

Abstract
Brain tumors—particularly glioblastoma multiforme (GBM)—pose an important public health problem in the US. Despite surgical and medical advances, the prognosis for patients with malignant gliomas remains grim: current therapy for is insufficient with nearly universal recurrence. A major reason for this failure is the difficulty of delivering therapeutic agents to the brain: better delivery approaches are needed to improve treatment. In this article, we summarize recent progress in drug delivery to the brain, with an emphasis on convection-enhanced delivery of nanocarriers. We examine the potential of new delivery methods to permit novel drug- and gene-based therapies that target brain cancer stem cells (BCSCs) and discuss the use of nanomaterials for imaging of tumors and drug delivery.
Keywords: Drug delivery, convection-based delivery, nanocarriers, gene therapy, brain cancer stem cells
Introduction
Improved methods for treating glioblastoma multiforme (GBM) and other malignant tumors in the brain are desperately needed. Malignant gliomas are the most common primary malignant brain tumors in adults. More than 15,000 new cases are diagnosed in the United States each year (1, 2). Despite surgical and medical advances, the five-year survival rate for GBM, the most common malignant glioma, has been a dismal 4% for the past few decades (3, 4). Current treatments for GBM are insufficient with a nearly universal recurrence after surgery, radiation therapy, and chemotherapy (5).
A major problem in therapy for GBM and other CNS diseases is getting drugs into the brain. The blood-brain barrier (BBB) prevents the transport of most systemically-delivered molecules into the brain. Recent research has created better methods for drug delivery to the brain. Here, we review the state-of-the-art in the development of new approaches for drug delivery to the brain, highlighting those methods that are most relevant to GBM.
Controlled release systems for delivery to the brain
The BBB limits the entry of systemically administered drugs to the brain, making delivery behind the BBB an appealing strategy. One method for accomplishing delivery behind the BBB is implantation of a drug-eluting material into the brain tissue. Controlled release systems for direct delivery of chemotherapy to brain tumors were first approved by the FDA in 1996 (6, 7). The rationale for this approach is that biocompatible materials can be introduced directly into the brain, perhaps during surgical resection of the tumor. If the materials are loaded with drugs and are engineered such that the drug is slowly released after implantation, then their implantation into the brain can provide long-term chemotherapy at the tumor site, circumventing the need for BBB penetration. In this approach, drug penetrates into the tissue near the implant by diffusion, producing the highest drug concentrations in the region most in need of treatment and eliminating side effects caused by delivery of drug to other tissues (Figure 1a).
Figure 1.

Representative drug distributions for a) diffusion-based implant systems, such as Gliadel®, b) CED of drug solutions, and c) CED of drug-loaded nanomaterials
The most successful controlled release systems are based on biocompatible polymers, particularly polymers that are also biodegradable. The first clinical trials of this approach used a 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)-loaded polyanhydride delivery system (8–11): BCNU is an alkylating agent, which was the most commonly used chemotherapy drug for GBM in the 1990s. An early clinical study demonstrated that the procedure was safe (12) and a subsequent placebo-controlled clinical trial showed that implantation of BCNU-loaded polymers was more effective than standard therapies in patients with recurrent GBM (13). This new method for brain tumor therapy was rapidly adopted (the clinical product is called Gliadel®), leading to dozens of clinical studies by investigators around the world, and improved lifespan for many brain tumor patients. Experimental controlled delivery systems have been produced with a variety of other compounds, including methotrexate (14), paclitaxel (10, 15), steroids (16), and camptothecin (CPT) (17), but so far none have been tested in humans.
The safety and effectiveness of this new approach for treating GBM has been established, but the increases in survival observed in clinical trials with Gliadel® are modest. However, diffusion-based implant systems—such as Gliadel®--can produce substantial increases in the duration of survival of animals, and optimal formulations can eliminate tumors in animal models. Implantation of a single polymer implant in the brain of an animal produces unprecedented concentrations of active BCNU within the tissue near the implant (10, 11). The contrast between these observations in animal models and clinical studies suggests that one of the limitations of Gliadel® is the small volume treated by diffusion-based delivery systems.
Convection-enhanced delivery to the brain
Convection-enhanced delivery (CED) is a promising approach to overcome the limited distribution volume associated with diffusion-based delivery systems. For diffusion-based delivery, drug molecules move passively from regions of high concentration to regions of lower concentration. As a result, large molecules such as antibodies diffuse no more than 1 mm in 3 days, and small drugs that may have better diffusion are often quickly eliminated by capillary clearance or metabolism (18). In contrast, for dispersion using convection, agents are delivered to the brain via flow through a cannula under constant pressure. In this scenario, the dispersion of agents is powered by bulk flow kinetics or gradients of pressure, in addition to gradient of concentrations. As a result, it is possible to distribute agents widely in the brain (Figure 1b). CED technology and factors affecting drug distribution by CED have been extensively reviewed (18, 19) and thus will not be discussed in detail here.
CED to the brain was first reported in the early 1990s (20). Since then, CED has been used in clinical trials, but this experience has revealed some limitations. Conventional CED of drug solutions results in an increased depth of penetration, but these results are transient. Free drugs are subject to high rates of elimination (i.e. they are transported into cerebrospinal fluid (CSF) or blood) or have short half-lives in the brain; therefore, they disappear soon after the infusion stops (18). Interstitial fluid flow might particularly impact CED of free agents to brain tumors. Mathematical models—and some experimental evidence—suggest that tumors maintain an elevated interstitial fluid pressure in their cores (21), which produces an outwardly-directed flow of extracellular fluid at their periphery (22). These limitations could explain the failure of the recent PRECISE trial, in which a potent targeted toxin, CB (cintredekin besudotox, IL13-PE38QQR) in aqueous suspension, was delivered to brain tumors via CED but failed to show advantage when compared to diffusion-based Gliadel® wafers (23).
To improve CED, agents can be loaded into nanocarriers, such as liposomes, micelles, dendrimers, or polymeric nanoparticles, which are small enough (typically 100–200 nm in diameter) to allow for infusion and penetration in brain. These carriers can protect therapeutic agents from loss and control their release for long periods after infusion (Figure 2). Nanoparticle delivery systems for drugs have been available for many years (24). Some research groups focus on the use of nanoparticles introduced systemically, with the hopes that some of these particles will enter the brain through the BBB. This approach appears to work in some cases, but the percentage of intravenously administered particles that enter the brain is very low (25–27). It is not yet clear whether sufficient quantities of drug can be delivered by systemically-administered nanoparticles to make this a useful method for treating tumors in the brain (although there is some evidence that systemically-administered nanoparticles may be useful for diagnostic purposes, such as iron oxide-containing nanoparticles that facilitate imaging of brain tumors (28)). An alternate approach is to deliver the nanoparticles directly into the brain, perhaps using CED to facilitate the distribution of the nanoparticles throughout the volume of the brain that needs therapy (Figure 1c).
Figure 2.

Nanomaterials used in drug delivery. A: Polymeric nanoparticles (NPs) are typically made of biodegradable polymers. B: Liposomes are composed of phospholipids. C: Polymeric micelles are formed in an aqueous environment from the association of block copolymers. D: Dendrimers are branched, tree-like polymers, allowing drugs to be attached to its many arms. E: Carbon nanotubes, made of benzene rings, can carry drugs inside the lumen of the tube or attached to the sides. Reproduced from Bennewitz and Saltzman (141), with permission
CED of lipid-based nanoparticle systems, such as liposomes, has been well studied. Liposomes are artificial phospholipid vesicles that form a “core-shell” structure and can be readily loaded with therapeutic agents. CED of liposomes has been evaluated in rat brain tumor xenografts (29–32) and canine spontaneous brain tumors (33); CED has also been tested in the normal brain of non-human primates (34, 35). Compared to CED of free drugs, CED of liposomes increases the distribution of delivered agents (36), reduces toxicity, and extends half-life (31, 33, 37). CED of liposomes has recently been tested in human GBM patients: it appears to be safe and to provide some therapeutic benefit (38, 39).
As an alternate to liposomes, polymer nanoparticles can be delivered via CED. In a recent study, CED of camptothecin-loaded nanoparticles led to longer survival in animals with intracranial tumors than CED of camptothecin alone (40). Previous efforts to deliver polymer nanoparticles via CED have achieved a limited volume of distribution, due to an inability to fabricate polymer nanoparticles of small enough size to allow for unhindered interstitial convective transport (40, 41). The pore size of the normal brain extracellular space is 38–64 nm (42) versus ~70–100 nm within a tumor in the brain (43), which suggests that nanoparticles for CED to intracranial tumors should be 60–80 nm in diameter to allow for access to tumorous tissue while sparing the normal brain. We recently developed methods for fabricating non-aggregrating PLGA nanoparticles of this size (44). Our data suggest that these nanoparticles are able to penetrate brain tissue with a mean volume of distribution that is at least 10-fold larger than previously published formulations (Zhou J. unpublished data).
New delivery technologies create opportunities for drug therapy
The most recent advance in drug therapy of GBM was the FDA approval in 2005 of temozolomide (TMZ), which has since replaced BCNU as standard therapy. Both TMZ and BCNU cross the BBB, but TMZ produces fewer side effects and is bioavailable after oral administration (BCNU is administered by infusion or through implantation of Gliadel®) (45–47). Even with these drugs, the median survival for GBM patients is less than 15 months: drug resistance and tumor recurrence remain major challenges.
In the past, drugs needed to cross the BBB to be candidates for use in treating GBM. New delivery technologies—such as implantable controlled release systems, CED, and nanoparticles—make it possible to use drugs that do not cross the BBB into the brain. These technologies create a new opportunity: drugs can be selected based solely on their potential for effectiveness against tumors, without regard for their ability to cross the BBB. For example, it is now clear that paclitaxel—which does not cross the BBB—is an effective agent for treating brain tumors when administered via paclitaxel-loaded implants or CED of paclitaxel nanoparticles or microparticles (15, 48). Likewise, CED permitted the clinical testing of immunotoxins, which do not cross the BBB, in the PRECISE trial (23).
Many experimental drugs are currently being examined for treatment of GBM (Table 1): these agents target a wide variety of molecular pathways important in glioma cell survival. But few of these agents will cross the BBB at doses sufficient to treat tumors in the brain without systemic side effects. Therefore, using conventional methods of drug delivery (infusion or oral administration), these novel agents will likely suffer from the same problem that has plagued drug therapy of brain tumors for decades: it will be difficult to deliver these agents to human tumors at a sufficient dose for effectiveness without also creating serious toxicity. New delivery systems may enhance the clinical value of some of these agents, by allowing sustained local delivery that concentrates the drug at the target.
Table 1.
Investigational drugs and drugs in clinical trials for GBM. Information was compiled from a variety of sources including clinicaltrials.gov (indicated by ClinicalTrials.gov Indentifiers such as NCT01051557), Adamson et al. (46), Visnyei et al (78), and individual references as listed.
| Molecular Target/Pathways |
Agents | References |
|---|---|---|
| mTORC1 inhibitors | Sirolimus, Everolimus, Temsirolimus, Deforolimus | (70, 142–145) |
| Dual mTORC1/mTORC2 inhibitors | OXA-01, OSI-027, AZD8055 | (146) |
| PI3 kinase inhibitors | XL765, LY294002, Wortmannin | (147–149) |
| Dual PI3K/mTOR inhibitors | NVP-BEZ235, PI-103 | (70, 148) |
| Raf inhibitors | Sorafenib | (150, 151) |
| AKT inhibitor | Perifosine | NCT01051557 |
| EGF RTK inhibitors | Erlotinib, Gefinib, lapatinib, BIBW2992, Nimotuzumab, Cetuximab, AEE788 | (145, 152–162) NCT00727506, NCT00753246 |
| Growth Factor inhibitor | Leflunomide, Suramin | (163–165) |
| Farnesyl transferase inhibitor | Tipifarnib, lonafarnib | (166–170) |
| Histone Deacetylase inhibitor | Vorinostat, Depsipeptide, Panobinostat | (171–173) |
| HSP90 inhibitor | NXD30001 | (174) |
| LDL receptor peptide | ANG1005 | (175, 176) |
| Met inhibitor | XL184 (cabozantinib) | (177, 178) |
| MMP inhibitor | Prinomastat (AG3340) | (179, 180) |
| Microtubule disruption | CYT997, MPC-6827, Epothilones | NCT01285414, NCT00892931, NCT00635557 |
| PDGF RTK inhibitor | Dasatinib, Imatinib, Tandutinib, Pazopanib | (152, 181–185) NCT00895960, NCT00609999, NCT00379080 |
| PKCB STK inhibitor | Enzastaurin | (186–188) |
| Proteosome inhibitor | Bortezomib | (189–193) |
| Sp1 inhibitor | Terameprocol | NCT00404248 |
| Topoisomerase inhibitor | RTA744, Etoposide, Topotecan, Irinotecan, AQ4N, Edotecarin, Rubitecan, Pyrazoloacridine, Karenitecin, Gimatecan | (78) |
| VEGF RTK inhibitor | PTK787 (Valatinib) | (194–196) |
| CDK4/6 inhibitor | PD-0332991 | (197, 198) |
| HIV Protease inhibitor | Nelfinavir | (199–201) |
| Capsaicin-like endocannabinoids | N-Arachidonoyldopamine, N-Oleoyldopamine, N-Palmitoyldopamine | (78, 202–207) |
| Anti-protozoal; 28S ribosome inhibitor | Emetine | (78, 208–210) |
| c-Jun-NH2-terminal kinase (JNK) activator | Anisomycin | (78, 208) |
| Uncharacterized Chembridge compounds | #5560509 #5256360, #5485415 | (78) |
| Angiogenesis Inhibitor | AGM 1470 | (211) |
| Hedgehog pathway inhibitor | Cyclopamine | (212) |
Further, it is becoming increasingly clear that certain cells within tumors play a major role in tumor progression, and that these cells are not sensitive to current drugs (49, 50). A small fraction of glioma cells has been identified as brain cancer stem cells (BCSCs), of which many are CD133+ (51–58). These cells have features similar to primitive neural stem cells, but they also have tumor-initiating functions (59). BCSCs appear to arise from deranged neural stem cell or glial cell progenitors (50–54): these cells have the ability to drive tumor formation, promote angiogenesis, and influence tumor cell migration (50, 55, 60). Current chemotherapeutic agents do not eliminate BCSCs effectively (49, 61, 62): BCSCs in culture are resistant to standard chemotherapy drugs, including temozolomide, carboplatin, cisplatin, paclitaxel, doxorubicin, vincristine, methotrexate, and etoposide (63–66). BCSCs are also resistant to standard radiotherapy (55). These observations suggest that current therapeutic regimens may produce short-term remissions, but are unlikely to cure GBM: long-term remissions require eradication of BCSCs (49, 50, 61, 67).
One promising approach to identifying novel therapies for BCSCs is to target key signaling pathways governing BCSC proliferation and self-renewal, such as the PTEN/PI3K/Akt pathway (68). Inhibition of the Akt pathway induces BCSC differentiation and inhibits self-renewal and tumorigenicity in vivo (69, 70). Developmental pathways, including the Notch (71, 72) and BMP pathways (73), are important for BCSC self-renewal and differentiation and can be targeted to preferentially eliminate BCSCs. In addition, several other signaling pathways, including EGFR (74), TGF-® (75) and HIF (76), are critical for BCSC proliferation, survival, and self-renewal. An alternate approach is to take advantage of recent advances in genomics and informatics to identify therapeutic agents via high-throughput screening. For example, an RNAi screen against a kinome library successfully identified TRRAP as a regulator of BCSC differentiation (77). More recently, eight small molecule drugs that preferentially inhibit BCSCs from GBM over tumor-matched non-BCSC GBM cells were identified from a library of over 30,000 (78). These early successes suggest that high-throughput screening is a promising approach for identifying novel BCSC-targeted therapeutics. Some of the most promising drugs now being examined for GBM therapy (Table 1) were identified through their activity on BCSCs.
Delivery remains a major obstacle for use of any agent that targets BCSC. The new delivery technologies described earlier will likely prove necessary for translating these agents to clinical practice.
Delivery systems for gene therapy
The promise of gene therapy in GBM has long been known, but the difficulty carrying it out is equally well recognized. An early gene therapy approach involved introduction of genes at the tumor site that would activate systemically administered prodrugs (79). One such system, which employed herpes simplex virus thymidine kinase and ganciclovir showed success in mouse models (80, 81), but in a phase III trial post-operative delivery failed to show benefit compared to post-operative radiotherapy (82). Other approaches have since emerged, particularly since the recognition of the role of microRNA (miRNA) in tumors and the power of RNA interference for silencing genes.
A variety of gene therapy approaches may be useful in GBM. Deletion of known tumor suppressors, such as PTEN, is a common mechanism of gliomagenesis (83). Rescued expression of PTEN in U87 glioma cells suppresses tumorigenicity in vivo and promotes entry into G1 phase of the cell cycle (84). Genes in the tumor-suppressing p53 pathway (85, 86), such as TP53, MDM2/4 and p14-ARF, and the retinoblastoma (Rb) pathway (85, 87), such as Rb, CDK4/6 and p16-INK4A, are also promising targets for treatment of GBM. Alternately, brain tumor growth can potentially be inhibited by blocking expression of genes, such as EGFR (88). High-throughput sequencing efforts and the compilation of data into The Cancer Genome Atlas, as well as the development of the Cancer Bioinformatics Grid have led to the identification of a number of highly novel chromosomal mutations, amplifications, and deletions that are important in GBM (85). Chief among them is mutation to isocitrate dehydrogenase (IDH) 1 and 2 (89). Mutated IDH1 leads to induction of the HIF pathway (90), which leads to an increased fraction of BCSCs (91).
MicroRNAs play important roles in gliomagenesis and can be targets for treatment. Reduced expression of a number of miRNAs has been noted in GBM (92). miR-7 is capable of repressing EGFR signaling (93), so rescue or ectopic expression of miR-7 in tumor cells may limit GBM progression. Expression of miR-221, an antiapoptotic factor, is markedly elevated in human GBM tissues (94), and inhibition of miR-221 decreases tumor growth in tumor xenograft (95). miR-10b, which inhibits cell cycle arrest and apoptosis while promoting growth through p16-INK4A and p21 targeting, is another miRNA known to be elevated in GBM tissue samples and cell lines (96). Several other miRNAs are important for brain tumor cell survival and represent potential targets for therapy (97, 98).
Delivery remains the major obstacle for gene-based therapies. Viral vectors—including retrovirus (82), adenovirus (99) and adeno-associated virus (100, 101)—are often used in evaluations in animals or humans, due to their high transduction efficiency. In addition to safety concerns, free viral particles in suspension may not reach disseminated tumor cells (102, 103). Synthetic non-viral vectors, such as nanoparticle-based gene delivery vehicles, might be a better approach due to their limited immunogenicity, ability to accommodate and deliver large pieces of genetic material, and potential for modification of their surface structures to allow targeting. Among non-viral vectors, cationic lipid-based systems, including liposome and solid lipid nanoparticles, are the most extensively studied (104, 105). Liposome-based gene delivery vehicles have been used for gene therapy to brain tumors in animals (106, 107) and have shown some promise in clinical trials (38, 39, 108). Cationic polymers, such as dendrimer- (109, 110) and polyethyleneimine-based nanocarriers (111), also have potential for gene delivery to brain tumors. But these highly charged vectors are usually toxic; because of excess positive charge at their surface, these nanocarriers inhibit normal cellular processes, such as clathrin-mediated endocytosis (112) and cell survival signaling (113), leading to substantial toxicity (112, 114, 115).
Most lipid and polymer gene delivery systems rely exclusively on cation density to form complexes with DNA. But it has recently been discovered that the ability of polymer nanocarriers to deliver genes depends on factors other than charge density, particularly polymer molecular weight and hydrophobicity. High molecular weight and increased hydrophobicity can compensate for low charge density to provide efficient gene delivery with minimal toxicity (116, 117). For example, poly(amine-co-ester) terpolymers, which have significantly lower charge density than traditional polycationic polymers, are able to deliver genes in vitro and in vivo with efficiency superior to existing commercially available gene delivery systems and with limited toxicity (118).
Another approach for developing safe and efficient polymer-based vehicles is to start with polymers that are known to be safe for clinical use and to modify them to enhance their ability to deliver genes or oligonucleotides. Poly(lactide-co-glycolide) (PLGA) was approved by FDA in 1969 and has been in continuous clinical use since that time. Octa-functional nanoparticles were produced by modification of PLGA nanoparticles through the use of chemical conjugation and physical formulation techniques. These nanoparticles exhibit limited toxicity but are able to efficiently deliver DNA or siRNA to tumor cells in vitro and in vivo (119), making them suitable for brain tumor gene therapy.
Imaging of drug delivery in the brain
Accurate measurement of the distribution of a delivered agent is needed to assess drug or gene delivery in GBM. Magnetic resonance imaging (MRI) allows for monitoring of delivery in certain cases. Recent studies have used diffusion-weighted imaging (DWI) as a means to monitor the administration of fluid into the brain; it is assumed that the location of changes in DWI correlates to the area occupied by infused drug. These studies find an area of decreased diffusion immediately after infusion that evolves into an area of increased fluid movement (120, 121). In the setting of CED, where many factors can influence the distribution of the infusion, this finding is particularly useful: optimizing fluid distribution is needed for effective treatment (121). Significantly, DWI is frequently used in the clinical setting and does not require the administration of contrast agents. But DWI must be used with caution: it relies upon water movement to produce signal changes, effectively only measuring the movement of the solvent, and measuring it indirectly. There is also debate as to the mechanisms producing the observed intensity changes, and whether they represent the infusate or reactive fluid movement (122).
Drug distribution can be quantified by MRI using gadolinium-based contrast agents. Contrast provided by gadolinium agents, such as gadolinium-diethylene triamine pentaacetic acid (Gd-DTPA), can be used to monitor disruption of the blood-brain barrier (123). When co-administered with a therapeutic agent or other macromolecule (Figure 3), Gd-DTPA can be used to assess the volume of drug delivery (123, 124). Gadolinium complexes can be conjugated to or encapsulated in nanocarriers, such as polymer nanoparticles (125, 126) or liposomes (29, 34) in order to assess the dispersion of drug-loaded particles. This approach is appealing, but it has limitations. Gadolinium contrast agents are smaller than many delivery vehicles. Thus, when used as surrogates, they can diffuse over a larger volume than the actual agent (although, in practice, this effect may be small) (123, 124, 127). Some groups have addressed this issue by conjugating therapies to materials that can be imaged directly, such as iron-oxide nanoparticles (128). But this approach can also present problems, because the addition of an imaging moiety can alter the agent in unforeseen ways, such that distribution behavior does not precisely mirror the natural, unmodified agent. Contrast agent-derived signals are more easily related to drug movement than diffuse signaling changes, such as those seen on DWI, but they have limitations.
Figure 3.
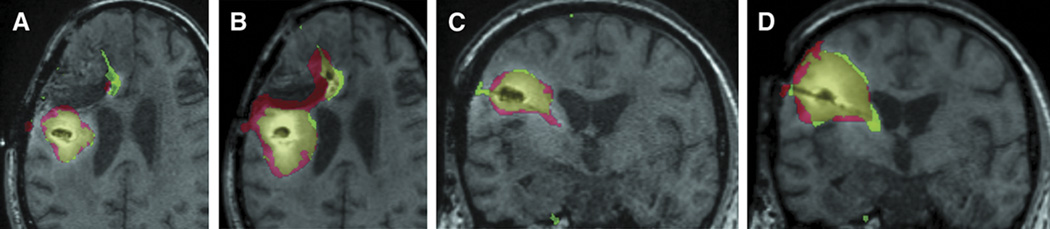
T1-MRI of Gd-DTPA (green) and 124I-labelled (red) human serum albumin coinfusion at 24 hours (A and C) and 72 hours (B and D). Note the large but imperfect area of overlap (yellow). Reproduced from (123), permission pending.
Nuclear medicine imaging can also be used to monitor drug distribution. In single-photon emission computed tomography (SPECT), a gamma-emitting tracer allows for three-dimensional visualization of the distribution area of an agent (123, 129). The radioisotope can be either co-administered with the drug of interest or directly bound to the biologically active molecule (such as siRNA) to allow for direct assessment of its volume of distribution (130). SPECT imaging can be coupled with CT or MRI for anatomic correlation (130). This approach is direct, and uses equipment that is significantly less expensive than those used in other nuclear medicine imaging techniques (131). The main drawback of conventional SPECT is resolution, although recent advances in hardware (such as pinhole-SPECT) allow for resolution at the millimeter level (132, 133).
Positron-emission-tomography (PET) is another promising modality for imaging drug delivery to tumors. As with gadolinium and SPECT contrast agents, PET tracers can be infused concurrently with drug or bound to the delivery system, such as nanoparticles (134–136). When PET is coupled with CT, molecular movement can be correlated with anatomy, allowing for measurement of the anatomical area of diffusion of tracer or tracer-laden substance (134, 135). Further, through the use of tracers that are derivatives of amino acids—such as O-(2-[18F]fluoroethyl)-L-tyrosine (FET)—PET imaging can estimate the borders of a tumor, allowing for more accurate assessment of drug distribution relative to tumor volume than MRI (134, 137, 138). Challenges for PET imaging include radiation exposure (which is also a challenge in SPECT), the high cost of the studies, and the short-lived nature of typical PET tracers (139). Another limitation is similar to the one mentioned for gadolinium agents in MRI: unless the tracer is directly coupled to the delivery agent, then the measurement of the area of diffusion is indirect. Direct radiolabeling of nanoparticles shows that it is possible to overcome this limitation (140).
Conclusions
New drug delivery strategies are already impacting treatment of GBM, and it seems clear that delivery systems will be needed for therapies of the future, which will be targeted to particular cells and depend on intracellular delivery of agents that do not readily cross the BBB or enter cells. Polymer implants, CED techniques, and degradable nanoparticles are powerful platform technologies for creation of new methods to treat GBM.
Acknowledgements
Our work on delivery systems for treating GBM is funded by the National Institutes of Health (Grant CA149128). We thank Nha Duong for preparation of Figure 1 and excellent editorial assistance.
Sources of support that require acknowledgment: National Institutes of Health (Grant CA149128 to WMS)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflicts of interest.
References
- 1.Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. doi: 10.1007/s00401-005-0991-y. [DOI] [PubMed] [Google Scholar]
- 2.Fisher PG, Buffler PA. JAMA. 2005;293:615–617. doi: 10.1001/jama.293.5.615. [DOI] [PubMed] [Google Scholar]
- 3.Scott CB, Scarantino C, Urtasun R, et al. Validation and predictive power of Radiation Therapy Oncology Group (RTOG) recursive partitioning analysis classes for malignant glioma patients: a report using RTOG 90-06. Int J Radiat Oncol Biol Phys. 1998;40:51–55. doi: 10.1016/s0360-3016(97)00485-9. [DOI] [PubMed] [Google Scholar]
- 4.National Brain Tumor Society. [accessed 28 October];2011 http://www.braintumor.org/get-involved/advocacy-public-policy/know-the-issues.html. [Google Scholar]
- 5.Sathornsumetee S, Rich JN. New treatment strategies for malignant gliomas. Expert Rev Anticancer. 2006;6:1087–1104. doi: 10.1586/14737140.6.7.1087. [DOI] [PubMed] [Google Scholar]
- 6.Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. 2002;41:403–419. doi: 10.2165/00003088-200241060-00002. [DOI] [PubMed] [Google Scholar]
- 7.Sawyer AJ, Piepmeier JM, Saltzman WM. New methods for direct delivery of chemotherapy for treating brain tumors. Yale J Biol Med. 2007;79:141–152. [PMC free article] [PubMed] [Google Scholar]
- 8.Yang M, Tamargo R, Brem H. Controlled delivery of 1,3-Bis(2-chloroethyl)-1-nitrosourea from ethylene-vinyl acetate copolymer. Cancer Res. 1989;49:5103–5107. [PubMed] [Google Scholar]
- 9.Tamargo RJ, Myseros JS, Epstein JI, et al. Interstitial chemotherapy of the 9L gliosarcoma: controlled release polymers for drug delivery in the brain. Cancer Res. 1993;53:329–333. [PubMed] [Google Scholar]
- 10.Fung LK, Ewend MG, Sills A, et al. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998;58:672–684. [PubMed] [Google Scholar]
- 11.Fung L, Shin M, Tyler B, et al. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res. 1996;13:671–682. doi: 10.1023/a:1016083113123. [DOI] [PubMed] [Google Scholar]
- 12.Brem H, Mahaley M, Vick N, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74:441–446. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- 13.Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet. 1995;345:1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 14.Dang WB, Colvin OM, Brem H, et al. Covalent coupling of methotrexate to dextran enhances the penetration of cytotoxicity into a tissue-like matrix. Cancer Res. 1994;54:1729–1735. [PubMed] [Google Scholar]
- 15.Walter KA, Cahan MA, Gur A, et al. Interstitial taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994;54:2207–2212. [PubMed] [Google Scholar]
- 16.Reinhard C, Radomsky ML, Saltzman WM, et al. Polymeric controlled release of dexamethasone in normal rat brain. J Control Release. 1991;16:331–340. [Google Scholar]
- 17.Fleming AB, Haverstick K, Saltzman WM. In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjug Chem. 2004;15:1364–1375. doi: 10.1021/bc034180o. [DOI] [PubMed] [Google Scholar]
- 18.Allard E, Passirani C, Benoit JP. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30:2302–2318. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Raghavan R, Brady ML, Rodriguez-Ponce MI, et al. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20:E12. doi: 10.3171/foc.2006.20.4.7. [DOI] [PubMed] [Google Scholar]
- 20.Bobo RH, Laske DW, Akbasak A, et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37:77–104. doi: 10.1016/0026-2862(89)90074-5. [DOI] [PubMed] [Google Scholar]
- 22.Smith JH, Humphrey JA. Interstitial transport and transvascular fluid exchange during infusion into brain and tumor tissue. Microvasc Res. 2007;73:58–73. doi: 10.1016/j.mvr.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Sampson JH, Archer G, Pedain C, et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial Clinical article. J Neurosurg. 2010;113:301–309. doi: 10.3171/2009.11.JNS091052. [DOI] [PubMed] [Google Scholar]
- 24.Fahmy TM, Fong PM, Goyal A, et al. Targeted nanoparticles for drug delivery. Nano Today. 2005;8:18–26. [Google Scholar]
- 25.Lockman PR, Oyewumi MO, Koziara JM, et al. Brain uptake of thiamine-coated nanoparticles. J Control Release. 2003;93:271–282. doi: 10.1016/j.jconrel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Koziara J, Lockman PR, Allen DD, et al. In situ blood-brain barrier transport of nanoparticles. Pharm Res. 2003;20:1772–1778. doi: 10.1023/b:pham.0000003374.58641.62. [DOI] [PubMed] [Google Scholar]
- 27.Kreuter J, Ramge P, Petrov V, et al. Direct evidence that polysorbate-80-coated poly(butylcyanoacrylate) nanoparticles delivery drugs to the CNS via specific mechanisms requiring prior binding of drug to the nanoparticles. Pharm Res. 2003;20:409–416. doi: 10.1023/a:1022604120952. [DOI] [PubMed] [Google Scholar]
- 28.Kircher MF, Mahmood U, King RS, et al. A multimodal nanoparticle for preoperative magnetic rsonance imaging and intraoperative optical brain tumor delineation. Cancer Res. 2003;63:8122–8125. [PubMed] [Google Scholar]
- 29.Saito R, Bringas JR, McKnight TR, et al. Distribution of liposomes into brain and rat brain tumor models by convection-enhanced delivery monitored with magnetic resonance imaging. Cancer Res. 2004;64:2572–2579. doi: 10.1158/0008-5472.can-03-3631. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita Y, Krauze MT, Kawaguchi T, et al. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts. Neuro-Oncology. 2007;9:20–28. doi: 10.1215/15228517-2006-016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krauze MT, Noble CO, Kawaguchi T, et al. Convection-enhanced delivery of nanoliposomal CPT-11 (irinotecan) and PEGylated liposomal doxorubicin (Doxil) in rodent intracranial brain tumor xenografts. Neuro-Oncology. 2007;9:393–403. doi: 10.1215/15228517-2007-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyata S, Kawabata S, Hiramatsu R, et al. Computed tomography imaging of transferrin targeting liposomes encapsulating both boron and iodine contrast agents by convection-enhanced delivery to F98 rat glioma for boron neutron capture therapy. Neurosurgery. 2011;68:1380–1387. doi: 10.1227/NEU.0b013e31820b52aa. discussion 1387. [DOI] [PubMed] [Google Scholar]
- 33.Dickinson PJ, LeCouteur RA, Higgins RJ, et al. Canine spontaneous glioma: a translational model system for convection-enhanced delivery. Neuro-Oncology. 2010;12:928–940. doi: 10.1093/neuonc/noq046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito R, Krauze MT, Bringas JR, et al. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp Neurol. 2005;196:381–389. doi: 10.1016/j.expneurol.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Krauze MT, McKnight TR, Yamashita Y, et al. Real-time visualization and characterization of liposomal delivery into the monkey brain by magnetic resonance imaging. Brain Res Protoc. 2005;16:20–26. doi: 10.1016/j.brainresprot.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Saito R, Krauze MT, Noble CO, et al. Tissue affinity of the infusate affects the distribution volume during convection-enhanced delivery into rodent brains: implications for local drug delivery. J Neurosci Methods. 2006;154:225–232. doi: 10.1016/j.jneumeth.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 37.Krauze MT, Forsayeth J, Yin D, et al. Convection-enhanced delivery of liposomes to primate brain. Method Enzymol. 2009;465:349–362. doi: 10.1016/S0076-6879(09)65018-7. [DOI] [PubMed] [Google Scholar]
- 38.Voges J, Reszka R, Gossmann A, et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann Neurol. 2003;54:479–487. doi: 10.1002/ana.10688. [DOI] [PubMed] [Google Scholar]
- 39.Ren H, Boulikas T, Lundstrom K, et al. Immunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene--a phase I/II clinical protocol. J Neuro-Oncol. 2003;64:147–154. doi: 10.1007/BF02700029. [DOI] [PubMed] [Google Scholar]
- 40.Sawyer AJ, Saucier-Sawyer JK, Booth CJ, et al. Convection-enhanced delivery of camptothecin-loaded polymer nanoparticles for treatment of intracranial tumors. Drug Deliv Transl Res. 2011;1:34–42. doi: 10.1007/s13346-010-0001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neeves KB, Sawyer AJ, Foley CP, et al. Dilation and degradation of the brain extracellular matrix enhances penetration of infused polymer nanoparticles. Brain Res. 2007;1180:121–132. doi: 10.1016/j.brainres.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel T, Zhou J, Piepmeier JM, et al. Ultrasmall polymeric nanoparticles improve distribution volume during convection-enhnaced delivery (abstract) Neuro-Oncology. 2010;12:91. [Google Scholar]
- 45.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 46.Adamson C, Kanu OO, Mehta AI, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert opinion on investigational drugs. 2009;18:1061–1083. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 47.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- 48.Marupudi NI, Han JE, Li KW, et al. Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf. 2007;6:609–621. doi: 10.1517/14740338.6.5.609. [DOI] [PubMed] [Google Scholar]
- 49.Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96:583–585. doi: 10.1093/jnci/djh095. [DOI] [PubMed] [Google Scholar]
- 50.Clarke MF. Neurobiology: at the root of brain cancer. Nature. 2004;432:281–282. doi: 10.1038/432281a. [DOI] [PubMed] [Google Scholar]
- 51.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 52.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 53.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 54.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 55.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 56.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 57.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 58.Gunther HS, Schmidt NO, Phillips HS, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2007 doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- 59.Fan X, Salford LG, Widegren B. Glioma stem cells: evidence and limitation. Semin Cancer Biol. 2007;17:214–218. doi: 10.1016/j.semcancer.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 60.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 61.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 62.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 63.Tang C, Chua CL, Ang BT. Insights into the cancer stem cell model of glioma tumorigenesis. Ann Acad Med Singapore. 2007;36:352–357. [PubMed] [Google Scholar]
- 64.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eramo A, Ricci-Vitiani L, Zeuner A, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006;13:1238–1241. doi: 10.1038/sj.cdd.4401872. [DOI] [PubMed] [Google Scholar]
- 66.Hirschmann-Jax C, Foster AE, Wulf GG, et al. A distinct "side population" of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A. 2004;101:14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abbott A. Cancer: the root of the problem. Nature. 2006;442:742–743. doi: 10.1038/442742a. [DOI] [PubMed] [Google Scholar]
- 68.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gallia GL, Tyler BM, Hann CL, et al. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther. 2009;8:386–393. doi: 10.1158/1535-7163.MCT-08-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sunayama J, Sato A, Matsuda K, et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro-Oncology. 2010;12:1205–1219. doi: 10.1093/neuonc/noq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fan X, Matsui W, Khaki L, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 72.Fan X, Khaki L, Zhu TS, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 74.Soeda A, Inagaki A, Oka N, et al. Epidermal growth factor plays a crucial role in mitogenic regulation of human brain tumor stem cells. J Biol Chem. 2008;283:10958–10966. doi: 10.1074/jbc.M704205200. [DOI] [PubMed] [Google Scholar]
- 75.Ikushima H, Todo T, Ino Y, et al. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504–514. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 76.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wurdak H, Zhu S, Romero A, et al. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell. 2010;6:37–47. doi: 10.1016/j.stem.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 78.Visnyei K, Onodera H, Damoiseaux R, et al. A molecular screening approach to identify and characterize inhibitors of glioblastoma multiforme stem cells. Mol Cancer Ther. 2011;10:1818–1828. doi: 10.1158/1535-7163.MCT-11-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller CR, Williams CR, Buchsbaum DJ, et al. Intratumoral 5-fluorouracil produced by cytosine deaminase/5-fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res. 2002;62:773–780. [PubMed] [Google Scholar]
- 80.Chambers R, Gillespie GY, Soroceanu L, et al. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a scid mouse model of human malignant glioma. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1411–1415. doi: 10.1073/pnas.92.5.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hamel W, Zirkel D, Mehdorn HM, et al. (E)-5-(2-bromovinyl)-2'-deoxyuridine potentiates ganciclovir-mediated cytotoxicity on herpes simplex virus-thymidine kinase--expressing cells. Cancer Gene Therapy. 2001;8:388–396. doi: 10.1038/sj.cgt.7700322. [DOI] [PubMed] [Google Scholar]
- 82.Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 83.Zhang W, Fine H. Mechanisms of Gliomagenesis. In: Janigro O, editor. The Cell Cycle in the Central Nervous System. VI. Totowa, NJ: Humana Press; 2006. pp. 449–462. [Google Scholar]
- 84.Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci USA. 1998;95:15406–15411. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Symonds H, Krall L, Remington L, et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell. 1994;78:703–711. doi: 10.1016/0092-8674(94)90534-7. [DOI] [PubMed] [Google Scholar]
- 87.Schmidt EE, Ichimura K, Reifenberger G, et al. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–6324. [PubMed] [Google Scholar]
- 88.Rao SK, Edwards J, Joshi AD, et al. A survey of glioblastoma genomic amplifications and deletions. J Neuro-Oncol. 2010;96:169–179. doi: 10.1007/s11060-009-9959-4. [DOI] [PubMed] [Google Scholar]
- 89.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. New Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mathieu J, Zhang Z, Zhou W, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ciafre SA, Galardi S, Mangiola A, et al. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Bioph Res Co. 2005;334:1351–1358. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 93.Kefas B, Godlewski J, Comeau L, et al. microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma. Cancer Res. 2008;68:3566–3572. doi: 10.1158/0008-5472.CAN-07-6639. [DOI] [PubMed] [Google Scholar]
- 94.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 95.Corsten MF, Miranda R, Kasmieh R, et al. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 96.Gabriely G, Yi M, Narayan RS, et al. Human glioma growth is controlled by microRNA-10b. Cancer Res. 2011;71:3563–3572. doi: 10.1158/0008-5472.CAN-10-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Turner JD, Williamson R, Almefty KK, et al. The many roles of microRNAs in brain tumor biology. Neurosurg Focus. 2010;28:E3. doi: 10.3171/2009.10.FOCUS09207. [DOI] [PubMed] [Google Scholar]
- 98.Kim T-M, Huang W, Park R, et al. A developmental taxonomy of glioblastoma defined and maintained by MicroRNAs. Cancer Res. 2011;71:3387–3399. doi: 10.1158/0008-5472.CAN-10-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vecil GG, Lang FF. Clinical trials of adenoviruses in brain tumors: a review of Ad-p53 and oncolytic adenoviruses. J Neuro-Oncol. 2003;65:237–246. doi: 10.1023/b:neon.0000003653.45635.32. [DOI] [PubMed] [Google Scholar]
- 100.Chiu T-L, Lin S-Z, Hsieh W-H, et al. AAV2-mediated interleukin-12 in the treatment of malignant brain tumors through activation of NK cells. Int J Oncol. 2009;35:1361–1367. doi: 10.3892/ijo_00000454. [DOI] [PubMed] [Google Scholar]
- 101.Harding TC, Lalani AS, Roberts BN, et al. AAV serotype 8-mediated gene delivery of a soluble VEGF receptor to the CNS for the treatment of glioblastoma. Mol Ther. 2006;13:956–966. doi: 10.1016/j.ymthe.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 102.Nilaver G, Muldoon LL, Kroll RA, et al. Delivery of herpesvirus and adenovirus to nude rat intracerebral tumors after osmotic blood-brain barrier disruption. Proc Natl Acad Sci USA. 1995;92:9829–9833. doi: 10.1073/pnas.92.21.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pulkkanen KJ, Yla-Herttuala S. Gene therapy for malignant glioma: current clinical status. Mol Ther. 2005;12:585–598. doi: 10.1016/j.ymthe.2005.07.357. [DOI] [PubMed] [Google Scholar]
- 104.Templeton NS, Lasic DD, Frederik PM, et al. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997;15:647–652. doi: 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]
- 105.Chen Y, Bathula SR, Yang Q, et al. Targeted nanoparticles deliver siRNA to melanoma. J Invest Dermatol. 2010;130:2790–2798. doi: 10.1038/jid.2010.222. [DOI] [PubMed] [Google Scholar]
- 106.Kato T, Natsume A, Toda H, et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide in GBM-initiating cells. Gene Ther. 2010;17:1363–1371. doi: 10.1038/gt.2010.88. [DOI] [PubMed] [Google Scholar]
- 107.Gupta B, Levchenko TS, Torchilin VP. TAT peptide-modified liposomes provide enhanced gene delivery to intracranial human brain tumor xenografts in nude mice. Oncology research. 2007;16:351–359. doi: 10.3727/000000006783980946. [DOI] [PubMed] [Google Scholar]
- 108.Jacobs A, Voges J, Reszka R, et al. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet. 2001;358:727–729. doi: 10.1016/s0140-6736(01)05904-9. [DOI] [PubMed] [Google Scholar]
- 109.Han L, Zhang A, Wang H, et al. Tat-BMPs-PAMAM conjugates enhance therapeutic effect of small interference RNA on U251 glioma cells in vitro and in vivo. Hum Gene Ther. 2010;21:417–426. doi: 10.1089/hum.2009.087. [DOI] [PubMed] [Google Scholar]
- 110.Huang R, Ke W, Han L, et al. Targeted delivery of chlorotoxin-modified DNA-loaded nanoparticles to glioma via intravenous administration. Biomaterials. 2011;32:2399–2406. doi: 10.1016/j.biomaterials.2010.11.079. [DOI] [PubMed] [Google Scholar]
- 111.Hwang DW, Son S, Jang J, et al. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials. 2011;32:4968–4975. doi: 10.1016/j.biomaterials.2011.03.047. [DOI] [PubMed] [Google Scholar]
- 112.Gao X, Yao L, Song Q, et al. The association of autophagy with polyethylenimine-induced cytotoxity in nephritic and hepatic cell lines. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 113.Kafil V, Omidi Y. Cytotoxic Impacts of Linear and Branched Polyethylenimine Nanostructures in A431 Cells. BioImpacts. 2011;1:23–30. doi: 10.5681/bi.2011.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tros de Ilarduya C, Sun Y, Duzgunes N. Gene delivery by lipoplexes and polyplexes. Eur J Pharm Sci. 2010;40:159–170. doi: 10.1016/j.ejps.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 115.Lv H, Zhang S, Wang B, et al. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 116.Sunshine JC, Akanda MI, Li D, et al. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules. 2011;12:3592–3600. doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu J, Jiang Z, Zhou J, et al. Enzyme-synthesized poly(amine-co-esters) as nonviral vectors for gene delivery. J Biomed Mater Res A. 2011;96:456–465. doi: 10.1002/jbm.a.32994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou J, Liu J, Jiang Z, et al. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nature Mater. 2011 doi: 10.1038/nmat3187. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou J, Patel T, Fu M, et al. Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials. 2011 doi: 10.1016/j.biomaterials.2011.09.061. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mardor Y, Roth Y, Lidar Z, et al. Monitoring response to convection-enhanced taxol delivery in brain tumor patients using diffusion-weighted magnetic resonance imaging. Cancer Res. 2001;61:4971–4973. [PubMed] [Google Scholar]
- 121.Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100:472–479. doi: 10.3171/jns.2004.100.3.0472. [DOI] [PubMed] [Google Scholar]
- 122.Vogelbaum MA. Convection enhanced delivery for the treatment of malignant gliomas: symposium review. J Neuro-Oncol. 2005;73:57–69. doi: 10.1007/s11060-004-2243-8. [DOI] [PubMed] [Google Scholar]
- 123.Sampson JH, Brady M, Raghavan R, et al. Colocalization of gadolinium-diethylene triamine pentaacetic acid with high-molecular-weight molecules after intracerebral convection-enhanced delivery in humans. Neurosurgery. 2011;69:668–676. doi: 10.1227/NEU.0b013e3182181ba8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mardor Y, Rahav O, Zauberman Y, et al. Convection-enhanced drug delivery: increased efficacy and magnetic resonance image monitoring. Cancer Res. 2005;65:6858–6863. doi: 10.1158/0008-5472.CAN-05-0161. [DOI] [PubMed] [Google Scholar]
- 125.Doiron AL, Chu K, Ali A, et al. Preparation and initial characterization of biodegradable particles containing gadolinium-DTPA contrast agent for enhanced MRI. Proc Natl Acad Sci U S A. 2008;105:17232–17237. doi: 10.1073/pnas.0710205105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ratzinger G, Agrawal P, Korner W, et al. Surface modification of PLGA nanospheres with Gd-DTPA and Gd-DOTA for high-relaxivity MRI contrast agents. Biomaterials. 2010;31:8716–8723. doi: 10.1016/j.biomaterials.2010.07.095. [DOI] [PubMed] [Google Scholar]
- 127.Bass LA, Wang M, Welch MJ, et al. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug Chem. 2000;11:527–532. doi: 10.1021/bc990167l. [DOI] [PubMed] [Google Scholar]
- 128.Hadjipanayis CG, Machaidze R, Kaluzova M, et al. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010;70:6303–6312. doi: 10.1158/0008-5472.CAN-10-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sampson JH, Brady ML, Petry NA, et al. Intracerebral infusate distribution by convection-enhanced delivery in humans with malignant gliomas: descriptive effects of target anatomy and catheter positioning. Neurosurgery. 2007;60 doi: 10.1227/01.NEU.0000249256.09289.5F. ONS89-98; discussion ONS98-9. [DOI] [PubMed] [Google Scholar]
- 130.Merkel OM, Librizzi D, Pfestroff A, et al. In vivo SPECT and real-time gamma camera imaging of biodistribution and pharmacokinetics of siRNA delivery using an optimized radiolabeling and purification procedure. Bioconjug Chem. 2009;20:174–182. doi: 10.1021/bc800408g. [DOI] [PubMed] [Google Scholar]
- 131.Dye M. PET & SPECT: Happy Together. Imaging Economics; 2005. [cited 2011; Available from: http://www.imagingeconomics.com/issues/articles/MI_2005-06_01.asp. [Google Scholar]
- 132.van der Have F, Vastenhouw B, Ramakers RM, et al. U-SPECT-II: An Ultra-High-Resolution Device for Molecular Small-Animal Imaging. J Nucl Med. 2009;50:599–605. doi: 10.2967/jnumed.108.056606. [DOI] [PubMed] [Google Scholar]
- 133.Qian J, Bradley EL, Majewski S, et al. A small-animal imaging system capable of multipinhole circular/helical SPECT and parallel-hole SPECT. Nucl Instrum Methods Phys Res A. 2008;594:102–110. doi: 10.1016/j.nima.2008.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Popperl G, Goldbrunner R, Gildehaus FJ, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET for monitoring the effects of convection-enhanced delivery of paclitaxel in patients with recurrent glioblastoma. Eur J Nucl Med Mol Imaging. 2005;32:1018–1025. doi: 10.1007/s00259-005-1819-7. [DOI] [PubMed] [Google Scholar]
- 135.Schluep T, Hwang J, Hildebrandt IJ, et al. Pharmacokinetics and tumor dynamics of the nanoparticle IT-101 from PET imaging and tumor histological measurements. Proc Natl Acad Sci USA. 2009;106:11394–11399. doi: 10.1073/pnas.0905487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sirianni R, Carson R, Zheng M, et al. Development of dPET, a non-invasive imaging technique to measure the distribution of drugs after direct delivery to the brain. J Nucl Med. 2010;51:829–829. [Google Scholar]
- 137.Pauleit D, Floeth F, Hamacher K, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET combined with MRI improves the diagnostic assessment of cerebral gliomas. Brain. 2005;128:678–687. doi: 10.1093/brain/awh399. [DOI] [PubMed] [Google Scholar]
- 138.Plotkin M, Gneveckow U, Meier-Hauff K, et al. 18F-FET PET for planning of thermotherapy using magnetic nanoparticles in recurrent glioblastoma. Int J Hyperther. 2006;22:319–325. doi: 10.1080/02656730600734128. [DOI] [PubMed] [Google Scholar]
- 139.Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]
- 140.Zhou M, Zhang R, Huang M, et al. A chelator-free multifunctional [64Cu]CuS nanoparticle platform for simultaneous micro-PET/CT imaging and photothermal ablation therapy. JACS. 2010;132:15351–15358. doi: 10.1021/ja106855m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bennewitz M, Saltzman WM. Nanotechnology for delivery of drugs to the brain for epilepsy. Neurotherapeutics. 2009;6:323–336. doi: 10.1016/j.nurt.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Metcalf CA, Bohacek R, Rozamus LW, et al. Structure-based design of AP23573, a phosphorous-containing analog of rapamycin for anti-tumor therapy. Proc Am Assoc Cancer Res. 2004;45:2476. [Google Scholar]
- 144.Mita M, Sankhala K, Abdel-Karim I, et al. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Inv Drug. 2008;17:1947–1954. doi: 10.1517/13543780802556485. [DOI] [PubMed] [Google Scholar]
- 145.Reardon DA, Quinn JA, Vredenburgh JJ, et al. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- 146.Falcon BL, Barr S, Gokhale PC, et al. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 2011;71:1573–1583. doi: 10.1158/0008-5472.CAN-10-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Prasad G, Sottero T, Yang X, et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-Oncology. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 149.Schultz RM, Merriman RL, Andis SL, et al. In vitro and in vivo antitumor activity of the phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer Res. 1995;15:1135–1139. [PubMed] [Google Scholar]
- 150.Yang F, Brown C, Buettner R, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. 2010;9:953–962. doi: 10.1158/1535-7163.MCT-09-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Siegelin MD, Raskett CM, Gilbert CA, et al. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neuroscience Letters. 2010;478:165–170. doi: 10.1016/j.neulet.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wick W, Weller M, Weiler M, et al. Pathway inhibition: emerging molecular targets for treating glioblastoma. Neuro-Oncology. 2011;13:566–579. doi: 10.1093/neuonc/nor039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yung WK, Vredenburgh JJ, Cloughesy TF, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro-Oncology. 2010;12:1061–1070. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang MY, Lu KV, Zhu S, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- 155.Diaz Miqueli A, Rolff J, Lemm M, et al. Radiosensitisation of U87MG brain tumours by anti-epidermal growth factor receptor monoclonal antibodies. Brit J Cancer. 2009;100:950–958. doi: 10.1038/sj.bjc.6604943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Franceschi E, Cavallo G, Lonardi S, et al. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Brit J Cancer. 2007;96:1047–1051. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pfeffer MR, Levitt ML, Aderka D. Gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:2755–2756. doi: 10.1200/JCO.2004.99.299. author reply 2756. [DOI] [PubMed] [Google Scholar]
- 158.Hegi ME, Diserens AC, Bady P, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther. 2011;10:1102–1112. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 159.Goudar RK, Shi Q, Hjelmeland MD, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther. 2005;4:101–112. [PubMed] [Google Scholar]
- 160.Eller JL, Longo SL, Kyle MM, et al. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56:155–162. doi: 10.1227/01.neu.0000145865.25689.55. discussion 162. [DOI] [PubMed] [Google Scholar]
- 161.Thiessen B, Stewart C, Tsao M, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol. 2010;65:353–361. doi: 10.1007/s00280-009-1041-6. [DOI] [PubMed] [Google Scholar]
- 162.Guo D, Prins RM, Dang J, et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci Signal. 2009;2:ra82. doi: 10.1126/scisignal.2000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Strawn LM, Kabbinavar F, Schwartz DP, et al. Effects of SU101 in combination with cytotoxic agents on the growth of subcutaneous tumor xenografts. Clin Cancer Res. 2000;6:2931–2940. [PubMed] [Google Scholar]
- 164.Grossman SA, Phuphanich S, Lesser G, et al. Toxicity, efficacy, and pharmacology of suramin in adults with recurrent high-grade gliomas. J Clin Oncol. 2001;19:3260–3266. doi: 10.1200/JCO.2001.19.13.3260. [DOI] [PubMed] [Google Scholar]
- 165.Laterra JJ, Grossman SA, Carson KA, et al. Suramin and radiotherapy in newly diagnosed glioblastoma: phase 2 NABTT CNS Consortium study. Neuro-Oncology. 2004;6:15–20. doi: 10.1215/S1152851703000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Glass TL, Liu TJ, Yung WK. Inhibition of cell growth in human glioblastoma cell lines by farnesyltransferase inhibitor SCH66336. Neuro-Oncology. 2000;2:151–158. doi: 10.1093/neuonc/2.3.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chaponis D, Barnes JW, Dellagatta JL, et al. Lonafarnib (SCH66336) improves the activity of temozolomide and radiation for orthotopic malignant gliomas. Journal of neurooncology. 2011;104:179–189. doi: 10.1007/s11060-010-0502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cloughesy TF, Wen PY, Robins HI, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24:3651–3656. doi: 10.1200/JCO.2006.06.2323. [DOI] [PubMed] [Google Scholar]
- 169.Lustig R, Mikkelsen T, Lesser G, et al. Phase II preradiation R115777 (tipifarnib) in newly diagnosed GBM with residual enhancing disease. Neuro-Oncology. 2008;10:1004–1009. doi: 10.1215/15228517-2008-070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Moyal EC, Laprie A, Delannes M, et al. Phase I trial of tipifarnib (R115777) concurrent with radiotherapy in patients with glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2007;68:1396–1401. doi: 10.1016/j.ijrobp.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 171.Sarcar B, Kahali S, Chinnaiyan P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J Neuro-Oncol. 2010;99:201–207. doi: 10.1007/s11060-010-0127-7. [DOI] [PubMed] [Google Scholar]
- 172.Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sawa H, Murakami H, Kumagai M, et al. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004;107:523–531. doi: 10.1007/s00401-004-0841-3. [DOI] [PubMed] [Google Scholar]
- 174.Zhu H, Woolfenden S, Bronson RT, et al. The novel Hsp90 inhibitor NXD30001 induces tumor regression in a genetically engineered mouse model of glioblastoma multiforme. Mol Cancer Ther. 2010;9:2618–2626. doi: 10.1158/1535-7163.MCT-10-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Régina A, Demeule M, Ché C, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Brit J Pharmacol. 2008;155:185–197. doi: 10.1038/bjp.2008.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Thomas FC, K T, V R, et al. Uptake of ANG1005, a novel paclitaxel derivative, through the blood-brain barrier into brain and experimental brain metastases of breast cancer. Pharm Res. 2009;26:2486–2494. doi: 10.1007/s11095-009-9964-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.De Groot JF, Prados M, Urqhart T, et al. A phase II study of XL184 in patients with progressive glioblastoma multiforme (GBM) in first or second relapse. J Clin Oncol. 2009;27:2047. [Google Scholar]
- 178.Wen PY, Prados M, Schiff D, et al. Phase 2 Study of XL184 (BMS-907351), an Inhibitor of MET, VEGFR2, and RET in Patients with Progressive Glioblastoma. J Clin Oncol. 2010;28:15s. [Google Scholar]
- 179.Price A, Shi Q, Morris D, et al. Marked inhibition of tumor growth in a malignant glioma tumor model by a novel synthetic matrix metalloproteinase inhibitor AG3340. Clin Cancer Res. 1999;5:845–854. [PubMed] [Google Scholar]
- 180.Levine PA, Phuphanich S, Glantz MJ, et al. Randomized phase II study of temozolomide (TMZ) with and without the metallopro-tease (MMP) inhibitor prinomastat in patients with glioblastoma multiforme (GBM) following best surgery and radiation therapy. Proc Annu Meet Am Soc Clin Oncol. 2002;21:100. [Google Scholar]
- 181.Milano V, Piao Y, LaFortune T, et al. Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther. 2009;8:394–406. doi: 10.1158/1535-7163.MCT-08-0669. [DOI] [PubMed] [Google Scholar]
- 182.Lu-Emerson C, Norden AD, Drappatz J, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. J Neuro-Oncol. 2010;104:287–291. doi: 10.1007/s11060-010-0489-x. [DOI] [PubMed] [Google Scholar]
- 183.Iwamoto FM, Lamborn KR, Robins HI, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02) Neuro-Oncology. 2010;12:855–861. doi: 10.1093/neuonc/noq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Razis E, Selviaridis P, Labropoulos S, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. 2009;15:6258–6266. doi: 10.1158/1078-0432.CCR-08-1867. [DOI] [PubMed] [Google Scholar]
- 185.Dresemann G. Imatinib and hydroxyurea in pretreated progressive glioblastoma multiforme: a patient series. Ann Oncol. 2005;16:1702–1708. doi: 10.1093/annonc/mdi317. [DOI] [PubMed] [Google Scholar]
- 186.Butowski N, Chang SM, Lamborn KR, et al. Enzastaurin plus temozolomide with radiation therapy in glioblastoma multiforme: a phase I study. Neuro-Oncology. 2010;12:608–613. doi: 10.1093/neuonc/nop070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kreisl TN, Kim L, Moore K, et al. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin Cancer Res. 2009;15:3617–3623. doi: 10.1158/1078-0432.CCR-08-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168–1174. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Unterkircher T, Cristofanon S, Vellanki SH, et al. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res. 2011;17:4019–4030. doi: 10.1158/1078-0432.CCR-11-0075. [DOI] [PubMed] [Google Scholar]
- 190.Balyasnikova IV, Ferguson SD, Han Y, et al. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 2011;310:148–159. doi: 10.1016/j.canlet.2011.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pedeboscq S, L'Azou B, Passagne I, et al. Cytotoxic and apoptotic effects of bortezomib and gefitinib compared to alkylating agents on human glioblastoma cells. J Exp Ther Oncol. 2008;7:99–111. [PubMed] [Google Scholar]
- 192.Kardosh A, Golden EB, Pyrko P, et al. Aggravated Endoplasmic Reticulum Stress as a Basis for Enhanced Glioblastoma Cell Killing by Bortezomib in Combination with Celecoxib or Its Non-Coxib Analogue, 2,5-Dimethyl-Celecoxib. Cancer Res. 2008;68:843–851. doi: 10.1158/0008-5472.CAN-07-5555. [DOI] [PubMed] [Google Scholar]
- 193.Phuphanich S, Supko JG, Carson KA, et al. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neuro-Oncol. 2010;100:95–103. doi: 10.1007/s11060-010-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gerstner ER, Eichler AF, Plotkin SR, et al. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. J Neuro-Oncol. 2011;103:325–332. doi: 10.1007/s11060-010-0390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Morabito A, De Maio E, Di Maio M, et al. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist. 2006;11:753–764. doi: 10.1634/theoncologist.11-7-753. [DOI] [PubMed] [Google Scholar]
- 196.Homsi J, Daud AI. Spectrum of activity and mechanism of action of VEGF/PDGF inhibitors. Cancer Control. 2007;14:285–294. doi: 10.1177/107327480701400312. [DOI] [PubMed] [Google Scholar]
- 197.Michaud K, Solomon DA, Oermann E, et al. Pharmacologic inhibition of cdk4/6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228–3238. doi: 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wiedemeyer WR, Dunn IF, Quayle SN, et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci USA. 2010;107:11501–11506. doi: 10.1073/pnas.1001613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pore N, Gupta AK, Cerniglia GJ, et al. HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia (New York, N Y) 2006;8:889–895. doi: 10.1593/neo.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tian X, Ye J, Alonso-Basanta M, et al. Modulation of CCAAT/enhancer binding protein homologous protein (CHOP)-dependent DR5 expression by nelfinavir sensitizes glioblastoma multiforme cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) J Biol Chem. 2011;286:29408–29416. doi: 10.1074/jbc.M110.197665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pyrko P, Kardosh A, Wang W, et al. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007;67:10920–10928. doi: 10.1158/0008-5472.CAN-07-0796. [DOI] [PubMed] [Google Scholar]
- 202.Guzman M, Sanchez C, Galve-Roperh I. Control of the cell survival/death decision by cannabinoids. J Mol Med (Berl) 2001;78:613–625. doi: 10.1007/s001090000177. [DOI] [PubMed] [Google Scholar]
- 203.Galve-Roperh I, Sanchez C, Cortes ML, et al. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- 204.Sanchez C, Galve-Roperh I, Canova C, et al. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- 205.Hu SS, Bradshaw HB, Benton VM, et al. The biosynthesis of N-arachidonoyl dopamine (NADA), a putative endocannabinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot Essent Fatty Acids. 2009;81:291–301. doi: 10.1016/j.plefa.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Davies JW, Hainsworth AH, Guerin CJ, et al. Pharmacology of capsaicin-, anandamide-, and N-arachidonoyl-dopamine-evoked cell death in a homogeneous transient receptor potential vanilloid subtype 1 receptor population. Brit J Anaesth. 2010;104:596–602. doi: 10.1093/bja/aeq067. [DOI] [PubMed] [Google Scholar]
- 207.Chu CJ, Huang SM, De Petrocellis L, Bisogno T, Ewing Sa, Miller JD, Zipkin RE, et al. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J Biol Chem. 2003;278:13633–13639. doi: 10.1074/jbc.M211231200. [DOI] [PubMed] [Google Scholar]
- 208.Xia S, Li Y, Rosen EM, et al. Ribotoxic stress sensitizes glioblastoma cells to death receptor induced apoptosis: requirements for c-Jun NH2-terminal kinase and Bim. Mol Cancer Res. 2007;5:783–792. doi: 10.1158/1541-7786.MCR-06-0433. [DOI] [PubMed] [Google Scholar]
- 209.Chau N-M, Rogers P, Aherne W, et al. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1alpha induction in response to hypoxic stress and growth factors. Cancer Res. 2005;65:4918–4928. doi: 10.1158/0008-5472.CAN-04-4453. [DOI] [PubMed] [Google Scholar]
- 210.Kong H, Lee S, Beebe K, et al. Emetine Promotes von Hippel-Lindau-Independent Degradation of Hypoxia-Inducible Factor-2 in Clear Cell Renal Carcinoma. Molecular Pharmacology. 2010;78:1072–1078. doi: 10.1124/mol.110.066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Takamiya Y, Brem H, Ojeifo J, et al. AGM-1470 inhibits the growth of human glioblastoma cells in vitro and in vivo. Neurosurgery. 1994;34:869–875. doi: 10.1227/00006123-199405000-00013. discussion 875. [DOI] [PubMed] [Google Scholar]
- 212.Bar EE, Chaudhry A, Lin A, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]