

Abstract
We present a case of a 40-year-old female from Turkey, who was referred to our outpatient clinic for an undetermined thalassemia and sickle cell trait. At first consultation hemoglobin was decreased (71 g/L) with microcytosis (MCV 55.1 fL), and hypochromia (MCHC 239 g/L). The patient had severe iron deficiency. Brilliant cresyl blue staining showed >50% of the erythrocytes with typical Hemoglobin H (HbH) inclusions. High-performance liquid chromatography (HPLC) revealed normal levels of HbA2 and Hemoglobin F (HbF), and additionally a hemoglobin S (19%). Molecular diagnostics revealed the mutations α2 IVS-I donor site −5nt and a -- MED II deletion in the alpha gene complex and confirmed the heterozygote mutation of the beta-gene at codon 6 (HBB:c.20A>T; HbS). In conclusion, we present an extremely rare combination of HbH disease and sickle cell trait. This combination may explain the mild form of the HbH disease, with moderate anemia, splenomegaly but iron deficiency, rather than iron overload, as usually observed in HbH disease.
Key words: alpha-Thalassemia, HbH disease, iron deficiency, sickle cell trait.
Introduction
α-Thalassemia is a common genetic disorder that is characterized by deficient or absent synthesis of α-globin chains of the hemoglobin (Hb) molecule.1 The human α-globin gene cluster has two α genes (α2 and α1) on each chromosome 16. A normal person has four functional α-globin genes and is designated as αα/αα.2 Most α-thalassemia determinants are deletions involving one ( –α/αα) or both: loss of either both α-genes from a single chromosome ( - - /αα) or one gene from both chromosomes ( - α/ - α). An individual with three α genes (–α/αα) is not anemic but a heterozygote who inherits two functional α-globin genes (– –/αα) has mild hypochromic microcytic anemia. Patients with HbH disease inherit only one functional α-globin gene (– –/–α). They usually produce less than 30% of the normal amount of α-globin. The patients usually have splenomegaly (which may be severe) and occasionally this is complicated by hypersplenism.1,3
Sickle-cell anemia is an autosomal recessive genetic red cell disorder with a worldwide distribution that results from a point mutation at codon 6 of the beta-globin gene, with the replacement of glutamic acid to valine, leading to the production of a defective beta globin chain (βS, 6V) of the hemoglobin.4 While the coinheritance of another beta-globin gene anomaly increases the proportion of HbS and therefore the risk of sickling, the combination of a deletion of an alpha-gene anomaly reduces intraerythrocytic HbS concentration, attenuating therefore the problems due to the sickle cell trait.5 Since sickle cell trait is mainly seen in black populations, the coinheritance of HbH disease and a sickle cell trait has only been rarely described.
Usually, in patients with HbH disease, ineffective erythropoiesis in an expanded marrow stimulates iron absorption even if iron stores are adequate, and this stimulation increases the risk of iron excess when transfusions are given.6 We show here a rare case of a coinheritance of a HbH disease together with a sickle cell trait, presenting with iron deficiency.
Case Report
A 40-year-old Turkish woman was referred to our outpatient clinic for evaluation and treatment of an undetermined thalassemia and sickle cell trait. The patient suffered from fatigue and splenomegaly. Additionally, she had hepatitis B virus infection most probably transfusion transmitted. The patient has two healthy children.
At first consultation hemoglobin (Hb) was decreased (71 g/L) with microcytosis (MCV 55.1 fL), hypochromia (MCHC 239 g/L), anisocytosis (RDW 20.5 %) and hypochromic reticulocytes (CHr 16.2 pG) (Table 1). Leucocyte and platelet counts were within the normal range. On the blood smear we found anisocytosis, poikilocytosis, anisochromia, microcytes and target cells. The patient had severe iron deficiency (ferritin level 7 ng/mL; transferrin saturation 7%). In brilliant cresyl blue staining more than 50% of the erythrocytes showed typical HbH inclusions. The sickle cell test was positive. HPLC (High-performance liquid chromatography) revealed a normal HbA2 (2.3%) and HbF (0.7%), an abnormal hemoglobin S (19%) and persistence of HbA (>50%) (Table 2 and Figure 1). Genomic DNA was isolated using manual or automated extraction protocols. Using a reverse-hybridization of biotinylated PCR products (ViennaLab) a heterozygous mutation of the β gene at codon 6 (HBB:c.20A>T; corresponding to HbS), was detected. The α-globin genes were amplified by PCR and sequenced using the BigDye™ Terminator Cycle Sequencing Kit, and the ABI 3130 automated capillary sequencer (Applied Biosystems Inc., Foster City, CA, USA). We revealed a 5 nucleotide deletion in the IVS-I donor site (GAGGTGAGG>GAGG-----) of the α2-globin gene (Figure 2). This deletion was pseudo-homozygote since we showed later that one allele of the alpha gene complex contained a 6.1kb deletion eliminating both alpha genes. Multiplex Ligation-dependent Probe Amplification (MLPA) reactions were performed for detection of copy number variation in the α-globin gene clusters. The reaction was performed according to the manufacturers protocol using the SALSA MLPA kit P140B2 HBA and the ServiceXS HBA kit described by Harteveld et al., 2005.7 Products were separated by capillary electrophoresis on the ABI 3130 (Applied Biosystems) and data analysed using GeneMarker (SoftGenetics). Threshold ratios for deletion and duplication were set at >0.75 and >1.3, respectively. We found a heterozygous deletion of probes 5–25 with the P140B2 kit and of probes 8–15 with the ServiceXS kit. This corresponds to a more than 6.1kb long deletion of one allele in the alpha gene complex including the zeta and both alpha genes, leaving the theta gene intact. This type of deletion is called - - Med-II and is rather common in the Mediterranean area and was frequently found in the Turkish/Greek population.8
Table 1. Hematological data from the patient.
| Parameter, Units | At 1st presentation | 3 months after 1500 mg Ferric carboxy-maltose infusion | Reference values |
|---|---|---|---|
| Hb, g/L | 71 | 83 | 120–160 |
| HCT, L/L | 0.30 | 0.33 | 0.36–0.46 |
| MCV, fL | 55.1 | 55.1 | 79–95 |
| MCH, pG | 13.2 | 13.7 | 27.0–33.2 |
| MCHC, g/L | 239 | 250 | 320–360 |
| hypoRBC, % | 76.1 | 69.8 | <5.0 |
| RDW, % | 20.5 | 19.9 | 11.5–14.5 |
| Platelets, ×109/L | 214 | 162 | 150–450 |
| Reticulocytes, ‰ | 20 | 22 | 10–27 |
| Reticulocytes, ×109/L | 108 | 112 | 40–140 |
| WBC, ×109/L | 4.85 | 6.02 | 3.50–10.00 |
| Neutrophils, ×109/L | 3.42 | 4.38 | 1.300–6.700 |
| Ferritin, ng/mL | 7 | 33 | 10–200 |
| Transferrin saturation, % | 7 | 14 | 16–45% |
| sTR, mg/L | 23.11 | 11.64 | 2.2–4.5 |
Hb, hemoglobin; HCT, hematocrit; MCV, mean corpuscolar volume; MCHC, mean corpuscolar hemoglobin concentration; RDW, red cell distribution width; hypoRBC, hypochromic erythrocyte; sTR, soluble transferrin receptor; WBC, white blood cells.
Table 2. Quantitave values of hemoglobin components from the patient obtained by High-performance liquid chromatography and results of molecular analysis.
| Parameter | Value | References |
|---|---|---|
| Hb A2, % | 2.3 | 2.0–3.5 |
| Hb F,% | 0.7 | <2 |
| Hb H, % | 11 | 0 |
| Abnormal Hb | HbS | None |
| Perc. abnormal Hb, % | 19 | 0 |
| Sickle cell test | Positive | Negative |
| Mutation analysis | 1.α2 IVS-I donor site −5nt / -- MED II deletion | |
| 2. heterozygous Hb S (HBB:c.20A>T) |
Hb, hemoglobin.
Figure 1.
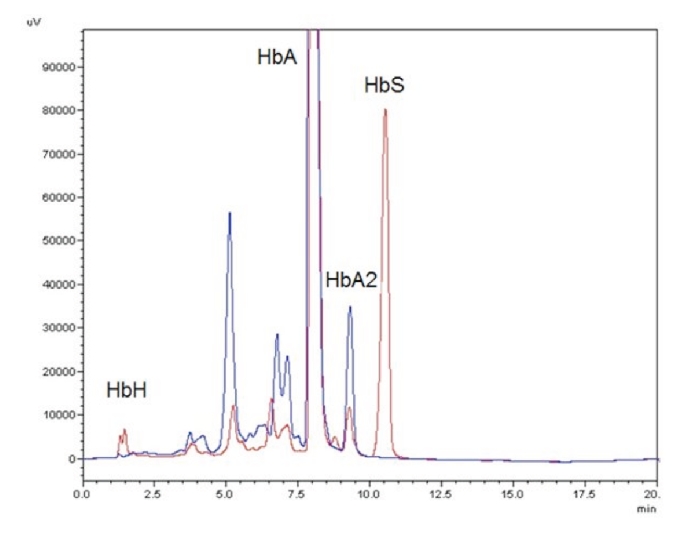
High-performance liquid chromatography pattern of our patient with Hemoglobin H (HbH) disease. Red graph represents our patient with clear HbH/HbBart's peak at 1.5 minutes and a HbS peak at 11 minutes retention time. Blue graph is a control.
Figure 2.

Sequence of the alpha 2 gene. Hemizygote deletion of 5 nucleotides at the IVS-I donorsite of the alpha2 gene, HBA2:c.95+2_95+6delTGAGG.
Thus this patient suffers from HbH disease and a sickle cell trait; the HbH disease was due to an α0-thalassemia (- -Med-II) / α+-thalassemia (α2 IVS-I donor site −5nt). The symptomatic anemia with iron deficiency was treated with iron infusion with a total dose of 1500 mg ferric carboxy-maltose. Thereafter, the Hb levels increased up to 83 g/L, the ferritin level to 33 µg/L and the transferrin saturation to 14% (Table 1).
Discussion
We show here a patient with a rare hereditary constellation of three different genetic anomalies of the haemoglobin genes: the patients presented HbH disease with two deletions on the alpha genes and a sickle cell anomaly on the beta gene. The coinheritance of HbH and sickle cell trait is rare.9 There are only few published case reports about the coincidence of HbH disease with a -- MED II deletion and a Hb S heterozygosity.10–11
HbH disease is a severe form of α-thalassemia but is compatible with life. Patients with HbH disease have a severe hypochromia with low MCV and MCH values, but increased iron stores.
In our case, the moderate, microcytic hyochromic anemia and the splenomegaly were the consequences of the HbH disease, and could not be reversed by iron substitution. However, we assume that the structural beta-globin chain variant in our patient leads to lower quantities of unstable HbH, and therefore to a milder form of HbH disease with less ineffective hematopoiesis and only minimal increase of gastrointestinal iron absorption. Hypermenorrhoea and two pregnancies were additional reasons for the unusual iron deficiency in this case of thalassemia.
References
- 1.Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13–13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colosimo A, Gatta V, Guida V, et al. Application of MLPA assay to characterize unsolved α-globin gene rearrangements. Blood Cells Mol Dis. 2011;46:139–44. doi: 10.1016/j.bcmd.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364:710–8. doi: 10.1056/NEJMoa1010174. [DOI] [PubMed] [Google Scholar]
- 4.Franceschi LD, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost. 2011;37:226–36. doi: 10.1055/s-0031-1273087. [DOI] [PubMed] [Google Scholar]
- 5.Ballas SK. Effect of α-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med. 2001;20:107–21. [PubMed] [Google Scholar]
- 6.Tichelli A, Rovó A. Clinical management of thalassemia in adults. Ther Umsch. 2010;67:237–43. doi: 10.1024/0040-5930/a000043. [DOI] [PubMed] [Google Scholar]
- 7.Harteveld CL, Voskamp A, Phylipsen M, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet. 2005;42:922–31. doi: 10.1136/jmg.2005.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutlar F, Gonzalez-Redondo JM, Kutlar A, et al. The levels of zeta, gamma, and delta chains in patients with HbH disease. Hum Genet. 1989;82:179–86. doi: 10.1007/BF00284054. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg MH, Coleman MB, Adams JG, 3rd, et al. A new gene deletion in the alpha-like globin gene cluster as the molecular basis for the rare alpha-thalassemia-1(--/alpha alpha) in blacks: HbH disease in sickle cell trait. Blood. 1986;67:469–73. [PubMed] [Google Scholar]
- 10.Cürük MA. HbH (beta4) disease in Cukurova, Southern Turkey. Hemoglobin. 2007;31:265–71. doi: 10.1080/03630260701297279. [DOI] [PubMed] [Google Scholar]
- 11.Yüregir GT, Aksoy K, Cürük MA, et al. HbH disease in a Turkish family resulting from the interaction of a deletional alpha-tha-lassaemia-1 and a newly discovered poly A mutation. Br J Haematol. 1992;80:527–32. doi: 10.1111/j.1365-2141.1992.tb04568.x. [DOI] [PubMed] [Google Scholar]