

Abstract
A new enantioselective α-oxidation of aldehydes has been accomplished using TEMPO and a synergistic combination of copper and organic catalysis. Expanding upon recently reported mechanistic studies, these mild catalytic conditions provide stable aldehyde products bearing a wide array of electronically and sterically diverse substructures. The utility of these oxidized products is highlighted by subsequent derivatization to a variety of common chiral synthons, without loss in enantiopurity.
Introduction
Found among many natural and non-natural medicinal agents, oxygen-bearing stereocenters are a fundamental structural motif that is broadly represented throughout the realm of organic architecture.1–3 While traditional synthetic approaches to enantioselective C–O bond formation are strikingly powerful (olefin epoxidation/dihydroxylation, ketone reduction), new strategies continue to expand the range of starting materials or functional groups from which this important stereogenicity can be created. Recently, the enantioselective α-oxidation of aldehydes has garnered substantial attention as a novel catalytic approach to asymmetric oxygen-bearing stereocenters. In particular, the capacity to selectively install C–O bonds in direct proximity to high value carbonyl systems allows for a diverse range of post synthetic elaboration steps (e.g. oxidation, reduction, Wittig olefination, alkyl or aryl nucleophilic addition etc.).4 A number of enantioselective aldehyde α-oxidation reactions have recently been reported; however, there are no direct methods that demonstrate high reaction efficiency and enantioselectivity, while generating stable aldehyde products with an expansive substrate scope. Given the demonstrated value of α-oxy carbonyl synthons, we recently sought to address this need for a broadly applicable and highly selective protocol. Herein we report the successful execution of these ideals via the exploitation of mechanistic insight gained in studies of the organocatalytic Sibi oxyamination reaction.
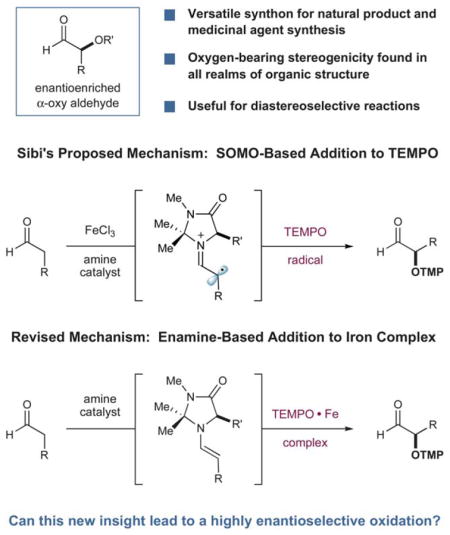
In 2003, along with Zhong, we reported the first catalytic enantioselective α-oxidation of aldehydes, utilizing nitro-sobenzene as an electrophilic source of oxygen.5,6 While the aldehyde products of this protocol were formed in high yield and with excellent enantioselectivity, their structural ground state was that of oligomers, rendering their isolation and derivatization troublesome. Later, Sibi disclosed an alternative method for preparing a number of stable α-oxy aldehydes using the commercially available 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) with an amine catalyst and FeCl3.7 In this report, it was proposed that an outer-sphere single electron oxidation of a transiently generated enamine (using FeCl3) produced a radical cation, which trapped the oxygen radical at the α-position. While on first inspection this mechanism seemed quite reasonable, we have recently demonstrated that this protocol does not involve SOMO-activation8 but instead an enamine addition pathway.9 More specifically, we found that the metal catalyst employed in the Sibi studies does not participate as an oxidant, but as a coordinating metal for the nitroxyl radical of TEMPO, generating a well-precedented η2 complex that is electrophilic at oxygen.10 As a result, enantioselective C–O bond formation in the Sibi oxyamination reaction occurs via addition of the transient enamine to an electrophilic iron-TEMPO complex.
Design plan
Inspired by these new mechanistic insights, we hypothesized that the use of alternative metal salts, which are known to form stable complexes with TEMPO,10 could lead to a more general and highly enantioselective method for the α-oxidation of aldehydes. To provide an electrophile optimally suited for coupling with an enamine, the electronic nature of the η2 complex could easily be tested by the choice of TEMPO framework11 and metal, while selection of a refined organocatalyst should increase the inherent enantioselectivity of the process. Since products of this nature are stable to racemization, the incorporation of an OTMP group offers a valuable handle for further structural elaboration, while the resulting N–O bond is readily cleaved to the free hydroxyl using mild yet chemically orthogonal conditions to that of carbonyl functionalization.7,12
Results and discussion
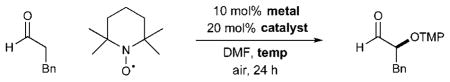
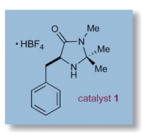
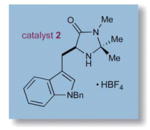
To evaluate the efficiency of this synergistic metal and amine catalyzed process, we began our investigations with TEMPO, 3-phenylpropanal, imidazolidinone catalyst 1, and [Fe(DMF)3Cl2][FeCl4] under ambient atmosphere in DMF (Table 1). We were delighted to find that the desired product13 was isolated in 70% yield, albeit with modest enantioselectivity in accord with Sibi’s studies (73% ee). Moreover, we observed that ambient air served as a stoichiometric oxidant, avoiding the need for additional equivalents of TEMPO as required in the original method (entry 1).14 In contrast to the use of iron-based catalysts,7 switching to CuCl2 provided the desired product with excellent yield and good selectivity at room temperature (entry 2). The pendant phenyl group of catalyst 1 was then replaced with a larger benzyl indole moiety (entry 3), leading to a further increase in enantiocontrol (93% yield, 87% ee). Moreover, lowering the reaction temperature to 0 °C afforded a useful increase in asymmetric induction (entry 4), while subsequent replacement of DMF with ethyl acetate or acetone (entries 5–6) promoted facile oxidation at −30 °C, (74–89% yield, 91–92% ee). Conveniently, ethyl acetate and acetone were taken from benchtop solvent bottles and simply stored for 1 h over oven-dried 4 Å molecular sieves prior to use.
Table 1.
Development of general enamine oxidation protocol
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Metal | T/°C | %yielda | %eeb |
| 1c | 1 | Fe3+ | 23 | 70 | 73 |
| 2 | 1 | CuCl2 | 23 | 95 | 85 |
| 3 | 2 | CuCl2 | 23 | 93 | 87 |
| 4 | 2 | CuCl2 | 0 | 78 | 90 |
| 5d | 2 | CuCl2 | −30 | 74 | 91 |
| 6e | 2 | CuCl2 | −30 | 89 | 92 |
|
|
||||
Determined by 1H NMR.
Determined by chiral HPLC of the corresponding alcohol; absolute configuration determined by correlation.
[Fe(DMF)3Cl2][FeCl4].
Ethyl acetate as solvent.
Acetone as solvent.
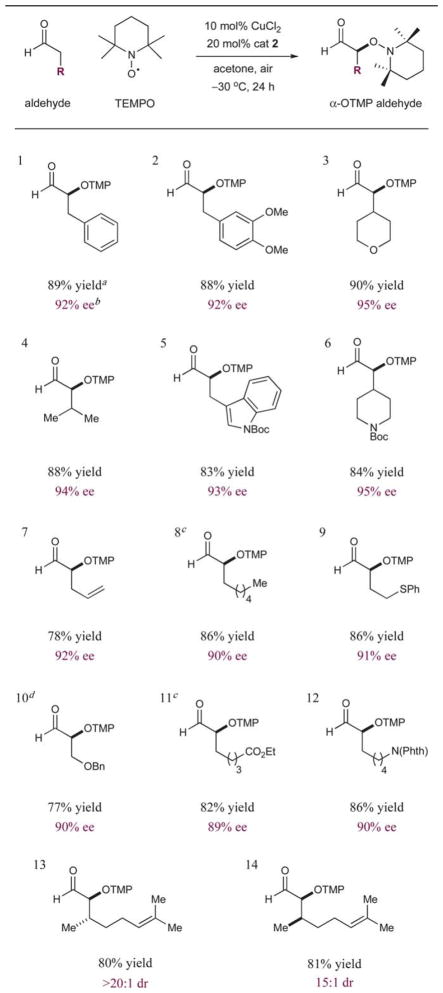
We next performed a series of experiments to define the scope of functional groups and steric constraints that are readily tolerated on the aldehydic component. As revealed in Table 2, ethers, esters, carbamates, and phthalimides can be incorporated without significant impact on yield or selectivity (entries 3, 6, and 10–12). Surprisingly, aldehydes bearing phenyl sulfides do not undergo oxidation at sulfur under these mild conditions or interfere with the inorganic catalyst, allowing the desired α-formyl oxidation product to be formed in 86% yield and 91% ee (entry 9). Aliphatic aldehydes are functionalized effectively (entries 4 and 8), while the incorporation of unsaturation in the form of olefins and aromatic rings is not detrimental to the process (entries 1–2, 5, 7, and 13–14). Notably, significant variation in steric environment on the aldehyde component is readily accommodated, with the more demanding frameworks furnishing the highest levels of asymmetric induction (entries 3–4, 6, and 13–14). Finally, subjection of (S)- and (R)-citronellal to oxidation yields the desired anti and syn products, respectively (entries 13–14). These experiments clearly demonstrate the value of catalyst-induced stereocontrol in preference to substrate-directed induction.
Table 2.
Enantioselective formyl α-oxidation: aldehyde scope
|
Isolated yields of aldehyde product (1.0 mmol scale).
Enantiomeric excess determined by chiral HPLC on corresponding alcohol or m-nitrobenzoyl ester.
Performed at −40 °C.
Ethyl acetate as solvent.
As shown in Scheme 1, numerous carbonyl transformations can be readily employed with these α-oxyamination products without any detectable loss in enantiopurity.15 For example, oxidation or reduction of the formyl group leads to α-oxygenated carboxylic acids or terminal alcohols, respectively. Reductive amination leads to 1,2-aminoalcohols, while Wittig olefination provides allylic alcohols with good cis-olefin selectivity (10 : 1 Z : E). Carbonyl addition using lithium enolates or Grignard reagents furnishes the corresponding ketones or substituted diols with good to excellent diastereomeric induction (13 : 1 and 5 : 1 dr respectively). As noted above, there is no loss in enantiopurity observed during any of these transformations, further highlighting the versatility and value of this enantioselective oxidation protocol and the resulting α-oxygenated formyl products.
Scheme 1.

Elaboration to common enantioenriched synthons.
Conclusions
On the basis of mechanistic insight into the Sibi oxyamination reaction, we have developed a widely applicable, highly enantioselective oxidation protocol that combines imidazolidinones and CuCl2 in a synergistic catalysis transform. The robust nature of this oxidation is highlighted by the use of ambient air and benchtop solvents. In contrast to existing methods, a broad range of heteroatoms, olefins, arenes, and steric environments are tolerated to provide stable aldehyde products in high yield and with excellent enantioselectivity. This protocol allows access to α-oxygenated systems in a broad sense that can be readily translated to many well-known synthons without loss in enantiopurity.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 01 GM093213-01) and gifts from Merck and Amgen. J. F. VH. thanks the Natural Sciences and Engineering Research Council (NSERC) for a predoctoral fellowship (PGS D).
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, and spectral data for all new compounds are provided (46 pages) (PDF). See DOI: 10.1039/c1sc00556a
Notes and references
- 1.Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Wiley-VCH; Weinheim, Germany: 2007. [Google Scholar]
- 2.Hollingsworth R, Wang G. Chem Rev. 2000;100:4267–4282. doi: 10.1021/cr990374e. [DOI] [PubMed] [Google Scholar]
- 3.Martin SF. J Nat Prod. 1992;55:1718–1731. doi: 10.1021/np50090a002. [DOI] [PubMed] [Google Scholar]
- 4.(a) Marigon M, Jørgensen KA. Chem Commun. 2006:2001–2011. doi: 10.1039/b517090g. [DOI] [PubMed] [Google Scholar]; (b) Guillena G, Raóon DJ. Tetrahedron: Asymmetry. 2006;17:1465–1492. [Google Scholar]; (c) Janey JM. Angew Chem, Int Ed. 2005;44:4292–4300. doi: 10.1002/anie.200462314. [DOI] [PubMed] [Google Scholar]; (d) Kano T, Mii H, Maruoka K. Angew Chem, Int Ed. 2010;49:6638–6641. doi: 10.1002/anie.201002965. [DOI] [PubMed] [Google Scholar]; (e) Bui NN, Ho XH, Mho SI, Jang HY. Eur J Org Chem. 2009:5309–5312. [Google Scholar]; (f) Akagawa K, Kudo K. Org Lett. 2011;13:3498–3501. doi: 10.1021/ol2012956. [DOI] [PubMed] [Google Scholar]; (g) Córdova A, Sudén H, Engqvist M, Ibrahem I, Casas J. J Am Chem Soc. 2004;126:8914–8915. doi: 10.1021/ja047930t. [DOI] [PubMed] [Google Scholar]
- 5.Brown SP, Brochu MP, Sinz CJ, MacMillan DWC. J Am Chem Soc. 2003;125:10808–10809. doi: 10.1021/ja037096s. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhong G. Angew Chem, Int Ed. 2003;42:4247–4250. doi: 10.1002/anie.200352097. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y, Yamaguchi J, Hibino K, Shoji M. Tetrahedron Lett. 2003;44:8293–8296. [Google Scholar]
- 7.Sibi MP, Hasagawa M. J Am Chem Soc. 2007;129:4124–4125. doi: 10.1021/ja069245n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For examples of our group’s development of SOMO catalysis, see: Beeson TD, Mastracchio A, Hong JB, Ashton K, MacMillan DWC. Science. 2007;316:582–585.Amatore M, Beeson TD, Brown SP, MacMillan DWC. Angew Chem, Int Ed. 2009;48:5121–5124. doi: 10.1002/anie.200901855.Wilson JE, Casarez AD, MacMillan DWC. J Am Chem Soc. 2009;131:11332–11334. doi: 10.1021/ja904504j.Rendler S, MacMillan DWC. J Am Chem Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p.
- 9.Van Humbeck JF, Simonovich SP, Knowles RR, MacMillan DWC. J Am Chem Soc. 2010;132:10012–10014. doi: 10.1021/ja1043006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Laugier J, Latour J, Caneschi A, Rey P. Inorg Chem. 1991;30:4474–4477. [Google Scholar]; (b) Pervukhina NV, Romanenko GV, Podberezskaya NV. J Struct Chem. 1994;35:367–390. [Google Scholar]; (c) Romanenko GV, Podberezskaya NV, Pervukhina NV. J Struct Chem. 1993;34:440–468. [Google Scholar]
- 11.A variety of TEMPO derivatives were evaluated; however, any alteration to the nitroxyl radical framework resulted in decreased reaction efficiency.
- 12.(a) Kirchberg S, Frohlich R, Studer A. Angew Chem, Int Ed. 2010;49:6877–6880. doi: 10.1002/anie.201002214. [DOI] [PubMed] [Google Scholar]; (b) Jahn U. J Org Chem. 1998;63:7130–7131. doi: 10.1021/jo981180m. [DOI] [PubMed] [Google Scholar]
- 13.OTMP = 1-oxy-2,2,6,6-tetramethylpiperidine.
- 14.(a) Geißlmei D, Jay WG, Falk H. Monatsh Chem. 2005;136:1591–1599. [Google Scholar]; (b) Bobbitt JM, Guttermuth MCF, Ma Z, Tang H. Heterocycles. 1990;30:1131–1140. [Google Scholar]
- 15.The enantioenriched aldehyde product was isolated with 93% ee on a large scale (8.3 mmol) for derivatization but in 92% ee on a 1.0 mmol scale.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.