

Abstract
Rheumatoid arthritis (RA) is a chronic autoimmune disease which primarily affects the synovial joints leading to inflammation, pain and joint deformities. Nonsteroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids, both of which inhibit cyclooxygenase (COX), have been extensively used for treating RA patients. Prostaglandin E synthase (PGES) is a specific biosynthetic enzyme that acts downstream of COX and converts prostaglandin (PG) H2 to PGE2. Among PGES isozymes, microsomal PGES-1 (mPGES-1) has been shown to be induced in a variety of cells and tissues under inflammatory conditions. The induction of mPGES-1 in the synovial tissue of RA patients is closely associated with the activation of the tissue by proinflammatory cytokines. Although selective mPGES-1 inhibitors have not yet been widely available, mice lacking mPGES-1 (mPGES-1–/– mice) have been generated to evaluate the physiological and pathological roles of mPGES-1 in vivo. Recent studies utilizing mPGES-1–/– mice have demonstrated the significance of mPGES-1 in the process of chronic inflammation and evocation of humoral immune response in autoimmune arthritis models. These recent findings highlight mPGES-1 as a novel therapeutic target for the treatment of autoimmune inflammatory diseases, including RA. Currently, both natural and synthetic chemicals are being tested for inhibition of mPGES-1 activity to produce PGE2. The present review focuses on the recent advances in understanding the role of mPGES-1 in the pathophysiology of RA.
Keywords: inflammation, microsomal prostaglandin E synthase-1, prostaglandin E2, rheumatoid arthritis, T-cell-dependent humoral immunity
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune inflammatory disease characterized by synovial inflammation, excessive synovial proliferation and progressive joint destruction. However, the etiological factors underlying RA are not yet fully understood. During active RA, inflammatory cells such as monocytes/macrophages, mast cells, T cells and B cells infiltrate the synovial joints. The immune cells interact in a complex and intricate manner leading to the release of pro-inflammatory mediators 1. High levels of proinflammatory cytokines including tumor necrosis factor-α (TNFα), interleukin (IL) -1 and IL-17 are detectable in the joint fluids and synovium of RA patients. These proinflammatory mediators have been demonstrated to play a vital role in the initiation and development of RA 2, 3. In addition, these cytokines are closely associated with the production of biologically active lipid mediators, including prostaglandin (PG) E2.
PGE2 is highly detectable in the synovial fluid of patients with RA 4. Proinflammatory cytokine-activated cells, including synovial cells, chondrocytes, macrophages and monocytes, are the primary sources of PGE2 in the inflamed joints of RA patients. Biosynthesis of the PGE2 is dependent on sequential enzymatic processes beginning with the release of arachidonic acid from membrane phospholipids by phospholipase. The arachidonic acid derived from the first step is further metabolized to PGH2 by cyclooxygenase (COX), which consists of two major isozymes, COX-1 and COX-2, as shown in Fig. 1. Under basal conditions, COX-1 is constitutively expressed in most tissues, while COX-2 is significantly upregulated in specific types of cells and tissues, including the synovial tissue. Since PGE2 production coincides with the up-regulation of COX-2 expression in cells of inflamed joint tissues 5, COX-2 has been targeted for the treatment of RA by using non-steroidal anti-inflammatory drugs (NSAIDs) including selective COX-2 inhibitors 6.
Fig. 1. Pathway of prostaglandin E2 biosynthesis.
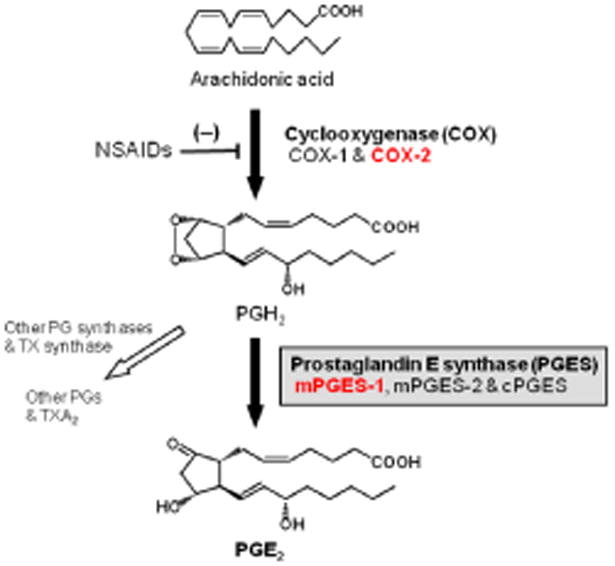
Arachidonic acid is converted to the unstable metabolite, prostaglandin (PG) H2 by two isozymes of cyclooxygenase (COX), constitutive COX-1 and inducible COX-2. PGH2 is then converted to PGE2 by prostaglandin E synthase (PGES). There are at least 3 isozymes of PGES, including microsomal PGES (mPGES) -1, mPGES-2 and cytosolic PGES (cPGES). PGH2 is also metabolized to other PGs by each specific PG terminal synthases (PGDS for PGD2, PGFS for PGF2α and PGIS for PGI2) and to thromboxane (TX) A2 by TXS. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit PGs and TXA2 production by inhibiting COXs.
Prostaglandin E synthase (PGES) is an enzyme that acts downstream of COX-2 and catalyzes the final step of PGE2 biosynthesis (Fig. 1). To date, three isoforms of PGES, cytosolic PGES (cPGES), microsomal PGES (mPGES) -1 and mPGES-2, have been cloned and characterized. Among the PGES isozymes, mPGES-1 was originally known as microsomal glutathione S-transferase 1-like 1 (MGST1-L1), and it is a glutathione-dependent enzyme. Most importantly, mPGES-1 shows coordinated induction with COX-2 under inflammatory conditions in various cells and tissues 7, 8. Although cPGES is also a glutathione dependent enzyme, it is expressed in most tissues under basal conditions and is not upregulated under inflammatory conditions 9. In contrast, mPGES-2 is not dependent on glutathione for its activity and is coupled to both COX-1 and COX-2 for PGE2 production. Similar to cPGES, it is constitutively expressed and its activity is not significantly increased during tissue inflammation 10. Thus, mPGES-1 could be considered to be the most promising PGES isozyme that might be utilized as a therapeutic target to treat inflammatory diseases. The present review focuses on the recent advances in understanding the roles of mPGES-1 in the pathophysiology of RA.
Induction of mPGES-1 in joint tissues and cells of RA patients
We have previously demonstrated that mPGES-1 and COX-2 are coordinately induced in cultured synovial fibroblasts obtained from RA patients, upon stimulation by pro-inflammatory cytokines such as IL-1β and TNF-α 11, 12. Interestingly, the low levels of mPGES-1 and COX-2 expression are observed in the untreated multi-passaged cells from RA patients, but fleshly isolated cells highly express both enzymes without stimulation with exogenous proinflammatory cytokines. In cytokine stimulated cells, mPGES-1 and COX-2 are co-localized in the perinuclear region and cooperate to increase PGE2 production 12. In contrast to mPGES-1, cPGES and mPGES-2 are expressed constitutively in RA synovial fibroblasts and their expressions are not affected by proinflammatory cytokine stimulation 13. Furthermore, these studies also indicated that mPGES-1 expression can be inhibited by glucocorticoids. Similar regulation of mPGES-1 expression has also been observed in chondrocytes 14.
In the synovium of RA patients, expression of mPGES-1 has been found to be localized in synovial lining cells, mononuclear and fibroblast-like cells present in the sublining, infiltrating synovial macrophages and vascular endothelial cells. Moreover, mPGES-1 expression was upregulated in synovium selectively in active RA and was minimally expressed in synovial tissue during inactive RA 10, 15. However, during both active and inactive RA, mPGES-2 expression was unchanged in the synovial tissue. In addition to synovium and infiltrating cells, mPGES-1 expression has also been detected in the chondrocytes obtained from articular cartilage 16. Although cPGES is also expressed in the synovial lining, sublining, and vascular endothelium, the number of cPGES-positive cells is significantly lower than that of mPGES-1-positive cells 15. These findings suggest that induction of mPGES-1 during active RA plays a major role in the production of PGE2, while mPGES-2 and/or cPGES may play a minor role in the production of PGE2 in RA.
We unexpectedly found that NSAIDs, including selective COX-2 inhibitors, downregulate mPGES-1 expression in IL-1β-stimulated RA synovial fibroblasts 13. This inhibition was relieved by the addition of PGE2. It has been well known that PGE2 exerts its role by binding different G-protein coupled receptors, EP1, EP2, EP3 and EP4. However, stimulation of only EP2 and EP4 receptors has been demonstrated to increase cyclic AMP via activation of adenylate cyclase 17. We demonstrated expression of both EP2 and EP4 receptors in synovial fibroblasts obtained from RA patients. Stimulation of EP2 and EP4 receptors on synovial fibroblasts by either PGE2 or selective agonists for the EP2 and EP4 or an activator of adenylate cyclase, forskolin, results in significant upregulation of mPGES-1 expression. This suggests that PGE2 enhances the expression of mPGES-1 in RA synovial fibroblasts by increasing cAMP through activation of EP2 and EP4. We also obtained similar results using chondrocytes. Hence, PGE2 may be a strong enhancer of mPGES-1 expression in synovial fibroblasts and chondrocytes. This positive feedback between mPGES-1 expression and PGE2 may play an important role in amplifying inflammation associated with RA. Fig. 2 demonstrates the positive feedback mechanism regulating mPGES-1 upregulation and PGE2 production.
Fig. 2. Regulation of mPGES-1 expression in rheumatoid synovial fibroblasts and chondrocytes.
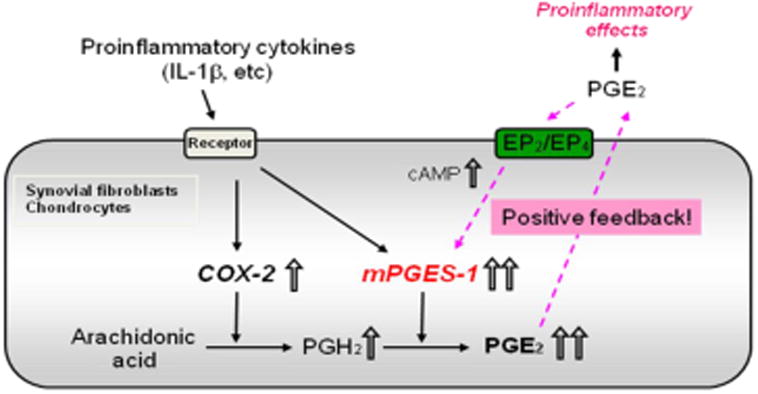
COX-2 and mPGES-1 are coordinately induced in cultured synovial fibroblasts and chondrocytes by stimulation of pro-inflammatory cytokines such as interleukin1β (IL-1β). Abundant PGE2 production at the inflammation sites of rheumatoid arthritis (RA) is caused by the coordinated up-regulation of mPGES-1 and COX-2. PGE2 further enhances mPGES-1 expression associated with increase of cyclic AMP (cAMP) via the EP2 and EP4 receptors. The positive feedback regulation of mPGES-1 expression by PGE2 may play an important role in the vicious circle of inflammation associated with RA 11-14.
Interestingly, Kusunoki et al demonstrated that adiponectin, an important adipokine, also induces the expression of mPGES-1 as well as COX-2, resulting in the increase of PGE2 production by RA synovial fibroblasts 18. This indicates that adiponectin may play a role in the pathogenesis of synovitis in RA patients by regulating PGE2 biosynthesis. The expression of mPGES-1 has been also reported to be regulated by several intracellar factors. Naraba et al demonstrated that the induction of mPGES-1 expression requires the presence of tandem GC boxes adjacent to the transcription initiation site. The group further demonstrated that the binding of the transcription factor Egr-1 to the proximal GC box is essential for transcriptional regulation of the human and murine mPGES-1 gene 19, 20. The induction of mPGES-1 is also positively regulated by a MAP kinases such as ERK-1/2 and p38 21. The induction of mPGES-1 may therefore be regulated by multiple signaling factors in RA.
Effect of genetic deletion of mPGES-1 on experimental autoimmune inflammatory arthritis in mice
Mice lacking mPGES-1 (mPGES-1–/– mice) have been generated and have provided novel insights into the role of mPGES-1 in eicosanoid biology 22, 23. We and other groups have independently shown that mPGES-1 deficiency results in a reduction in the severity of arthritis, along with decreased synovial hyperplasia, tissue destruction and infiltration of inflammatory cells in type II collagen (CII) induced arthritis (CIA), a representative animal model of RA 22, 24. We have also shown a significant reduction in pain perception after mechanical stimulation in CIA. Previous studies have demonstrated that mice lacking COX-2 (COX-2–/– mice) also display a significant reduction in both clinical and histological arthritis in CIA 25. Likewise, administration of a COX-2 inhibitor, but not a COX-1 inhibitor, reduces paw inflammation 26. In addition, another CIA study, utilizing mice lacking the EP2 receptor and pharmacological inhibition of EP4, supports the essential role of PGE2 mediated by EP2/EP4 signaling in the development of autoimmune arthritis 27. Taken together, the data indicates that COX-2/mPGES-1-derived PGE2 may play a role in the development of RA, predominantly via EP2/EP4 signaling.
Role of mPGES-1 in T-cell dependent humoral immune response
RA is strongly dependent on the immunologic events associated with autoimmunity. Generation of a humoral immune response is a critical event in the development of autoimmune diseases, including RA. Humoral immune response can be divided into T-cell dependent (TD) and T-cell independent (TI) response, based on the requirement for T-cell activation (Fig. 3). TD response requires T-cell/B-cell interaction via interaction between cell surface receptors and ligands to initiate antibody production. In contrast, TI response does not require T-cell interaction and antibody production occurs after B-cell activation, following binding of antigens to B-cell receptors (BCR).
Fig. 3. mPGES-1 mediates T-cell dependent humoral immune response.
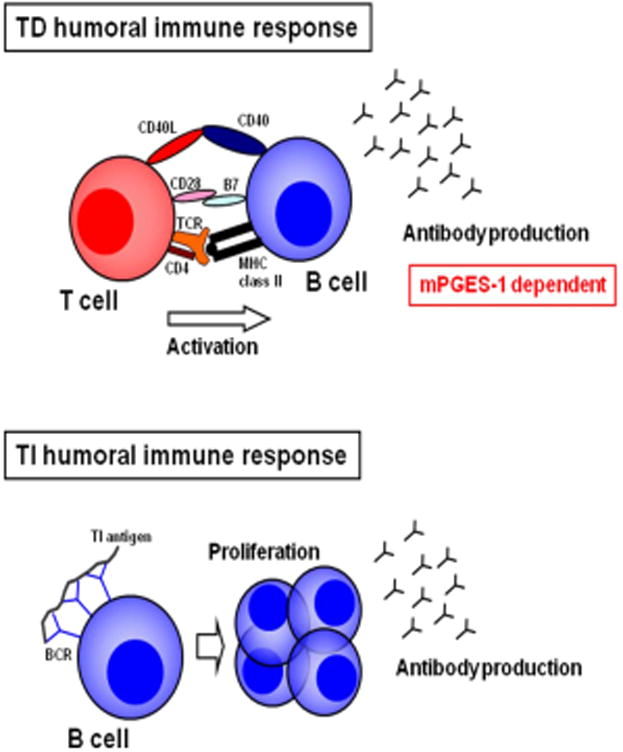
The humoral immune responses, a critical event for autoimmune diseases, are divided into T-cell dependent (TD) and T-cell independent (TI) response, according to their requirement of T-cells. TD response requires T-cell/B-cell contact via interaction of cell surface molecules and ligands to develop antibody production by B-cells. In contrast, TI response does not require T-cells and the antibody production directly occurs after B-cell activation followed by binding of antigens to B-cell receptor (BCR) on B-cells. mPGES-1 and PGE2-dependent action plays a key role in evocation of TD humoral response.
PGE2 has been reported to modulate immunologic events including dendritic cell maturation, macrophage activation, B-cell and T-cell function 28. However, the immunomodulatory role of mPGES-1 was unclear. We have shown that the levels of type II collagen (CII) -specific IgG subclasses (total IgG, IgG1, IgG2a, IgG2b, IgG2c and IgG3) are significantly reduced in mPGES-1–/– mice during the development of CIA, when compared to WT mice 24. The reduction of CII-specific antibody production correlates with the reduction in the incidence and severity of arthritis, suggesting an important role of mPGES-1 and its derived PGE2 in the development of acquired immune response in CIA. The changes observed in the immunological responses of mPGES-1–/– mice in our study are consistent with previous observations in COX-2–/– mice during CIA 25, suggesting the pivotal role of COX-2/mPGES-1/PGE2 axis in the development of an antibody response to CII, a classic TD antigen that induces humoral and cellular immunity. These findings provide novel insights relevant to the therapeutic potential for pharmacologic inhibition of mPGES-1 in autoimmune inflammatory diseases, including RA.
Role of mPGES-1 in altering the cytokine profile
Recent studies have demonstrated the key role of Th17, a major subset of T-cells producing IL-17, in the pathogenesis of arthritis 29. PGE2 has been shown to facilitate expansion of the Th17 subset of T helper cells via its specific receptor subtypes 30, 31. Hence, we examined the effect of mPGES-1 gene deletion on the production of IL-17 in cultured splenocytes obtained on day 10 following immunization with CII in the CIA model. Levels of IL-17 were significantly increased after CII stimulation in splenocytes of WT mice. However, significantly lower levels of IL-17 were observed in mPGES-1–/– splenocytes compared to WT on exposure to CII (Fig. 4). Likewise, the genetic deletion of mPGES-1 resulted in a failure to increase PGE2 production in mPGES-1–/– splenocytes compared with WT after stimulation with CII, demonstrating the essential role of mPGES-1 in producing PGE2 in this model 32. The relative decrease in production of IL-17 associated with the reduction of PGE2 production from CII-stimulated splenocytes in mPGES-1–/– mice vs. WT mice in vitro suggests that the mPGES-1 and its derivative PGE2 may alter the differentiation and/or maintenance of IL-17-producing Th17 subset of T-cells in CIA. This altered cytokine environment may be one of the mechanisms by which immunoglobulin levels and arthritis severity are reduced in mPGES-1–/– mice in vivo in the CIA model 24.
Fig. 4. mPGES-1-PGE2 mediates Th17 responses in CIA.
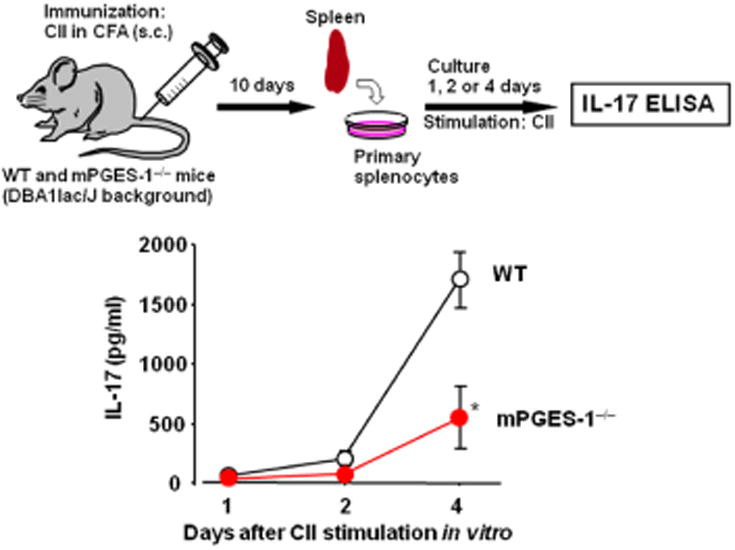
Splenocytes were isolated from WT and mPGES-1–/– mice at day 10 after immunization with bovine type II collagen (CII) in complete Freund's adjuvant (CFA). The cells were then cultured with CII (100 μg/ml) for 1, 2 or 4 days in vitro. The levels of interleukin-17 (IL-17) in culture medium were measured by ELISA. The production of IL-17 in CII-stimulated mPGES-1–/– splenocytes was significantly lower than that in WT cells (n=3; *P<0.05). mPGES-1-PGE2 system may alter the differentiation and/or maintenance of Th17 lymphocytes in collagen-induced arthritis (CIA) 32.
The effects of PGE2 on dendritic cell phenotype and function have also been the subject of investigation. Our recent study with mPGES-1–/– mice showed that mPGES-1 is required for PGE2 production after lipopolysaccharide (LPS) stimulation in dendritic cells derived from bone marrow in vitro 33. Although mPGES-1–/– dendritic cells exhibit normal maturation and migration, the level of IL-12, a critical Th1 polarizing cytokine, after LPS stimulation is significantly lower when compared to WT cells. Interestingly, we observed that mPGES-1 deletion results in diversion of prostanoid production from PGE2 to PGD2, suggesting shunting of prostanoid biosynthesis in dendritic cells associated with mPGES-1 deficiency. In addition, the increase in PGD2, and not decreased PGE2, may be the likely cause of diminished IL-12 production in LPS-stimulated mPGES-1–/– dendritic cells. Taken together, mPGES-1 may be an attractive target for the treatment of autoimmune inflammatory diseases such as RA by altering cytokine production of immune effector cells.
Inhibition of mPGES-1 and its future prospect
Selective COX-2 inhibitors are extensively used for the treatment of RA. However, several clinical trials demonstrated that some of selective COX-2 inhibitors increase the risk of cardiovascular events in RA patients 34. One of the possible mechanisms to explain the cardiovascular side effects due to COX-2 inhibition may be the concomitant decrease in production of PGE2 and anti-thrombotic PGI2 35. In order to minimize the side effect of selective COX-2 inhibitors, selective suppression of mPGES-1-derived PGE2 production will be an attractive therapeutic alternative. In fact, loss of mPGES-1 leads to significant reduction in PGE2 production and increase in PGI2 production with no alterations in blood pressure or thrombosis in mice, when fed with normal or high salt diet 36, 37. In addition, the deletion of mPGES-1 delays the development of atherosclerosis in parallel with reduction in PGE2 synthesis and increased PGI2 production 38.
To date, selective mPGES-1 inhibitors are not widely available, but some synthetic and natural chemicals that inhibit mPGES-1 have been investigated in vivo and in vitro by several groups. The chemicals that have been reported for their specific and/or non-specific inhibitory effects on mPGES-1 activity to produce PGE2 are shown in Fig. 539-46. Notably, several studies have shown that some of these compounds are potent inhibitors of human mPGES-1, but do not inhibit murine mPGES-1. Hence, human-mPGES-1 knock-in mice have been generated and are being utilized to determine the efficacy of these compounds in vivo. It will be very important to assess if mPGES-1 inhibitors can mimic the results observed in mPGES-1–/– mice. In addition, therapeutic efficacy and safety of mPGES-1 inhibitors need to be critically studied in humanized animal models.
Fig. 5. Synthetic inhibitors targeting mPGES-1 activity.
Synthetic chemicals that have been reported their specific and/or non-specific inhibitory effects on mPGES-1 activity to produce PGE2 are indicated in this figure. The name and chemical structure of each inhibitor are from references 39-46.
Acknowledgments
We would like to thank Dr. Jerold G. Woodward (University of Kentucky), Dr. Mohit Kapoor (University of Montreal), Dr. Lorenzo Polenzani (Angelini Research Center) and Dr. Natsuko Kusunoki (Toho University School of Medicine) for their suggestions. This work was supported by the NIH/NIAMS R01 AR049010, Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and grant from the Hokkaido Heart Association.
References
- 1.Szekanecz Z, Koch AE. Update on synovitis. Curr Rheumatol Rep. 2001;3:53–63. doi: 10.1007/s11926-001-0051-0. [DOI] [PubMed] [Google Scholar]
- 2.Kuligowska M, Odrowaz-Sypniewska G. Role of interleukin-17 in cartilage and bone destruction in rheumatoid arthritis. Ortop Traumatol Rehabil. 2004;6:235–241. [PubMed] [Google Scholar]
- 3.Zwerina J, Redlich K, Schett G, Smolen JS. Pathogenesis of rheumatoid arthritis: targeting cytokines. Ann N Y Acad Sci. 2005;1051:716–729. doi: 10.1196/annals.1361.116. [DOI] [PubMed] [Google Scholar]
- 4.Egg D, Gunther R, Herold M, Kerschbaumer F. Prostaglandins E2 and F2 alpha concentrations in the synovial fluid in rheumatoid and traumatic knee joint diseases. Z Rheumatol. 1980;39:170–175. [PubMed] [Google Scholar]
- 5.Crofford LJ. COX-2 in synovial tissues. Osteoarthritis Cartilage. 1999;7:406–408. doi: 10.1053/joca.1999.0226. [DOI] [PubMed] [Google Scholar]
- 6.Crofford LJ, Lipsky PE, Brooks P, Abramson SB, Simon LS, van de Putte LB. Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum. 2000;43:4–13. doi: 10.1002/1529-0131(200001)43:1<4::AID-ANR2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 7.Murakami M, Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 8.Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J Biol Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 10.Murakami M, Nakashima K, Kamei D, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 11.Stichtenoth DO, Thoren S, Bian H, Peters-Golden M, Jakobsson PJ, Crofford LJ. Microsomal prostaglandin E synthase is regulated by proinflammatory cytokines and glucocorticoids in primary rheumatoid synovial cells. J Immunol. 2001;167:469–474. doi: 10.4049/jimmunol.167.1.469. [DOI] [PubMed] [Google Scholar]
- 12.Kojima F, Naraba H, Sasaki Y, Okamoto R, Koshino T, Kawai S. Coexpression of microsomal prostaglandin E synthase with cyclooxygenase-2 in human rheumatoid synovial cells. J Rheumatol. 2002;29:1836–1842. [PubMed] [Google Scholar]
- 13.Kojima F, Naraba H, Sasaki Y, Beppu M, Aoki H, Kawai S. Prostaglandin E2 is an enhancer of inter-leukin-1beta-induced expression of membrane-associated prostaglandin E synthase in rheumatoid synovial fibroblasts. Arthritis Rheum. 2003;48:2819–2828. doi: 10.1002/art.11261. [DOI] [PubMed] [Google Scholar]
- 14.Kojima F, Naraba H, Miyamoto S, Beppu M, Aoki H, Kawai S. Membrane-associated prostaglandin E synthase-1 is upregulated by proinflammatory cytokines in chondrocytes from patients with osteoarthritis. Arthritis Res Ther. 2004;6:R355–365. doi: 10.1186/ar1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westman M, Korotkova M, af Klint E, et al. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 2004;50:1774–1780. doi: 10.1002/art.20286. [DOI] [PubMed] [Google Scholar]
- 16.Kojima F, Kato S, Kawai S. Prostaglandin E synthase in the pathophysiology of arthritis. Fundam Clin Pharmacol. 2005;19:255–261. doi: 10.1111/j.1472-8206.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 17.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 18.Kusunoki N, Kitahara K, Kojima F, et al. Adiponectin stimulates prostaglandin E2 production in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010;62:1641–1649. doi: 10.1002/art.27450. [DOI] [PubMed] [Google Scholar]
- 19.Naraba H, Yokoyama C, Tago N, et al. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J Biol Chem. 2001;277:28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- 20.Subbaramaiah K, Yoshimatsu K, Scherl E, et al. Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease. Evidence for involvement of the transcription factor Egr-1. J Biol Chem. 2004;279:12647–12658. doi: 10.1074/jbc.M312972200. [DOI] [PubMed] [Google Scholar]
- 21.Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004;50:2829–2838. doi: 10.1002/art.20437. [DOI] [PubMed] [Google Scholar]
- 22.Trebino CE, Stock JL, Gibbons CP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E2 production is regulated by the glutathione-dependent prostaglandin E2 synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 24.Kojima F, Kapoor M, Yang L, et al. Defective generation of a humoral immune response is associated with a reduced incidence and severity of collagen-induced arthritis in microsomal prostaglandin E synthase-1 null mice. J Immunol. 2008;180:8361–8368. doi: 10.4049/jimmunol.180.12.8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myers LK, Kang AH, Postlethwaite AE, et al. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 2000;43:2687–2693. doi: 10.1002/1529-0131(200012)43:12<2687::AID-ANR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Ochi T, Ohkubo Y, Mutoh S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem Pharmacol. 2003;66:1055–1060. doi: 10.1016/s0006-2952(03)00420-9. [DOI] [PubMed] [Google Scholar]
- 27.Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J Exp Med. 2006;203:325–335. doi: 10.1084/jem.20051310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. J Pharmacol Sci. 2010;112:1–5. doi: 10.1254/jphs.09r03cp. [DOI] [PubMed] [Google Scholar]
- 29.Lubberts E. Th17 cytokines and arthritis. Semin Immunopathol. 2010;32:43–53. doi: 10.1007/s00281-009-0189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao C, Sakata D, Esaki Y, et al. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 31.Boniface K, Bak-Jensen KS, Li Y, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kojima F, Kottangada PC, Yang L, Wang X, Fleishaker EL, Crofford LJ. Keystone Symposia, TH17 Cells in Health and Disease. Vancouver; British Columbia, Canada: 2009. [Google Scholar]
- 33.Monrad SU, Kojima F, Kapoor M, et al. Genetic deletion of mPGES-1 abolishes PGE2 production in murine dendritic cells and alters the cytokine profile, but does not affect maturation or migration. Prostaglandins Leukot Essent Fatty Acids. 2011;84:113–121. doi: 10.1016/j.plefa.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 35.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapoor M, Kojima F, Qian M, Yang L, Crofford LJ. Shunting of prostanoid biosynthesis in microsomal prostaglandin E synthase-1 null embryo fibroblasts: regulatory effects on inducible nitric oxide synthase expression and nitrite synthesis. FASEB J. 2006;20:2387–2389. doi: 10.1096/fj.06-6366fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci U S A. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koeberle A, Siemoneit U, Buhring U, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- 40.Xu D, Rowland SE, Clark P, et al. MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326:754–763. doi: 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]
- 41.Gosselin F, Lau S, Nadeau C, Trinh T, O'Shea PD, Davies IW. A practical synthesis of m-prostaglandin E synthase-1 inhibitor MK-7285. J Org Chem. 2009;74:7790–7797. doi: 10.1021/jo901798d. [DOI] [PubMed] [Google Scholar]
- 42.Koeberle A, Haberl EM, Rossi A, et al. Discovery of benzo[g]indol-3-carboxylates as potent inhibitors of microsomal prostaglandin E2 synthase-1. Bioorg Med Chem. 2009;17:7924–7932. doi: 10.1016/j.bmc.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 43.Koeberle A, Rossi A, Zettl H, et al. The molecular pharmacology and in vivo activity of 2-(4-chloro-6-(2,3-dimethylphenylamino)pyrimidin-2-ylthio) octanoic acid (YS121), a dual inhibitor of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Pharmacol Exp Ther. 2010;332:840–848. doi: 10.1124/jpet.109.160663. [DOI] [PubMed] [Google Scholar]
- 44.Bruno A, Di Francesco L, Coletta I, et al. Effects of AF3442 [N-(9-ethyl-9H-carbazol-3-yl)-2-(trifluoromethyl)benzamide], a novel inhibitor of human microsomal prostaglandin E synthase-1, on prostanoid biosynthesis in human monocytes in vitro. Biochem Pharmacol. 2010;79:974–981. doi: 10.1016/j.bcp.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 45.Pawelzik SC, Uda NR, Spahiu L, et al. Identification of key residues determining species differences in inhibitor binding of microsomal prostaglandin E synthase-1. J Biol Chem. 2010;285:29254–29261. doi: 10.1074/jbc.M110.114454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mbalaviele G, Pauley AM, Shaffer AF, et al. Distinction of microsomal prostaglandin E synthase-1 (mPGES-1) inhibition from cyclooxygenase-2 inhibition in cells using a novel, selective mPGES-1 inhibitor. Biochem Pharmacol. 2010;79:1445–1454. doi: 10.1016/j.bcp.2010.01.003. [DOI] [PubMed] [Google Scholar]