The estimated mean difference in QTcSS time-matched change from baseline between both 2 mg prucalopride and placebo as well as between 10 mg prucalopride and placebo was <5 ms at all time points studied on day 5 and day 13, respectively. These ranged from 0.89 ms (90% CI −3.43, 5.21 ms) at 12 h post dose to 3.83 ms (90% CI −0.28, 7.94 ms) at 3.5 h post dose for 2 mg prucalopride. For 10 mg prucalopride, they ranged from −0.75 ms (90% CI −4.11, 2.60 ms) at 12 h post dose to 3.03 ms (90% CI −0.33, 6.38 ms) at 2.5 h post dose. All lower limits were below zero and each corresponding upper limit of the two-sided 90% CI was below the 10 ms non-inferiority margin (Figure 1).
Figure 1.
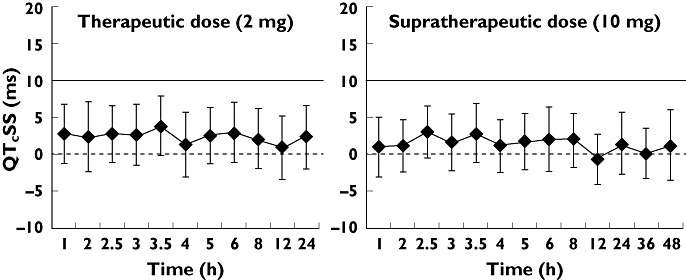
Mean QTcSS differences (ms) in time-matched change from baseline between A) prucalopride (2 mg) and placebo, and B) prucalopride (10 mg) and placebo, by time point. Error bars represent 90% CIs. The solid line indicates the usual 10 ms regulatory threshold
Moxifloxacin vs. placebo (assay sensitivity)
At all individual time points, except at 1 h post dose, the lower limit of the 90% CI of the difference in QTcSS time-matched change from baseline between moxifloxacin and placebo was >5 ms. Mean QTcSS changes from baseline for moxifloxacin increased up to +12.7 ms (5 h post dose), while the mean QTcSS change from baseline for placebo fluctuated post dose between −0.7 ms and +0.8 ms. This confirms that the assay was sufficiently sensitive to detect a QTc prolongation tendency associated with a single dose of moxifloxacin.
The mean QTcSS during treatment with moxifloxacin was overall somewhat higher in group 2a (day 1) compared with group 2b (day 15). A difference between the groups was already observed pretreatment. Differences were considered not clinically relevant.
Heart rate, PR and QRS
Differences in time-matched changes in HR from baseline between 2 mg prucalopride (day 5) and placebo were positive at all time points studied and were of the same order at the supratherapeutic dose of 10 mg (day 13). Changes were mostly statistically significant, of the order of 2–5 beats min−1, but did not exceed 6 beats min−1 following either 2 mg prucalopride (4.71 beats min−1, 90% CI 2.18, 7.24 beats min−1, 12 h post dose), or 10 mg prucalopride (5.80 beats min−1, 90% CI 3.54, 8.06 beats min−1, 3.5 h post dose).
Consistent with the change in HR, non-clinically relevant decreases in mean PR interval were observed: 2 mg range −3.8 ms (5 h) to −10.7 (2 h) ms, and 10 mg range −2.2 ms (36 h) to −10.1 (2 h) ms. Despite an increase in dose, the mean differences remained of the same order.
Mean changes from baseline in QRS for 2 mg prucalopride ranged from −0.3 ms (24 h) to 1.8 ms (0 h), and for 10 mg prucalopride from −0.4 ms (24 h) to 1.4 ms (5 h and 36 h).
Outlier analysis
There were no outlying QTcSS values >450 ms at any time point following either 2 mg or 10 mg prucalopride. The number of occurrences of QTcSS increases of 30–60 ms from baseline was low in the prucalopride treated subjects: seven for 2 mg prucalopride (five for placebo) (day 5), 12 for 10 mg prucalopride (eight for placebo) (day 13), while those for moxifloxacin were 21 (five for placebo). There were only isolated occurrences of an increase >60 ms and the number did not exceed two in any group.
ECG morphology analysis
No ventricular arrhythmias were observed and no ECG abnormalities were considered to be of clinical significance or recorded as an AE. There were no significant differences between prucalopride and placebo in ECG morphology changes or arrhythmia.
Pharmacokinetics of prucalopride
Following a dose of 2 mg daily, prucalopride was rapidly absorbed, reaching a mean Cmax of 7.37 ng ml−1 at a median time of 2 h post dose on day 5 (at steady state). At the 10 mg dose, Cmax reached a mean of 42.7 ng ml−1 2 h post dose on day 13, a 5.8-fold increase in exposure for a 5-fold increase in dose. AUC over the dosing interval increased from 108 ng ml−1 h following daily dosing with 2 mg to 590 ng ml−1 h following 10 mg daily dosing, a 5.5-fold increase.
Safety
Overall, 105 (88%) volunteers in groups 1, 2a and 2b reported a total of 450 treatment-emergent AEs. These were more common on the first day of treatment with prucalopride (2 mg). The incidence of diarrhoea, headache and nausea was 57%, 80% and 22%, respectively, on prucalopride vs. 2%, 7% and 2%, respectively, with placebo. From day 2 to 5, there were considerably fewer reports of diarrhoea (15%), headache (10%) and nausea (7%) with prucalopride treatment. The corresponding incidences for placebo were 10%, 7% and 0%, respectively. Most AEs were mild or moderate and related to study medication. There were no severe AEs. Only one serious AE (spontaneous abortion during placebo treatment) and two AEs leading to withdrawal (see earlier section in Results). There was no observable dose-related trend in treatment-emergent AEs in the prucalopride group.
There were no observable dose-related trends, clinically relevant changes or notable differences in treatment groups in any vital signs parameters, ECG safety parameters or laboratory safety parameters.
Discussion
This thorough QT study, which was performed in compliance with the ICH E14 Guidance on Clinical Evaluation of QT/QTc Interval Prolongation, and which met the requirements for assay sensitivity, confirmed that prucalopride at both therapeutic (2 mg) [4–6] and supratherapeutic doses (10 mg) had no detectable effect on cardiac repolarization. A randomized, two-arm parallel-group design was chosen because of the need to titrate the doses up to a supratherapeutic level for tolerability reasons.
The design chosen for this study is novel, reducing the number of subjects required to exclude a repolarization effect, by combining the two active groups (therapeutic and supratherapeutic) and by nesting the positive control treatment within the placebo arm of the study, resulting in effectively two study groups, active and control. This is possible because the positive control (moxifloxacin) has a relatively short half-life (compared with prucalopride), and provided that there is a sufficient period of washout between positive control and placebo observations (1–2 days), the comparisons with prucalopride are not compromised.
By subdividing the placebo group, the design is arranged such that one subgroup receives the positive control at the beginning of the study (group 2a) and the other receives it at the end (group 2b, 2 days after the final supratherapeutic day for prucalopride). Thus, an AB/BA crossover design (positive control vs. placebo) can be used, which gives the advantage that any time or environmental related changes in the QTc interval which take place during the study can be isolated (and eliminated) in the positive control analysis.
Although the design proved efficient in this study, extra care was required with the study blinding. In particular, to maintain blinding, dummy ECGs were collected for the prucalopride group on days where ECG measurements were required to assess the positive control in the placebo groups. Although, in general, assay sensitivity can be achieved with open label moxifloxacin [18], in a study with a nested crossover design blinding of the active control is required, as open-label moxifloxacin would effectively unblind the placebo group.
The use of universal QT correction methods, such as Bazett's (QTcB) and Fridericia's (QTcF), has been questioned as these may introduce errors in estimating drug-induced QT prolongation, secondary to changes in heart rate [22]. The use of customized QT correction methods, such as the individual QT correction (QTcI), based on individual regression models computed from a set of QT/RR measurements collected during drug free periods, has been promoted as the best alternative to the universal correction methods [23]. However, based on practical and theoretical considerations, a large number of drug free ECGs are required to ensure reliable calculations of QTcl [24].
Alternatively, population specific or study specific QT correction methods (QTcSS) have been suggested, which utilize the mean or median of all individual QT/RR slopes to generate a common slope for all study participants [25]. However such an approach introduces a possible bias due to gender differences if the allocation of males and females to the groups is imbalanced. In this study, this was avoided by the use of separate medians for males and females. Furthermore, when QTcl is derived from a limited number of ECG data points it shows marginally increased variance (and hence lower power) compared with QTcSS. Given the limited number of drug free ECGs available for the derivation of a customized QT correction, we preferred to avoid the QTcI method in this study for the reasons discussed above. The study specific correction yielded gender specific exponents of 0.296 and 0.357, respectively. Consequently, for this study, the QTcSS results were similar to the QTcF results.
For both therapeutic and supratherapeutic doses, the upper limit of the two-sided 90% CI for the difference in time-matched QTcSS change from baseline between prucalopride and placebo was below the non-inferiority margin of 10 ms at all time points. In addition, no subject (male or female) had a QTcSS increase greater than 60 ms, resulting in QTcSS greater than 500 ms (the threshold of clinical and regulatory concern) with either dose of prucalopride. There were also no significant differences between prucalopride and placebo in ECG morphology changes or arrhythmias, and no clinically relevant abnormalities were observed.
Importantly, this study confirmed the expected QTc effect associated with the positive control moxifloxacin. During the first 6 h after administration, the moxifloxacin baseline and placebo adjusted QTcSS mean effect and lower 90% CI limit were above the 5 ms regulatory threshold at all time points, except at 1 h post dose for the lower 90% CI. For this analysis, the QTcSS values of the parallel groups 2a and 2b were combined. It is therefore of interest to compare the QTcSS values of groups 2a (day 1) and 2b (day 15). The values were similar although the mean values for group 2a were somewhat lower than for group 2b. However the differences remained well within the standard deviation. Also on non-treatment days the QTcSS values were around 5 ms lower for group 2a, except at 1 h where they were 1–2 ms higher. These findings reinforce confidence in the validity of the results obtained with prucalopride.
Both doses of prucalopride were associated with a small and, in most cases, statistically significant increase in HR, which is not considered to be clinically relevant. A small, transient increase in HR with prucalopride treatment has also been observed in animals. An extended series of safety pharmacology studies with special emphasis on cardiovascular parameters showed no relevant changes in haemodynamic or ECG-derived parameters (QTc), with the exception of a modest increase in heart rate and blood pressure in conscious dogs after bolus intravenous administration, which was not observed either in anaesthetized dogs or after oral administration in dogs reaching similar plasma concentrations [26].
Consistent with the change in HR, non-clinically relevant decreases in mean PR interval were observed, which were similar for 2 and 10 mg prucalopride. QRS interval showed the same pattern.
The pharmacokinetics of prucalopride are linear and the supratherapeutic dose of 10 mg represents a 5-fold increase in exposure of that of the therapeutic dose of 2 mg. Metabolism is not a major route of elimination of prucalopride and a large fraction of the drug is excreted unchanged. Prucalopride has a low potential for drug–drug interactions. It is not expected to alter the pharmacokinetics of drugs metabolized by the cytochrome P450 system, nor are these drugs expected to affect the disposition of prucalopride. Although prucalopride may be a weak substrate for P-glycoprotein (P-gp), it is not an inhibitor of P-gp at clinically relevant concentrations. No effect on the plasma concentrations of prucalopride was observed with erythromycin, paroxetine, cimetidine and probenecid [26]. Ketoconazole, at a high dose of 200 mg twice daily, increased the area under the curve of prucalopride by approximately 40% [26] which is likely attributable to inhibition of P-gp mediated renal transport. Prucalopride may also be secreted via other renal transporters and inhibition of all transporters involved in the active secretion of prucalopride may theoretically increase its exposure by up to 75%. Assuming this worst case scenario, the supra-therapeutic dose of 10 mg still yields an almost 3-fold increase of this exposure. Therefore the choice of 10 mg once daily as the supratherapeutic dose for this study appears appropriate.
Treatment with prucalopride at doses up to 10 mg once daily was safe and well tolerated from day 2 onwards. The high incidence of gastrointestinal-related treatment-emergent AEs in the prucalopride group is due to the pharmacodynamic action of the drug and is more often seen on days 1–2 of treatment, consistent with findings previously reported [4–6].
In conclusion, this thorough QT study, conducted in accordance with the ICH E14 Guidance on Clinical Evaluation of QT/QTc Interval Prolongation, found no evidence of QT prolongation or pro-arrhythmic potential for prucalopride in healthy volunteers, at doses up to five times the recommended therapeutic dose. At both therapeutic (2 mg) and supratherapeutic (10 mg) doses, the upper CI for the time-matched changes from baseline QTcSS values were below the 10 ms regulatory threshold at all time points, confirming the absence of any clinically relevant effect of prucalopride on cardiac repolarization. The results with the positive control, moxifloxacin, demonstrated that this study was sufficiently sensitive to detect a QT prolongation of the magnitude of regulatory and clinical concern. At doses of 2 mg and 10 mg prucalopride, a small and clinically irrelevant increase in HR was observed, but there were no significant differences between prucalopride and placebo in ECG morphological changes or arrhythmias.
Acknowledgments
Medical writing support in the preparation of the manuscript was provided by Delia Randall and Victoria Harvey and was funded by Shire-Movetis NV. The authors would also like to thank Joanne Zhang, PhD, of the Office of Biostatistics, CDER, FDA, for her original contribution to the nested crossover design for assay sensitivity assessment in parallel group thorough QT studies, on which this study design is based.
Competing Interests
Boaz Mendzelevski, MD (employed by CoreLab Partners) and Patricia Robinson are consulting for Shire-Movetis NV. There is no other financial information to disclose.
Dennis Chanter, DPhil is contracted by CoreLab Partners. There is no other financial information to disclose.
John Camm, MD is an advisor to Shire-Movetis NV regarding drug-induced QT effects.
Jannie Ausma, Rene Kerstens and Lieve Vandeplassche are employees of Shire-Movetis NV. There is no other financial information to disclose.
REFERENCES
- 1.Bouras EP, Camilleri M, Burton DD, McKinzie S. Selective stimulation of colonic transit by the benzofuran 5HT4 agonist, prucalopride, in healthy humans. Gut. 1999;44:682–86. doi: 10.1136/gut.44.5.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Maeyer JH, Lefebvre RA, Schuurkes JA. 5-HT4 receptor agonists: similar but not the same. Neurogastroenterol Motil. 2008;20:99–112. doi: 10.1111/j.1365-2982.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 3.De Schryver AM, Andriesse GI, Samsom M, Smout AJ, Gooszen HG, Akkermans LM. The effects of the specific 5HT(4) receptor agonist, prucalopride, on colonic motility in healthy volunteers. Aliment Pharmacol Ther. 2002;16:603–12. doi: 10.1046/j.1365-2036.2002.01195.x. [DOI] [PubMed] [Google Scholar]
- 4.Camilleri M, Kerstens R, Rykx A, Vandeplassche L. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med. 2008;358:2344–54. doi: 10.1056/NEJMoa0800670. [DOI] [PubMed] [Google Scholar]
- 5.Quigley EM, Vandeplassche L, Kerstens R, Ausma J. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation – a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther. 2009;29:315–28. doi: 10.1111/j.1365-2036.2008.03884.x. [DOI] [PubMed] [Google Scholar]
- 6.Tack J, van Outryve M, Beyens G, Kerstens R, Vandeplassche L. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut. 2009;58:357–65. doi: 10.1136/gut.2008.162404. [DOI] [PubMed] [Google Scholar]
- 7.Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, Siegl PK, Strang I, Sullivan AT, Wallis R, Camm AJ, Hammond TG. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- 8.Chapman H, Pasternack M. The action of the novel gastrointestinal prokinetic prucalopride on the HERG K+ channel and the common T897 polymorph. Eur J Pharmacol. 2007;554:98–105. doi: 10.1016/j.ejphar.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 9.European Medicines Agency. CHMP assessment report for Resolor (procedure number EMEA/H/C/1012). Document reference EMEA/664892/2009.2009. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001012/WC500053997.pdf (last accessed 8 September 2011)
- 10.Emmanuel AV, Kamm MA, Roy AJ, Antonelli K. Effect of a novel prokinetic drug, R093877, on gastrointestinal transit in healthy volunteers. Gut. 1998;42:511–16. doi: 10.1136/gut.42.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouras EP, Camilleri M, Burton DD, Thomforde G, McKinzie S, Zinsmeister AR. Prucalopride accelerates gastrointestinal and colonic transit in patients with constipation without a rectal evacuation disorder. Gastroenterology. 2001;120:354–60. doi: 10.1053/gast.2001.21166. [DOI] [PubMed] [Google Scholar]
- 12.Krogh K, Jensen MB, Gandrup P, Laurberg S, Nilsson J, Kerstens R, De Pauw M. Efficacy and tolerability of prucalopride in patients with constipation due to spinal cord injury. Scand J Gastroenterol. 2002;37:431–6. doi: 10.1080/003655202317316060. [DOI] [PubMed] [Google Scholar]
- 13.Sloots CE, Poen AC, Kerstens R, Stevens M, De Pauw M, Van Oene JC, Meuwissen SG, Felt-Bersma RJ. Effects of prucalopride on colonic transit, anorectal function and bowel habits in patients with chronic constipation. Aliment Pharmacol Ther. 2002;16:759–67. doi: 10.1046/j.1365-2036.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- 14.Coremans G, Kerstens R, De Pauw M, Stevens M. Prucalopride is effective in patients with severe chronic constipation in whom laxatives fail to provide adequate relief. Results of a double-blind, placebo-controlled clinical trial. Digestion. 2003;67:82–9. doi: 10.1159/000070202. [DOI] [PubMed] [Google Scholar]
- 15.Emmanuel AV, Roy AJ, Nicholls TJ, Kamm MA. Prucalopride, a systemic enterokinetic, for the treatment of constipation. Aliment Pharmacol Ther. 2002;16:1347–56. doi: 10.1046/j.1365-2036.2002.01272.x. [DOI] [PubMed] [Google Scholar]
- 16.Camilleri M, Beyens G, Kerstens R, Robinson P, Vandeplassche L. Safety assessment of prucalopride in elderly patients with constipation: a double-blind, placebo-controlled study. Neurogastroenterol Motil. 2009;21:1256–e117. doi: 10.1111/j.1365-2982.2009.01398.x. [DOI] [PubMed] [Google Scholar]
- 17.European Medicines Agency. ICH Harmonised Tripartite Guideline (E14) (CHMP/ICH/2/04). The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non Antiarrhythmic Drugs. 2005. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000429.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580029590&jsenabled=true (last accessed 8 September 2011)
- 18.European Medicines Agency. ICH Harmonised Tripartite Guideline (E14) (EMEA/CHMP/ICH/310133/2008). The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs: questions and answers. 2008. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000429.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580029590&jsenabled=true (last accessed 8 September 2011)
- 19.Fridericia LS. Die Systolendauer im Elektrokardiogramm bei normalen Menschen und bei Herzkranken [Systolic period in the electrocardiogram in normal humans and in cardiac patients] Acta Med Scand. 1920;53:489–506. [Google Scholar]
- 20.Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;70:353–70. [Google Scholar]
- 21.Bloomfield DM, Kost JT, Ghosh K, Hreniuk D, Hickey LA, Guitierrez MJ, Gottesdiener K, Wagner JA. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84:475–80. doi: 10.1038/clpt.2008.33. [DOI] [PubMed] [Google Scholar]
- 22.Indik JH, Pearson EC, Fried K, Woosley RL. Bazett and Fridericia QT correction formulas interfere with measurement of drug-induced changes in QT interval. Heart Rhythm. 2006;3:1003–7. doi: 10.1016/j.hrthm.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 23.Malik M. Problems of heart rate correction in assessment of drug-induced QT interval prolongation. J Cardiovasc Electrophysiol. 2001;12:411–20. doi: 10.1046/j.1540-8167.2001.00411.x. [DOI] [PubMed] [Google Scholar]
- 24.Couderc JP, Xiaojuan X, Zareba W, Moss AJ. Assessment of the stability of the individual-based correction of QT interval for heart rate. Ann Noninvasive Electrocardiol. 2005;10:25–34. doi: 10.1111/j.1542-474X.2005.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van de Water A, Verheyen J, Xhonneux R, Reneman RS. An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J Pharmacol Methods. 1989;22:207–17. doi: 10.1016/0160-5402(89)90015-6. [DOI] [PubMed] [Google Scholar]
- 26.Movetis. 2009. pp. 1–9. Summary of product characteristics, Resolor (prucalopride). October.