

Abstract
AIM
According to product information, montelukast is extensively metabolized by CYP3A4 and CYP2C9. However, CYP2C8 was also recently found to be involved. Our aim was to study the effects of selective CYP2C8 and CYP3A4 inhibitors on the pharmacokinetics of montelukast.
METHODS
In a randomized crossover study, 11 healthy subjects ingested gemfibrozil 600 mg, itraconazole 100 mg (first dose 200 mg) or both, or placebo twice daily for 5 days, and on day 3, 10 mg montelukast. Plasma concentrations of montelukast, gemfibrozil, itraconazole and their metabolites were measured up to 72 h.
RESULTS
The CYP2C8 inhibitor gemfibrozil increased the AUC(0,∞) of montelukast 4.3-fold and its t1/2 2.1-fold (P < 0.001). Gemfibrozil impaired the formation of the montelukast primary metabolite M6, reduced the AUC and Cmax of the secondary (major) metabolite M4 by more than 90% (P < 0.05) and increased those of M5a and M5b (P < 0.05). The CYP3A4 inhibitor itraconazole had no significant effect on the pharmacokinetic variables of montelukast or its M6 and M4 metabolites, but markedly reduced the AUC and Cmax of M5a and M5b (P < 0.05). The effects of the gemfibrozil-itraconazole combination on the pharmacokinetics of montelukast did not differ from those of gemfibrozil alone.
CONCLUSIONS
CYP2C8 is the dominant enzyme in the biotransformation of montelukast in humans, accounting for about 80% of its metabolism. CYP3A4 only mediates the formation of the minor metabolite M5a/b, and is not important in the elimination of montelukast. Montelukast may serve as a safe and useful CYP2C8 probe drug.
Keywords: CYP2C8, CYP3A4, gemfibrozil, interaction, itraconazole, montelukast pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The elimination of montelukast occurs mainly via metabolism and, according to its product information, CYP3A4 and 2C9 are the main metabolizing enzymes in vitro.
Recent studies, however, suggest that CYP2C8 may have a role in the metabolism of montelukast.
WHAT THIS STUDY ADDS
The CYP3A4 inhibitor itraconazole markedly reduces the formation of a minor metabolite of montelukast, but has no significant effect on the total elimination of montelukast.
Montelukast is predominantly metabolized by CYP2C8, and can thus be used as a sensitive CYP2C8 probe drug.
Introduction
Montelukast is a leukotriene receptor antagonist frequently used in the treatment of asthma [1–3], being among the top 10 drugs in the United States by the amount of prescriptions dispensed in 2008. The oral bioavailability of montelukast is 60–70%, it is over 99% bound to plasma proteins and its elimination half-life is 4–5 h. Montelukast is extensively metabolized to one major and several minor metabolites that are mainly excreted into the bile [4, 5]. The major biliary metabolite is a dicarboxylic acid (M4), resulting from further oxidation of a primary metabolite, M6 (Figure 1).
Figure 1.
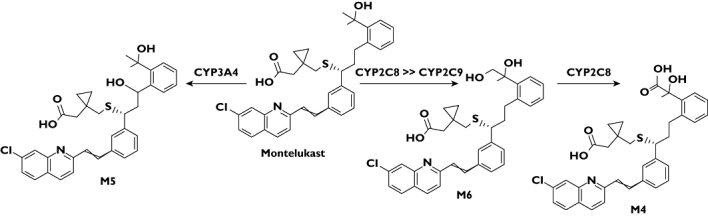
The chemical structures of montelukast and its metabolites M6, M4 and M5, and the CYP enzymes participating in their formation in humans. The diastereomers of metabolite M5 are indicated with a or b in this article, based on their chromatographic retention times
According to the product information of montelukast, the in vitro formation of M6 is catalyzed by cytochrome P450 (CYP) 2C9 and the formation of M5 by CYP3A [6]. In our recent in vivo study, the CYP2C8 inhibitor gemfibrozil greatly increased the area under the plasma concentration–time curve (AUC) of montelukast, which together with our in vitro findings suggests that CYP2C8 is of major importance in the elimination of montelukast [7, 8].
The major metabolite of gemfibrozil, gemfibrozil 1-O-β glucuronide, is a potent and selective mechanism-based inhibitor of CYP2C8 with a rapid but long-lasting CYP2C8 inhibitory effect [9–13]. In vivo, gemfibrozil has increased the AUC values of many CYP2C8 substrates, e.g. cerivastatin and repaglinide [14, 15]. In vitro gemfibrozil is also a CYP2C9 inhibitor [16], but it has not inhibited the CYP2C9-mediated metabolism of warfarin in vivo[17] and neither has it had any effect on the pharmacokinetics of zafirlukast in vivo[18]. The antimycotic itraconazole is a strong and selective CYP3A4 inhibitor, and it has greatly increased the plasma concentrations of numerous CYP3A4 probe substrates, such as midazolam, simvastatin, buspirone and felodipine [19–22]. Concomitant administration of itraconazole and gemfibrozil can lead to drastic increases in the plasma concentrations of drugs, which are metabolized by both CYP3A4 and CYP2C8 [15, 23]. In order to study the contributions of CYP3A4 and CYP2C8 to the metabolism of montelukast, we investigated the effects of itraconazole, gemfibrozil and their combination on the pharmacokinetics of montelukast in healthy subjects.
Methods
Subjects
Eleven healthy volunteers (eight men, three women, aged 21–29 years, weight 65–113 kg) participated in the study after each gave written informed consent and were ascertained to be healthy by medical history, clinical examination, and routine laboratory tests. None received continuous medication or herbal medicines, used hormonal contraception or was a tobacco smoker. The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District and by the Finnish Medicines Agency.
Study design
A randomized, placebo-controlled, four phase crossover study with a washout period of 4 weeks between the phases was carried out. Each subject took either 600 mg gemfibrozil (Lopid 600 mg tablets, Gödecke, Karlsruhe, Germany), 100 mg itraconazole (first dose 200 mg, Sporanox 100 mg capsules, Janssen-Cilag, Borgo San Michele, Latina, Italy), or both, or unmatched placebo (University Pharmacy placebo tablets, University Pharmacy, Helsinki, Finland) twice daily at 09.00 h and 21.00 h for 5 days. On day 3, the morning dose of these medications was taken at 08.00 h and, 1 h later, a single dose of 10 mg montelukast (Singulair 10 mg, Merck Sharp Dohme, B.V. Haarlem, Netherlands) was administered orally with 150 ml water. The subjects had fasted for 9 h before montelukast intake and received a standardized meal 3 h and standardized light meals 7 h and 11 h after montelukast intake.
Blood sampling
On days of montelukast administration, timed blood samples (4 or 9 ml) were drawn from a cannulated forearm vein before and 0.5, 1, 2, 3, 4, 5, 7, 9, 12, 24, 48 and 72 h after administration of montelukast. Blood samples were collected into tubes containing ethylenediaminetetraacetic acid (EDTA). Plasma was separated immediately and stored at −70°C until analysis.
Determination of drug concentrations
For the determination of plasma montelukast and its metabolites, plasma samples were spiked with internal standard (IS, montelukast-d6), and the concentrations (montelukast, metabolite M6) or the metabolite to IS peak height ratios (metabolites M4, M5a and M5b) were measured by use of a SCIEX API 2000 liquid chromatography-tandem mass spectrometry system (Sciex Division of MDS Inc, Toronto, ON, Canada) after protein precipitation (montelukast) or solid phase extraction (metabolites), as described earlier. M4, M5a and M5b were identified by their ion transitions and the different retention times of M5 diastrereomers (a and b), as described earlier [7]. Montelukast sodium, montelukast-d6 sodium and montelukast metabolite M6 were purchased from Toronto Research Chemicals (North York, Ontario, Canada).
The lower limit of quantification (LLQ) was 0.3 ng ml−1 for plasma montelukast and 1 ng ml−1 for M6. The signal to noise ratio <10 was used as the LLQ for the other metabolites, for which no authentic compounds were available. The detector response (metabolite to IS peak height ratio) of M4, M5a and M5b was shown to be linear over the relevant concentration range by using a plasma dilution series. The between-day coefficient of variation (CV) for montelukast was 6.3% at 1.5 ng ml−1, 4.6% at 15 ng ml−1, 7.6% at 150 ng ml−1 and 4.7% at 1500 ng ml−1, and for M6 14.4% at 1.5 ng ml−1, 11.9% at 15 ng ml−1 and 8.5% at 150 ng ml−1. Gemfibrozil, its glucuronide, itraconazole or hydroxy-itraconazole (OH-itraconazole) did not interfere with the assays.
The plasma concentrations of gemfibrozil and gemfibrozil 1-O-β glucuronide in samples taken before and at 1, 3, 5, 12, 24, 48 and 72 h after montelukast administration were determined by the use of a SCIEX API 2000 QTRAP liquid chromatography-tandem mass spectrometry system (Sciex Division of MDS Inc, Toronto, ON, Canada) as described previously [7, 24]. The LLQs for gemfibrozil and gemfibrozil 1-O-β glucuronide were 0.1 mg l−1 and 0.05 mg l−1, respectively and the between day CVs were 2.1–6.2% and 3.0–9.7% at relevant concentrations, respectively. Gemfibrozil 1-O-β glucuronide was purchased from Toronto Research Chemicals (North York, Ontario, Canada).
The plasma concentrations of itraconazole and OH-itraconazole in samples taken before and at 1, 3, 5, 12, 24, 48 and 72 h after montelukast administration were determined by high performance liquid chromatography as described before [21, 25]. The LLQ for itraconazole and OH-itraconazole was 20 ng ml−1 and the between-day CVs were 0.8–3.3% and 1.3–2.1% at relevant concentrations, respectively.
Pharmacokinetic assessments
The pharmacokinetics of montelukast and its metabolites M4, M5a, M5b and M6 were characterized by observed peak plasma concentration (Cmax), time to reach Cmax (tmax), elimination half-life (t1/2), and AUC from 0 to 7 or 72 h or infinity (AUC 0,7 h; 0,72 h; 0,∞). The Cmax and tmax values were taken directly from the original data. The terminal log-linear part of each plasma concentration–time curve was identified visually, and the elimination rate constant (λz) was determined by linear regression analysis of the log-linear part of the concentration–time curve. The t1/2 was calculated by the equation t1/2 = ln2/λz. The AUC values were calculated by the linear trapezoidal rule for the rising phase and the log-linear trapezoidal rule for the descending phase, with extrapolation to infinity by division of the last measured concentration by λz. The pharmacokinetic calculations were performed with MK-Model, version 5.0 (Biosoft, Cambridge, UK).
Statistical analysis
For montelukast and metabolite M6, the pharmacokinetic variables are expressed as geometric mean and CV in the text and Table 1, except for tmax. Because some of the metabolite concentrations were below their LLQ in some of the study phases, the results for metabolites M4, M5a and M5b are expressed as median and range in the text and Table 1. For clarity, all concentrations in Figures are given as mean values (standard error (SEM)). For montelukast and metabolite M6 all pharmacokinetic variables, except tmax, were log transformed and the statistical comparisons between the four phases were performed with repeated measures anova, followed by a posteriori testing with the paired t-test with the Bonferroni correction. All tmax values and all pharmacokinetic variables of M4, M5a and M5b were compared with the Friedman two-way anova followed by the Wilcoxon signed rank test with the Bonferroni correction. The level of statistical significance was P < 0.05. The analysis was performed with SPSS for Windows version 17.0 (SPSS Inc, Chicago, IL, USA).
Table 1.
Pharmacokinetic variables of montelukast and its metabolites in 11 healthy volunteers after a single oral dose of 10 mg montelukast on day 3 of a 5 day treatment with 600 mg gemfibrozil, 100 mg itraconazole (first dose 200 mg), or both, or placebo, twice daily
| Variable | Placebo phase (control) | Gemfibrozil phase | Itraconazole phase | Gemfibrozil and itraconazole phase |
|---|---|---|---|---|
| Montelukast | ||||
| Cmax (ng ml−1) | 457 (0.49) | 665 (0.23) | 511 (0.46) | 550 (0.69) |
| Ratio to control (95% CI) | 1.5 (0.93, 2.3) | 1.1 (0.66, 1.9) | 1.2 (0.79, 1.8) | |
| tmax (h) | 2 (1, 4) | 5 (2,5) | 4 (1, 5) | 5 (3, 9)*,‡ |
| t1/2 (h) | 6.4 (0.28) | 13 (0.23)*** | 7.0 (0.24)‡‡‡ | 16 (0.31)***,‡‡‡ |
| Ratio to control (95% CI) | 2.1 (1.7,2.5) | 1.1 (0.90, 1.3) | 2.4 (2.0, 3.0) | |
| AUC(0,∞) (ng h ml−1) | 2780 (0.30) | 11 900 (0.30)*** | 3250 (0.31)‡‡‡ | 11 200 (0.56)***,‡‡‡ |
| Ratio to control (95% CI) | 4.3 (2.9,6.3) | 1.2 (0.78, 1.8) | 4.0 (2.7, 6.0) | |
| M6 | ||||
| Cmax (ng ml−1) | 24 (0.63) | 19 (0.37) | 27 (0.75) | 21 (0.91) |
| Ratio to control (95% CI) | 0.80 (0.50,1.3) | 1.2 (0.63, 2.1) | 0.88 (0.44, 1.7) | |
| tmax (h) | 4 (2, 5) | 12 (9,12)* | 4 (2, 5)† | 12 (7, 24)*,‡ |
| t1/2 (h) | 3.9 (0.28) | 16 (0.25)*** | 4.1 (0.20)‡‡‡ | 21 (0.43)***,‡‡‡ |
| Ratio to control (95% CI) | 4.1 (3.4,4.8) | 1.0 (0.91, 1.2) | 5.3 (3.8, 7.5) | |
| AUC(0,7 h) (ng ml−1 h) | 101 (0.56) | 57 (0.46) | 113 (0.66)† | 37 (1.2)*,‡ |
| Ratio to control (95% CI) | 0.56 (0.29,1.1) | 1.1 (0.67, 1.9) | 0.36 (0.15, 0.90) | |
| AUC(0,∞) (ng ml−1 h) | 160 (0.42) | 555 (0.42)*** | 200 (0.54)‡‡‡ | 749 (0.72)***,‡‡‡ |
| Ratio to control (95% CI) | 3.5 (2.4,5.0) | 1.3 (0.85, 1.8) | 4.7 (2.9, 7.6) | |
| M6 : montelukast AUC(0,72 h) ratio | 0.06 (0.35) | 0.05 (0.21) | 0.06 (0.36)† | 0.06 (0.29)‡‡‡ |
| Ratio to control (95% CI) | 0.78 (0.54,1.1) | 1.1 (0.78, 1.5) | 1.0 (0.73, 1.5) | |
| M4 | ||||
| Cmax (U ml−1) | 49 (0, 360) | 3.5 (0,32)* | 75 (0, 587)† | 0 (0, 31)*,‡ |
| tmax (h) | 5 (4, 9) | 12 (12,24) | 5 (3, 9) | 24 (12, 48) |
| AUC(0,72 h) (U ml−1 h) | 656 (0, 2930) | 26 (0,1130)* | 728 (0, 3340)† | 0 (0, 764),‡ |
| M4 : M6 AUC(0,72 h) ratio (U ng−1) | 4.5 (0, 13) | 0.05 (0,1.9)* | 3.7 (0, 10)† | 0 (0, 1.1)*,‡ |
| M5a | ||||
| Cmax (U ml−1) | 208 (62, 339) | 464 (236,1160)* | 15 (0, 43)*,† | 57 (8.9, 326)*,† |
| tmax (h) | 3 (2, 4) | 4 (3,9)* | 4 (2, 5) | 9 (4, 24)*,‡ |
| t1/2 (h) | 3.0 (2.1, 4.8) | 16 (12,23)** | – | – |
| AUC(0,72 h) (U ml−1 h) | 1210 (335, 2150) | 9640 (4600,16 500)* | 94 (0, 167)*,† | 1440 (88, 6640),†,‡ |
| M5a : M AUC(0, 72 h) ratio (U ng−1) | 0.37 (0.22, 0.68) | 0.76 (0.54,1.1)* | 0.03 (0, 0.04)*,† | 0.12 (0.02, 0.44)*,†,‡ |
| M5b | ||||
| Cmax (U ml−1) | 195 (59, 393) | 322 (142,1030)* | 16 (0, 78)*,† | 27 (0, 267)*,† |
| tmax (h) | 3 (2, 5) | 3 (2,5) | 4 (2, 12) | 5 (3, 12)* |
| t1/2 (h) | 2.4 (1.5, 3.0) | 16 (7.3,25)** | – | – |
| AUC(0,72 h) (U ml−1 h) | 938 (259, 1860) | 5100 (1550,8540)* | 80 (0, 693)*,† | 729 (0, 3880),†,‡ |
| M5b : M AUC(0,72 h) ratio (U ng−1) | 0.33 (0.16, 0.64) | 0.38 (0.26,0.70) | 0.02 (0, 0.16)*,† | 0.06 (0, 0.26)*,† |
For montelukast and M6, data are given as geometric means (geometric CV), except for tmax, which is given as median (range). For M4, M5a and M5b, data for all variables are given as median (range). The t1/2 values of M5a and M5b during the itraconazole and gemfibrozil-itraconazole phases are not given, because they could not be reliably determined for a majority of the subjects. Cmax, observed peak plasma concentration; CI, confidence interval; tmax, time to reach Cmax; t1/2, elimination half-life; AUC(0,7 h), area under the concentration vs. time curve from time 0 to 7 h; AUC(0,72 h), area under the concentration vs. time curve from time 0 to 72 h; AUC(0,∞), area under the concentration vs. time curve from time 0 to infinity; M, montelukast; U, arbitrary units (the ratio of the peak height of the metabolite to the peak height of the internal standard).
P < 0.05 vs. control
P < 0.005 vs. control
P < 0.001 vs. control
P < 0.05 vs. gemfibrozil
††P < 0.005 vs. gemfibrozil
P < 0.001 vs. gemfibrozil
P < 0.05 vs. itraconazole
‡‡P < 0.005 vs. itraconazole
P < 0.001 vs. itraconazole.
For M4, M5a and M5b 0 stands for median values that were under the lower limit of quantification.
Results
Effect of gemfibrozil
During the gemfibrozil phase, the geometric mean ratio to control of the AUC(0,∞) of montelukast was 4.3-fold (95% confidence interval (CI) 2.9, 6.3, P < 0.001), and that of its t1/2 was 2.1-fold (95% CI 1.7, 2.5, P < 0.001), i.e. the t1/2 was prolonged from 6.4 h to 13 h (Table 1, Figure 2). Gemfibrozil reduced the formation clearance of the primary metabolite M6 (Figure 2). However, the AUC(0,∞) of M6 was increased 3.5-fold (95% CI 2.4, 5.0, P < 0.001) by gemfibrozil, because the further metabolism of M6 to M4 was strongly inhibited by gemfibrozil. Gemfibrozil prolonged the t1/2 of M6 4.1-fold (95% CI 3.4, 4.8, P < 0.001), i.e. from 3.9 h to 16 h, and greatly reduced the plasma concentrations of the secondary metabolite M4; for example, the median Cmax of M4 was reduced from 49 U ml−1 (control phase) to 3.5 U ml−1 (gemfibrozil phase, P < 0.05). Also the AUC(0,72 h) of M4 and the M4 : M6 AUC(0,72 h) ratio were greatly reduced in the gemfibrozil phase compared with the placebo phase (P < 0.05, Table 1, Figure 3). On the other hand, the Cmax and AUC(0,72 h) of both M5a and M5b metabolites were increased in the gemfibrozil phase (P < 0.05, Table 1, Figure 3).
Figure 2.
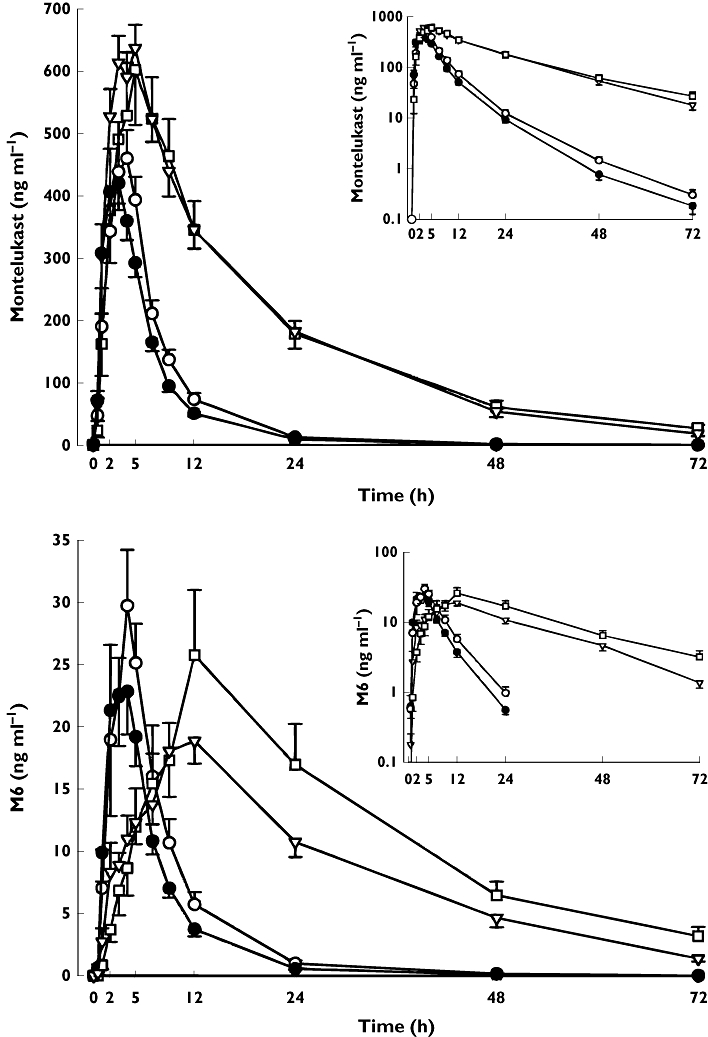
Mean ± SEM plasma concentrations of montelukast and metabolite M6 in 11 healthy volunteers after a single oral dose of 10 mg montelukast on day 3 of a 5 day treatment with 600 mg gemfibrozil (▿), 100 mg itraconazole (first dose 200 mg) (○), or both (□), or placebo (•), twice daily. Inset depicts the same data on a semi-logarithmic scale
Figure 3.
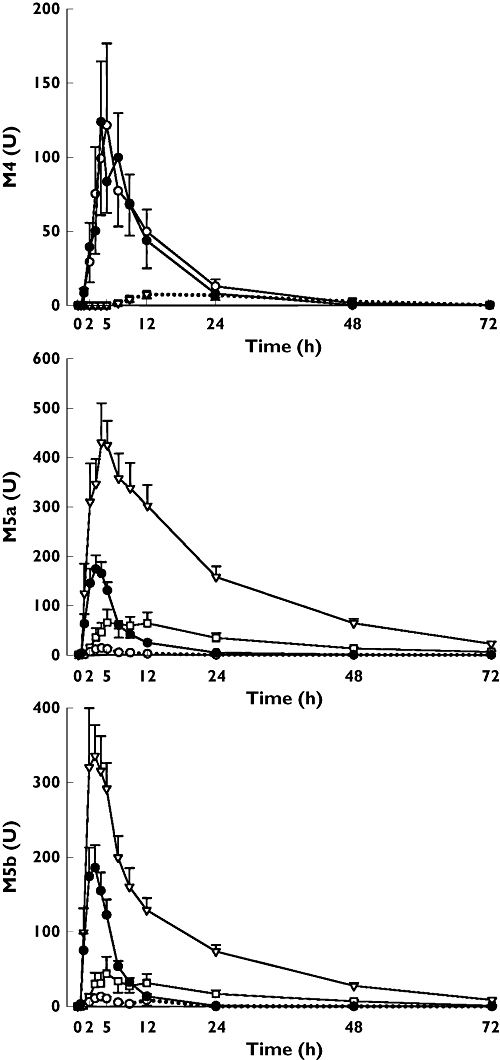
Mean ± SEM plasma concentrations of metabolites M4, M5a and M5b in 11 healthy volunteers after a single oral dose of 10 mg montelukast on day 3 of a 5 day treatment with 600 mg gemfibrozil (▿), 100 mg itraconazole (first dose 200 mg) (○), or both (□), or placebo (•), twice daily. The dotted lines depict plasma concentrations below the lower limit of quantification
Effect of itraconazole
Itraconazole alone, compared with placebo, had no significant effect on the pharmacokinetic variables of parent montelukast or the metabolites M6 or M4. However, the pharmacokinetic variables of M5a and M5b were considerably affected by itraconazole, compared with placebo (Table 1, Figure 3). The median Cmax and AUC(0,72 h) of both M5a and M5b were more than 90% smaller during the itraconazole phase than during the placebo phase (P < 0.05).
Effect of gemfibrozil-itraconazole combination
Compared with the placebo phase, the gemfibrozil-itraconazole combination had similar effects on the pharmacokinetics of montelukast and its M6 and M4 metabolites as gemfibrozil alone. The geometric mean ratio to control of montelukast AUC(0,∞) was 4.0-fold (95% CI 2.7, 6.0, P < 0.001) and that of its t1/2 was 2.4-fold (95% CI 2.0, 3.0, P < 0.001). The AUC(0,∞) and t1/2 of montelukast differed from those observed during the itraconazole phase, but not from those observed during the gemfibrozil phase (Table 1, Figure 2). Regarding the pharmacokinetic variables of the minor metabolites M5a and M5b, the effect of the gemfibrozil-itraconazole combination was similar to that of itraconazole alone (Table 1, Figure 3). However, the Cmax and AUC values of M5a and M5b were reduced to a lesser degree during the gemfibrozil-itraconazole phase than during the itraconazole alone phase, because of the opposite effects of itraconazole and gemfibrozil on the concentrations of these metabolites (Table 1, Figure 3).
Plasma gemfibrozil, gemfibrozil 1-O-glucuronide, itraconazole and OH-itraconazole
Concomitant use of itraconazole with gemfibrozil did not affect the plasma concentrations of gemfibrozil and gemfibrozil 1-O-β glucuronide, compared with the gemfibrozil alone phase (Figure 4). In contrast to this, the geometric mean plasma concentrations of itraconazole and OH-itraconazole were more than 50% lower during the gemfibrozil-itraconazole phase than during the itraconazole alone phase (Figure 4).
Figure 4.
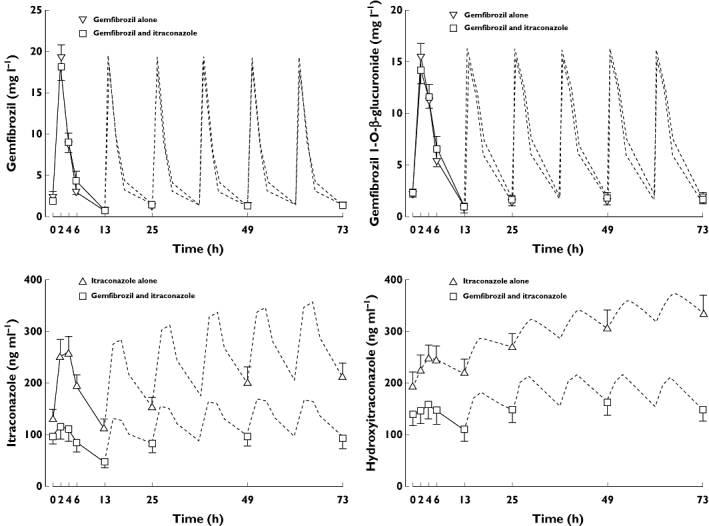
Mean ± SEM plasma concentrations of gemfibrozil and gemfibrozil-1-O-β-glucuronide, itraconazole and OH-itraconazole in 11 healthy volunteers on days 3 to 5 of a 5 day treatment with 600 mg gemfibrozil or 100 mg itraconazole (first dose 200 mg), or both, twice daily. Time 0 refers to administration of gemfibrozil and itraconazole, i.e. 1 h before the administration of montelukast. The dotted lines indicate dose intervals, where no plasma samples were available
Discussion
The primary aim of our present drug interaction study was to characterize the roles of CYP2C8 and CYP3A4 enzymes in the elimination of montelukast in humans. To this end, we administered gemfibrozil and itraconazole, both alone and in combination, and gave a single therapeutic dose of montelukast during each of these treatments, followed by plasma sampling over the next 72 h. This long sampling period and repeated administration of the CYP2C8 and CYP3A4 inhibitors were considered to be necessary, because our previous study had uncovered that gemfibrozil slows the elimination of montelukast and some of its metabolites [7].
Gemfibrozil and itraconazole are strong inhibitors of CYP2C8 and CYP3A4 in vivo, respectively, and they are recommended to be used as probe inhibitors of these enzymes in the US Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA) guidelines [26–29]. Although gemfibrozil is an inhibitor of CYP2C9 in vitro[16], it is not known to inhibit any other CYP enzyme than CYP2C8 in vivo in humans. For example, gemfibrozil does not inhibit CYP2C9 [17] or CYP3A4 [30] enzymes in vivo. The effect of gemfibrozil on CYP2C8 is principally mediated by the gemfibrozil 1-O-β-glucuronide metabolite, which is a selective irreversible, mechanism-based inhibitor of CYP2C8 [10, 11]. Itraconazole and some of its metabolites, including hydroxy-itraconazole, are potent inhibitors of CYP3A enzymes [31–33] and therefore administration of itraconazole leads to strong inhibition of CYP3A4 in vivo[19]. However, itraconazole does not significantly inhibit the CYP2C8 and CYP2C9 enzymes. For example, itraconazole does not affect the pharmacokinetics of the CYP2C8 substrate pioglitazone [34] or reduce the CYP2C9-mediated metabolism of losartan to its active metabolite in humans [35]. Gemfibrozil and itraconazole have long-persisting enzyme inhibitory effects in vivo, the former due to the irreversible CYP2C8 inhibition and the latter because of its long half-life [12, 13, 36, 37]. Thus, the twice daily dosing of these inhibitors caused a strong and uninterrupted inhibition of the CYP2C8 or/and CYP3A4 enzymes for the 72 h follow-up after montelukast ingestion. In addition, the results of the plasma concentration monitoring confirmed the compliance in the ingestion and absorption of gemfibrozil and itraconazole in all the volunteers.
According to the product information of montelukast and the early in vitro study of Chiba et al., CYP3A4, CYP2C9 and CYP2A6 can metabolize montelukast [6, 38]. The formation of the metabolite M5 was described to be catalyzed by CYP3A4 and the formation of M6 by CYP2C9 [38]. However, the in vitro study was performed using a montelukast concentration exceeding over 10 000 times its therapeutic free concentration in plasma [38]. This is likely to have resulted in saturation of the CYP2C8-mediated metabolism of montelukast, due to the potent CYP2C8 inhibitory effect of montelukast [39], leading to an overestimation of the significance of CYP3A4, CYP2C9 and CYP2A6 in the total metabolism of montelukast.
Our present study confirms that gemfibrozil greatly increases the plasma concentrations of montelukast, and inhibits its further metabolism to M6 and M4 [7]. The AUC(0,∞) of montelukast was increased about 4.3-fold and its t1/2 over two-fold by gemfibrozil, compared with placebo. The formation of the main metabolite M6 was impaired, and its further oxidation to M4 was almost abolished by gemfibrozil. Our in vivo findings are thus in a good agreement with the recent in vitro results of Filppula et al. [8]: in human liver microsomes gemfibrozil 1-O-β-glucuronide strongly inhibited the formation of M6 metabolite from therapeutic concentrations of montelukast, and recombinant CYP2C8 catalyzed the depletion of montelukast and the formation of M6 at a six times higher intrinsic clearance than did CYP2C9, while other CYP isoforms produced no M6. On the basis of these in vitro results, the authors estimated that on average, CYP2C8 accounts for more than 70% of the oxidative metabolism of montelukast in vivo, with a 16% contribution by CYP3A4 (M5 formation) and 12% by CYP2C9 [8]. Taken together, the results of the recent in vitro and our in vivo interaction studies in humans indicate that CYP2C8 plays a crucial role in the metabolism of montelukast.
We also studied the effect of the CYP3A4 inhibitor itraconazole alone and in combination with gemfibrozil, on the pharmacokinetics of montelukast. Itraconazole alone had only a minor, statistically non-significant effect on the Cmax, AUC and t1/2 of parent montelukast. Furthermore, the effect of the itraconazole-gemfibrozil combination on these variables was comparable with that of gemfibrozil alone, indicating that itraconazole has no relevant additive effects on these variables, compared with the effects of gemfibrozil alone. The only significant effect of itraconazole was a large reduction in the formation of the minor montelukast metabolites M5a and M5b (21-hydroxylation of montelukast). The median Cmax of M5a and M5b decreased to less than 10% of control during the itraconazole phase and to less than 30% of control even during the gemfibrozil-itraconazole phase. This effect of itraconazole is in good agreement with in vitro results suggesting a crucial role for CYP3A4 in the formation M5a/b [8, 38]. However, at therapeutic plasma concentrations of montelukast, the contribution of CYP3A4 to the total elimination of montelukast (via formation of M5a/b and other metabolites) is obviously negligible, even when the dominant CYP2C8-mediated route is strongly inhibited. This was demonstrated in the present study by the lack of difference in montelukast exposure between the gemfibrozil-itraconazole phase and the gemfibrozil phase.
Previously, the effects of gemfibrozil, itraconazole and their combination have been investigated on the pharmacokinetics of repaglinide, pioglitazone and loperamide using a similar study design as in the present study [15, 23, 34]. The mean AUC of repaglinide was increased 8.1-fold by gemfibrozil alone, 1.4-fold by itraconazole alone and even 19.4-fold by their combination [15]. Similarly, the AUC of loperamide was increased 2.2-fold by gemfibrozil, 3.8-fold by itraconazole and 12.6-fold by their combination. On the other hand, gemfibrozil increased the AUC of pioglitazone 3.2-fold, whereas itraconazole neither influenced pioglitazone AUC nor changed the effect of gemfibrozil on it [34]. Thus, both CYP2C8 and CYP3A4 have significant roles in the elimination of repaglinide and loperamide, and the concomitant inhibition of both enzymes causes a supra-additive increase in their AUC values. In contrast to these, CYP3A4 is only of marginal importance for montelukast and pioglitazone, and its ‘knock-out’ does not increase significantly the exposure to montelukast and pioglitazone.
As gemfibrozil and itraconazole do not inhibit CYP2C9 in vivo[17, 35], the exact role of CYP2C9 in the metabolism of montelukast in vivo remains unknown. However, as gemfibrozil alone increased the AUC of montelukast almost five-fold, it can be estimated that CYP2C8 accounts for about 80% of its metabolism, and only about 20% of its metabolism is mediated by other enzymes in humans. Moreover, the contribution of CYP2C9 to the total metabolism of montelukast is likely to be negligible, because its contribution in vitro at clinically relevant montelukast concentrations is smaller than that of CYP3A4 [8] and because the present data indicate that CYP3A4 also has an insignificant role in the total elimination of montelukast, despite its role in the formation of M5 from montelukast.
In the present study, gemfibrozil and gemfibrozil 1-O-β-glucuronide plasma concentrations were not affected by itraconazole, in line with those reported earlier [7]. However, the plasma concentrations of itraconazole and OH-itraconazole were over 50% lower during the gemfibrozil-itraconazole phase than during the itraconazole phase. This phenomenon has been observed already in earlier studies [15, 34], and can possibly be explained by reduced bioavailability of itraconazole or its displacement from plasma proteins by gemfibrozil. However, the individual concentrations in all phases were consistent with good compliance by all subjects.
In its draft guidance on drug interaction studies, the FDA lists repaglinide and rosiglitazone as suitable probe substrates of CYP2C8 for in vivo studies [26, 27, 29]. The EMA states in its draft guideline, that there is no well-documented CYP2C8 in vivo probe drug at present, and lists amodiaquine and cerivastatin as alternatives that may be used [28]. However, neither of them is on the market in the European Union (EU). In addition, rosiglitazone is not available in Europe, since EMA recommended suspension of its marketing authorization in the EU in September 2009 due to increased cardiovascular risks [40].
Repaglinide has been widely used as an in vivo probe for CYP2C8. As mentioned, gemfibrozil has increased the AUC of repaglinide about eight-fold (ranging from 5.5- to 15-fold) even after a single dose of gemfibrozil [12, 13, 15, 41]. However, repaglinide is also a substrate of organic anion-transporting polypeptide 1B1 (OATP1B1) and its pharmacokinetics are influenced by SLCO1B1 polymorphisms [42]. Moreover, it may, as an antidiabetic drug, induce hypoglycaemia even after a single dose in healthy volunteers. This risk can be seriously increased in some subjects and by CYP2C8 inhibition, and therefore its use as an in vivo probe requires frequent blood glucose monitoring and carbohydrate administration.
The usefulness of montelukast as an in vivo probe of CYP2C8 activity is easily defendable, as montelukast has proved relatively safe in clinical use, and even with 900 mg daily doses no clinically important adverse effects have been observed in short-term clinical studies [6]. Therefore, no specific safety monitoring is needed in its use. As shown in our previous and present studies [7], montelukast can be a sensitive and specific CYP2C8 probe drug: the effect of CYP2C8 inhibition by gemfibrozil on its AUC was almost five-fold, and strong CYP3A4 inhibition had no relevant effect on the elimination of montelukast. Moreover, although the exact role of CYP2C9 in the metabolism of montelukast in vivo is still unknown, CYP2C9 is likely to account only for a minor part of its biotransformation, thus not affecting the usefulness of montelukast as a CYP2C8 probe. However, apart from a possible role of OATP2B1 [43], the role of other membrane transporters in the pharmacokinetics of montelukast has not been elucidated.
It can be concluded that CYP2C8 is the dominant enzyme in the biotransformation of montelukast in humans, accounting for about 80% of its metabolism. CYP3A4 only mediates the formation of the minor metabolite M5a/b of montelukast and is not important in the elimination of montelukast. Montelukast may serve as a safe and useful CYP2C8 probe drug.
Acknowledgments
This study was supported by the Helsinki University Central Hospital Research Fund and the Sigrid Jusélius Foundation (Helsinki, Finland). We thank Kaisa Kurkinen, Jouko Laitila, Eija Mäkinen-Pulli and Lisbet Partanen for skillful technical assistance.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Reiss TF, Chervinsky P, Dockhorn RJ, Shingo S, Seidenberg B, Edwards TB. Montelukast, a once-daily leukotriene receptor antagonist, in the treatment of chronic asthma: a multicenter, randomized, double-blind trial. Montelukast Clinical Research Study Group. Arch Intern Med. 1998;158:1213–20. doi: 10.1001/archinte.158.11.1213. [DOI] [PubMed] [Google Scholar]
- 2.Altman LC, Munk Z, Seltzer J, Noonan N, Shingo S, Zhang J, Reiss TF. A placebo-controlled, dose-ranging study of montelukast, a cysteinyl leukotriene-receptor antagonist. Montelukast Asthma Study Group. J Allergy Clin Immunol. 1998;102:50–6. doi: 10.1016/s0091-6749(98)70054-5. [DOI] [PubMed] [Google Scholar]
- 3.Lipworth BJ. Leukotriene-receptor antagonists. Lancet. 1999;353:57–62. doi: 10.1016/S0140-6736(98)09019-9. [DOI] [PubMed] [Google Scholar]
- 4.Cheng H, Leff JA, Amin R, Gertz BJ, De Smet M, Noonan N, Rogers JD, Malbecq W, Meisner D, Somers G. Pharmacokinetics, bioavailability, and safety of montelukast sodium (MK-0476) in healthy males and females. Pharm Res. 1996;13:445–8. doi: 10.1023/a:1016056912698. [DOI] [PubMed] [Google Scholar]
- 5.Balani SK, Xu X, Pratha V, Koss MA, Amin RD, Dufresne C, Miller RR, Arison BH, Doss GA, Chiba M, Freeman A, Holland SD, Schwartz JI, Lasseter KC, Gertz BJ, Isenberg JI, Rogers JD, Lin JH, Baillie TA. Metabolic profiles of montelukast sodium (Singulair), a potent cysteinyl leukotriene1 receptor antagonist, in human plasma and bile. Drug Metab Dispos. 1997;25:1282–7. [PubMed] [Google Scholar]
- 6.Singulair [Label] Whitehouse Station, NJ: Merck&Co; 2010. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020829s055,020830s056,021409s031lbl.pdf (last accessed December 2010) [Google Scholar]
- 7.Karonen T, Filppula A, Laitila J, Niemi M, Neuvonen PJ, Backman JT. Gemfibrozil markedly increases the plasma concentrations of montelukast: a previously unrecognized role for CYP2C8 in the metabolism of montelukast. Clin Pharmacol Ther. 2010;88:223–30. doi: 10.1038/clpt.2010.73. [DOI] [PubMed] [Google Scholar]
- 8.Filppula AM, Laitila J, Neuvonen PJ, Backman JT. Reevaluation of the microsomal metabolism of montelukast – major contribution by CYP2C8 at clinically relevant concentrations. Drug Metab Dispos. 2011;39:904–11. doi: 10.1124/dmd.110.037689. [DOI] [PubMed] [Google Scholar]
- 9.Shitara Y, Hirano M, Sato H, Sugiyama Y. Gemfibrozil and its glucuronide inhibit the organic anion transporting polypeptide 2 (OATP2/OATP1B1:SLC21A6)-mediated hepatic uptake and CYP2C8-mediated metabolism of cerivastatin: analysis of the mechanism of the clinically relevant drug-drug interaction between cerivastatin and gemfibrozil. J Pharmacol Exp Ther. 2004;311:228–36. doi: 10.1124/jpet.104.068536. [DOI] [PubMed] [Google Scholar]
- 10.Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, Toren P, Parkinson A. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–7. doi: 10.1124/dmd.105.007633. [DOI] [PubMed] [Google Scholar]
- 11.Baer BR, Delisle RK, Allen A. Benzylic oxidation of gemfibrozil-1-O-beta-glucuronide by P450 2C8 leads to heme alkylation and irreversible inhibition. Chem Res Toxicol. 2009;22:1298–309. doi: 10.1021/tx900105n. [DOI] [PubMed] [Google Scholar]
- 12.Tornio A, Niemi M, Neuvonen M, Laitila J, Kalliokoski A, Neuvonen PJ, Backman JT. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h after the dose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther. 2008;84:403–11. doi: 10.1038/clpt.2008.34. [DOI] [PubMed] [Google Scholar]
- 13.Backman JT, Honkalammi J, Neuvonen M, Kurkinen KJ, Tornio A, Niemi M, Neuvonen PJ. CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: estimation of CYP2C8 half-life using repaglinide as an in vivo probe. Drug Metab Dispos. 2009;37:2359–66. doi: 10.1124/dmd.109.029728. [DOI] [PubMed] [Google Scholar]
- 14.Backman JT, Kyrklund C, Neuvonen M, Neuvonen PJ. Gemfibrozil greatly increases plasma concentrations of cerivastatin. Clin Pharmacol Ther. 2002;72:685–91. doi: 10.1067/mcp.2002.128469. [DOI] [PubMed] [Google Scholar]
- 15.Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–51. doi: 10.1007/s00125-003-1034-7. [DOI] [PubMed] [Google Scholar]
- 16.Wen X, Wang JS, Backman JT, Kivistö KT, Neuvonen PJ. Gemfibrozil is a potent inhibitor of human cytochrome P450 2C9. Drug Metab Dispos. 2001;29:1359–61. [PubMed] [Google Scholar]
- 17.Lilja JJ, Backman JT, Neuvonen PJ. Effect of gemfibrozil on the pharmacokinetics and pharmacodynamics of racemic warfarin in healthy subjects. Br J Clin Pharmacol. 2005;59:433–9. doi: 10.1111/j.1365-2125.2004.02323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karonen T, Neuvonen PJ, Backman JT. The CYP2C8 inhibitor gemfibrozil does not affect the pharmacokinetics of zafirlukast. Eur J Clin Pharmacol. 2011;67:151–5. doi: 10.1007/s00228-010-0908-0. [DOI] [PubMed] [Google Scholar]
- 19.Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55:481–5. doi: 10.1038/clpt.1994.60. [DOI] [PubMed] [Google Scholar]
- 20.Neuvonen PJ, Kantola T, Kivistö KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–41. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]
- 21.Jalava KM, Olkkola KT, Neuvonen PJ. Itraconazole greatly increases plasma concentrations and effects of felodipine. Clin Pharmacol Ther. 1997;61:410–5. doi: 10.1016/S0009-9236(97)90191-0. [DOI] [PubMed] [Google Scholar]
- 22.Kivistö KT, Lamberg TS, Kantola T, Neuvonen PJ. Plasma buspirone concentrations are greatly increased by erythromycin and itraconazole. Clin Pharmacol Ther. 1997;62:348–54. doi: 10.1016/S0009-9236(97)90038-2. [DOI] [PubMed] [Google Scholar]
- 23.Niemi M, Tornio A, Pasanen MK, Fredrikson H, Neuvonen PJ, Backman JT. Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur J Clin Pharmacol. 2006;62:463–72. doi: 10.1007/s00228-006-0133-z. [DOI] [PubMed] [Google Scholar]
- 24.Roadcap BA, Musson DG, Rogers JD, Zhao JJ. Sensitive method for the quantitative determination of gemfibrozil in dog plasma by liquid-liquid cartridge extraction and liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;791:161–70. doi: 10.1016/s1570-0232(03)00217-4. [DOI] [PubMed] [Google Scholar]
- 25.Gubbins PO, Gurley BJ, Bowman J. Rapid and sensitive high performance liquid chromatographic method for the determination of itraconazole and its hydroxy-metabolite in human serum. J Pharm Biomed Anal. 1998;16:1005–12. doi: 10.1016/s0731-7085(97)00062-9. [DOI] [PubMed] [Google Scholar]
- 26.Huang SM, Temple R, Throckmorton DC, Lesko LJ. Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clin Pharmacol Ther. 2007;81:298–304. doi: 10.1038/sj.clpt.6100054. [DOI] [PubMed] [Google Scholar]
- 27.US Food and Drug Administration, Draft Guidance for Industry. Drug interaction studies – study design, data analysis, and implications for dosing and labeling. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf (last accessed 8 April 2011)
- 28.European Medicines Agency. Guideline on the investigation of drug interactions (draft, 2010). Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/05/WC500090112.pdf (last accessed 8 April 2011)
- 29.Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ, Chung S, Colangelo P, Frucht D, Green MD, Hepp P, Karnaukhova E, Ko HS, Lee JI, Marroum PJ, Norden JM, Qiu W, Rahman A, Sobel S, Stifano T, Thummel K, Wei XX, Yasuda S, Zheng JH, Zhao H, Lesko LJ. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–70. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 30.Backman JT, Kyrklund C, Kivistö KT, Wang JS, Neuvonen PJ. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68:122–9. doi: 10.1067/mcp.2000.108507. [DOI] [PubMed] [Google Scholar]
- 31.Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32:1121–31. doi: 10.1124/dmd.104.000315. [DOI] [PubMed] [Google Scholar]
- 32.Templeton IE, Thummel KE, Kharasch ED, Kunze KL, Hoffer C, Nelson WL, Isoherranen N. Contribution of itraconazole metabolites to inhibition of CYP3A4 in vivo. Clin Pharmacol Ther. 2008;83:77–85. doi: 10.1038/sj.clpt.6100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang JS, Wen X, Backman JT, Taavitsainen P, Neuvonen PJ, Kivistö KT. Midazolam alpha-hydroxylation by human liver microsomes in vitro: inhibition by calcium channel blockers, itraconazole and ketoconazole. Pharmacol Toxicol. 1999;85:157–61. doi: 10.1111/j.1600-0773.1999.tb00085.x. [DOI] [PubMed] [Google Scholar]
- 34.Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;77:404–14. doi: 10.1016/j.clpt.2004.12.266. [DOI] [PubMed] [Google Scholar]
- 35.Kaukonen KM, Olkkola KT, Neuvonen PJ. Fluconazole but not itraconazole decreases the metabolism of losartan to E-3174. Eur J Clin Pharmacol. 1998;53:445–9. doi: 10.1007/s002280050405. [DOI] [PubMed] [Google Scholar]
- 36.Grant SM, Clissold SP. Itraconazole. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in superficial and systemic mycoses. Drugs. 1989;37:310–44. doi: 10.2165/00003495-198937030-00003. [DOI] [PubMed] [Google Scholar]
- 37.Backman JT, Kivistö KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration-time curve for oral midazolam is 400-fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54:53–8. doi: 10.1007/s002280050420. [DOI] [PubMed] [Google Scholar]
- 38.Chiba M, Xu X, Nishime JA, Balani SK, Lin JH. Hepatic microsomal metabolism of montelukast, a potent leukotriene D4 receptor antagonist, in humans. Drug Metab Dispos. 1997;25:1022–31. [PubMed] [Google Scholar]
- 39.Walsky RL, Obach RS, Gaman EA, Gleeson JP, Proctor WR. Selective inhibition of human cytochrome P4502C8 by montelukast. Drug Metab Dispos. 2005;33:413–8. doi: 10.1124/dmd.104.002766. [DOI] [PubMed] [Google Scholar]
- 40.European Medicines Agency Press release, 23 September. 2010. Available at http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/public_health_alerts/2010/09/human_pha_detail_000020.jsp&mid=&source=homeMedSearch&category=human (last accessed 17 October 2011)
- 41.Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Mechanism-based inactivation of CYP2C8 by gemfibrozil occurs rapidly in humans. Clin Pharmacol Ther. 2011;89:579–86. doi: 10.1038/clpt.2010.358. [DOI] [PubMed] [Google Scholar]
- 42.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, Eichelbaum M, Kivistö KT, Neuvonen PJ. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–78. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 43.Mougey EB, Feng H, Castro M, Irvin CG, Lima JJ. Absorption of montelukast is transporter mediated: a common variant of OATP2B1 is associated with reduced plasma concentrations and poor response. Pharmacogenet Genomics. 2009;19:129–38. doi: 10.1097/FPC.0b013e32831bd98c. [DOI] [PMC free article] [PubMed] [Google Scholar]