

Abstract
Background
In colorectal cancer (CRC), microsatellite instability (MSI) is a valuable marker of defective DNA mismatch repair that identifies cancers with distinct phenotypic properties, including favorable survival. However, the optimal assay for MSI status is unknown. We have evaluated a simplified 3-marker assay for MSI and compared it with the 5-marker (NCI) assay to see if technical variations in MSI testing are important.
Materials and Methods
DNA samples from 357 CRCs were evaluated for MSI using the 5 microsatellite markers recommended for the NCI assay (BAT 25, BAT26, D2S123, D5S346, and D17S250). Results were compared with a simplified 3-marker assay (BAT25, BAT26, and D2S123). CRCs identified as MSI were evaluated for their clinical, pathological, and genetic characteristics.
Results
The 5-marker assay identified 96 cancers as MSI. Only 56 of these were MSI by the 3-marker assay (3-marker+ group), leaving 40 cases identified as MSI only by NCI criteria (3-marker− group). The remaining 261 cancers were microsatellite stable (MSS). The 3-marker+ MSI tumors had features characteristic of MSI tumors: more proximal, poorly differentiated, associated with hereditary nonpolyposis colorectal cancer (HNPCC), more BRAF mutations, fewer KRAS mutations, better 5-year disease-specific survival, more frequent mismatch repair (MMR) protein loss, and less likely to be metastatic on presentation (P<.05). Chromosomal arm loss was observed only in 3-marker− MSI and MSS cancers (P<.05).
Conclusion
The 3-marker MSI assay outperforms the traditional 5-marker assay for identifying patients with favorable prognosis and homogeneous clinical and genetic features. More accurate MSI testing should improve prognostic and predictive scoring systems for colorectal cancer.
Because of its prevalence in colorectal cancer and associated favorable prognosis, microsatellite instability (MSI) is an important genetic marker in colorectal cancer that can be useful in providing personalized care to individual patients. This condition is found in 10–20% of colorectal cancers (CRC) and is an indicator of defective DNA mismatch repair (MMR).1 It can arise in either a sporadic or hereditary fashion. In the majority of MSI cancers, methylation silencing of the MLH1 gene occurs, often in older patients.2 This variety of colorectal cancer has also been described as the CpG Island Methylator Phenotype (CIMP), in which a variety of genes are methylated and often inactivated. In the heritable form, hereditary nonpolyposis colorectal cancer (HNPCC), there is a germline mutation in 1 of 5 mismatch repair genes (MLH1, MSH2, MSH6, PMS2, and PMS1). MSI cancers are diploid, arise predominantly in the proximal colon, are frequently poorly differentiated, and in general have an indolent biology and improved prognosis compared with microsatellite stable (MSS) cancers.1,3,4
Because of these strong biological associations, MSI is a useful genetic marker in managing colorectal cancer patients. It becomes useful in subdividing colorectal cancer patients into distinct groups that show survival differences. Tumors without MSI are MSS and exhibit chromosomal instability (CIN). Tumors with CIN tend to be aneuploid, have a less favorable prognosis, and often do not have the morphological and clinical characteristics of MSI tumors.3,4 Also, MSI is well established as a marker of favorable prognosis.1–4 It is currently in use in a trial of selective chemotherapy for stage II CRC and has been advocated as a routine prognostic test to supplement pathologic staging.5 In fact, several groups have proposed use of MSI to predict response to chemotherapy.6 Whether MSI tumors respond differently to 5-fluorouracil (5-FU) chemotherapy remains unclear because conflicting results have been reported in the literature.6–8
A major concern about using MSI status in patient management decisions is the technical reproducibility and accuracy of MSI testing. We and others have reported that the technical methods and scoring systems used in MSI testing can have a considerable impact on the accuracy of the test.4 In 1997, in an attempt to standardize methods, a consensus panel convened by the National Cancer Institute recommended the use of 5 microsatellite markers to determine MSI status in colorectal cancer.9 However, recent reports have shown that the 5-marker MSI assay has poor specificity and inferior accuracy compared with assays that use 5–7 microsatellite markers that contain only mononucleotide repeat sequences.10–12 Furthermore, an immunohistochemical test to evaluate the expression of MMR genes in tumor cells has emerged as another method of detecting defective MMR; this test has a high but imperfect correlation with MSI status.13 In this report, the authors evaluated: (1) the ability of a simplified 3-marker assay and the traditional 5-marker assay to accurately detect MSI tumors, (2) the clinical and molecular characteristics of patients identified as MSI by each assay, and (3) the accuracy of each assay in identifying MMR protein defects.
PATIENTS AND METHODS
Patients and Tumor Samples
Data were obtained from 357 snap-frozen tissue specimens collected prospectively between January 1990 and December 2004 following institutional guidelines and a protocol approved by the Institutional Review Board. All patients presented with primary adenocarcinoma of the colon or rectum and underwent surgical resection as initial treatment, either for curative or palliative intent. The tumors were from all sites of the large intestine (129 tumors proximal to the splenic flexure, 228 distal). Clinical and pathological information was collected prospectively in a database. The AJCC tumor stages for the patients were: 63 stage I, 104 stage II, 98 stage III, and 92 stage IV.
Staging information was incomplete for 2 patients. The patients were between the ages of 18 and 90 years with a slight male predominance. Median follow-up was 5.0 years, ranging from 6 weeks to 14.7 years. Tissue was snap frozen in liquid nitrogen within 30 min of resection and stored at −80°C. DNA was extracted using a proteinase K/LiCl/EtOH protocol and quantified using a GeneQuant Pro RNA/DNA calculator.
MSI Assay
Oligonucleotide primers for mononucleotide markers BAT25, BAT26 and dinucleotide markers D2S123, D5S346, and D17S250 were fluorescently labeled. The amplification of each was optimized for identical polymerase chain reaction (PCR) conditions using AmpliTaqGold DNA Polymerase. Every colon cancer specimen and its corresponding normal tissue (100 ng DNA per reaction) were analyzed for all 5 microsatellite markers (by uniplex PCR; 5-marker assay) and for only BAT25, BAT26, and D2S123 using multiplex PCR (3-marker assay), as previously described.1 PCR volume was 20 μL. The PCR products were resolved on an 8.3% polyacrylamide gel in an ABI PRISM 377 DNA Sequencer (Applied Biosystems). Allele size instability (ASI) was detected and distinguished from PCR stutter using Genotyper 2.5 software (Applied Biosystems). Following their detection during electrophoresis, peaks were ordered by size (smallest to largest) and individually labeled with their size in base pairs. Then, using parameters provided by ABI, labels were removed from peaks that were followed within 3 base pairs, or preceded within 1.6 base pairs, by a higher amplitude peak. Allelic size instability was defined as the identification in a tumor of an allele with a length not seen in normal colonic mucosa from the same individual.1 Tumor tissue from 18 patients with Lynch syndrome (LS) served as positive controls for MSI.1 MSI was scored as present when ≥2 of 3 markers showed size instability for the multiplex 3-marker assay, or ≥2 of 5 for the 5-marker (NCI) assay. Tumors identified as MSI by only the multiplex assay are referred to as 3-marker+ MSI, whereas those identified only by the 5-marker assay and not the 3-marker assay are referred to as 3-marker− MSI. The MSS tumors refer to those that were scored as such by both assays.
Histopathologic Studies
Using hematoxylin and eosin stained 3 μm sections taken from paraffin blocks, a pathologist evaluated the morphology of colorectal cancers from 16 of the 18 HNPCC patients and 349 of the 357 studied patients. Tumors were classified as poorly differentiated, moderately differentiated, or well differentiated based on established criteria and in comparing them to the LS tumors. The presence or absence of lymphovascular invasion (LVI) in the tumor and degree of tumor-infiltrating lymphocytes (TIL) in some specimens were also recorded.14–16 Immunohistochemistry was performed on 129 tumor samples to check for the presence or absence of mismatch repair proteins. Primary monoclonal antibodies against MLH1 (clone G168–728, diluted 1:250, PharMingen, San Diego, CA), MSH2 (clone FE11, diluted 1:50, Oncogene Research Products, Cambridge, MA), MSH6 (clone GRBP.P1/2.D4, diluted 1:200; Serotec Inc, Raleigh, NC), and PMS2 (clone A16–4, diluted 1:200, BD PharMingen, San Diego, CA) were applied to 5-μm thick 10% formalin fixed, paraffin-embedded tissue sections.17
The sections underwent a process of deparaffinization, rehydration, and washing in xylene, graded alcohols, and distilled water, respectively. Blockage of endogenous peroxide activity was performed after incubation with 3% H2O2. A 10 mM citrate buffer with a subsequent microwave antigen retrieval procedure was done.
Antigen-antibody reaction was visualized using the avidin-biotin peroxidase complex (LSAB kit, Dako) and diaminobenzidine as the chromogen. Slides were counter-stained with hematoxylin. Each tumor sample was rated on a scale (0–1) from complete absence of each of the 4 MMR proteins, to full expression. Non-neoplastic colonic mucosa and colorectal tumors known to be deficient of MLH1, MSH2, MSH6, and PMS2, respectively, were used as external controls. Negative protein expression was defined as complete absence of nuclear staining within tumor cells.
KRAS and BRAF Mutation Analysis
KRAS and BRAF mutation analysis was done using PCR/Ligase Detection Reaction (LDR) techniques. DNA was extracted from archived tumor samples as described previously. Primers for KRAS exon 1 were 5′-TTTCATT ATTTTTATTATAAGGCCTGCTGA-3′ and 5′-GAATGG TCCTGCACCAGTAATATGC-3′. Primers for BRAF were exon 15 forward primer 5′-TTCTAATGCTTGCTCT GATAGGA-3′ and reverse primer 5′-GGCCAAAAATT TAATCAGTGGA-3′. Fluorescently labeled LDR primers were used for the 7 common KRAS (Val, Asp, Ala, Arg, Ser, and Cys at codon 12 and Asp at codon 13) mutations.18 The LDR sequences for BRAF V600E mutation were: wild-type 5′-/56-FAM/-AGTAAAAATAGGTGATTTTGG TC TAGCTACAGT-3′, mutant 5′-/56-FAM/AAAAATAGGT GATTTTGGTCTAGCTACAGA-3′, and common 5′-/5Phos/GAAATCTCGATGGAGTGGGTCC-3′. PCR was carried out as previously described.19 Proteinase K inactivation step was subsequently used to inactivate any Taq polymerase. In brief, LDR was done in a final volume of 25 μL using 2 μL of PCR amplicon and thermocycled with the following program: 94°C for 1.5 minutes, followed by 10 cycles of 94°C for 1 minute, 10 cycles of 65°C for 4 minutes, and held at 4°C.
Array Comparative Genomic Hybridization (aCGH)
Array CGH analysis was done on Agilent 44 K arrays by the Genomic Core Laboratory of Sloan-Kettering Institute. In brief, 2 μg of DNA from tumor and from matched normal colon mucosa was labeled using biotin nick translation. Tumor DNA was labeled with a red fluorescent label and normal DNA with a green fluorescent label. Labeled tumor and matched normal DNA were mixed in equal concentrations and precipitated with human Cot-1 DNA, then hybridized to the 44 K CGH microarray chip. Agilent Genotyper software was used to measure chromosomal arm deletions and amplifications. We used a z-score of 2.0, log2 ratios of red fluorescence over green, and a moving average of 2 Mb. Chromosomal arm deletions and amplifications were defined as copy number loss or gain, respectively, in more than 90% of probes assigned to that chromosomal arm, with cutoff values set to ±2 median absolute deviation (MAD).
Statistics
The Fisher exact test was used to compare the 3 patient groups divided by the 3- and 5-marker assays in the categories of Lynch syndrome association, right-sided cancer, KRAS and BRAF mutations, differentiation, and stage IV disease. Survival curves were constructed using SPSS version 15.0 from SPSS Inc. (Fig. 2). Differences in survival of the 3 groups were compared using the log-rank test. Disease-specific survival, defined as death related to cancer and greater than 30 days postoperation, was evaluated. Patients that died of other causes or were lost to follow-up were censored. Differences in MMR staining were also examined using the Fisher exact test; occurrence of genetic aberrations was compared using the Wilcoxon rank test. The level of significance was set at P<.05.
FIG. 2.
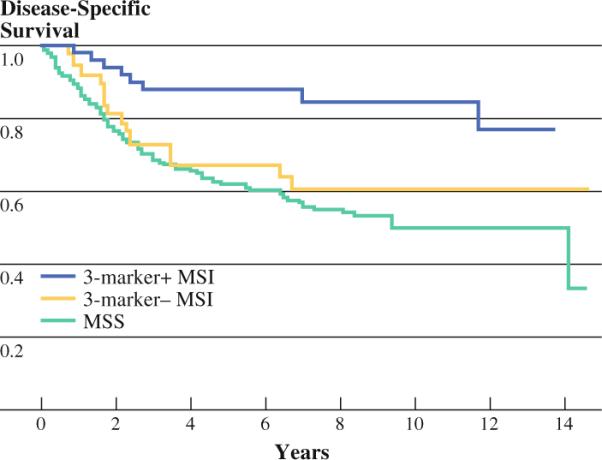
Disease-specific survival is shown for true MSI colorectal cancers (positive on 3-marker test), false MSI cancers (positive on NCI 5-marker test but negative on 3-marker test), and MSS cancers. True MSI cancers have favorable prognosis compared with the other groups (P < .01)
RESULTS
The MSI status of 357 colorectal tumors was: 56 cancers (16%) were MSI by the 3-marker assay, 40 (11%) were MSI only by the 5-marker assay, and 261 (73%) were MSS by both assays.
Clinical and genetic features of the 3 patient groups defined by MSI status are shown in Table 1. There were no significant differences in age or gender (53% of the overall study population was male, 47% female). However, in nearly every other category listed in Table 1, the 3-marker+ MSI patients showed characteristics that were distinct from the other 2 groups. The 3-marker+ MSI tumors were more often located in the right colon (P = .002), contained a much higher proportion of poorly differentiated cancers (P = .029), and showed a highly favorable rate of survival. Also, only 7% of those tumors were metastatic on presentation, as opposed to 28% for each of the other 2 groups (P = .020). Of the patients with MSI tumors identified by the 3-marker assay, 14% met clinical criteria for HNPCC (by the Amsterdam 2 criteria), compared with <1% of patients with 5-marker MSI or MSS cancers (P = .018). In addition, 21% of the patients in the 3-marker+ group developed colorectal cancer before age 50, as opposed to 13% in the 3-marker− and MSS groups each (P = .250).
TABLE 1 Clinical and geneticfeatures of patients according to the microsatellite instability (MSI) status of their primary colorectal cancers
| NCI 5-marker MSI+ (N = 96) |
MSS (N = 261) | |||
|---|---|---|---|---|
| 3-Marker+ cases (N = 56) | 3-Marker− cases (N = 40) | P value | ||
| Mean age | 65 | 65 | .636 | 64 |
| Male | 50% | 60% | .883 | 48% |
| Early-onset CRC (Age < 50) | 21% | 13% | .250 | 13% |
| Lynch syndrome (Amsterdam 2 criteria) | 14% | 0% | .018* | 0.4% |
| Right-sided CRC | 68% | 38% | .002* | 30% |
| High-grade histology | 25% | 8% | .029* | 8% |
| BRAF mutations | 34% | 3% | .000* | 3% |
| KRAS mutations | 21% | 45% | .013* | 39% |
| Stage IV | 7% | 28% | .020* | 29% |
| 5-yr disease-specific survival | 88% | 67% | .001* | 62% |
P < .001, for comparisons to 3-marker− cases
KRAS Codon 12/13 and BRAF V600E Mutations
We found the expected high proportion of KRAS mutations (45%) in MSS cancers and found a nearly equivalent proportion (39%) in the 3-marker− MSI tumors. In contrast, the KRAS mutation proportion was lower in 3-marker MSI tumors (21%) (P = .013), a finding consistent with published data showing a lower prevalence of KRAS mutations in sporadic MSI-H colorectal tumors. BRAF V600E mutations are known to occur at relatively high frequency in sporadic MSI cancers but are absent from HNPCC associated cancers. Our data show a high proportion of BRAF mutations (34%) in the 3-marker+ MSI group, confirming that a large proportion of these cancers are sporadic MSI tumors. In this group, BRAF mutations were enriched over 10-fold (34%) compared with MSS (3%) and 3-marker− MSI cases (3%) (P = .000).
MMR Staining
To verify the 3-marker MSI assay's ability to identify tumors with the absence of MMR activity, we evaluated the expression of 4 mismatch repair (MMR) genes in paraffin sections of tumor tissue. Normal tissues and MSS cancers, which are competent for mismatch repair, should retain expression of all 4 genes. Among true MSI cancers, approximately 60–70% will lose expression of 1 or 2 of these genes. In our series, 127 samples had sufficient paraffin-embedded tumor for assessment of MMR gene expression, and 126 yielded informative data (Table 2). All of the tumors with MLH1 and PMS2 deficiency were MSI 3-marker+ tumors, while the other groups had normal staining of MMR proteins. More than 70% of MSI 3-marker+ tumors had loss of MLH1 and/or PMS2 expression, whereas none of the MSI 3-marker− or MSS tumors had deficiency in any MMR proteins (P = .000).
TABLE 2 Immunohistochemistry for mismatch repair (MMR) proteins
| NCI 5-marker MSI+ (N = 96) |
MSS (N = 261)(%) | |||
|---|---|---|---|---|
| 3-Marker+ cases (N = 23) (%) | 3-Marker− cases (N = 19) (%) | P value | ||
| MLH1 deficient | 52 | 0 | .000* | 0 |
| MSH2 deficient | 4 | 0 | .012* | 0 |
| MSH6 deficient | 4 | 0 | .012* | 0 |
| PMS2 deficient | 57 | 0 | .000* | 0 |
| Any MMR protein deficient | 71 | 0 | .000* | 0 |
P < .001, for comparisons to 3-marker− cases
Comparative Genomic Hybridization (CGH) Array
A total of 25 tumors were subjected to chromosomal analysis using Agilent CGH 44 K arrays. Of these, 16 were MSI by the 3-marker assay, 4 were MSI 3-marker− tumors, and 5 were MSS (Fig. 1). Three of the four 3-marker− MSI tumors had loss of at least 1 chromosomal arm (P = .000) as did two of the five MSS tumors (P = .012). This relatively high prevalence of chromosomal instability and aneuploidy in the 3-marker− group is unusual for tumors classified as MSI. On the other hand, none of the 3-marker+ MSI tumors had chromosomal arm loss, indicating that they are diploid tumors (Table 3). When looking at total genomic instability, 9 of the 16 3-marker+ MSI tumors had no identifiable gene amplification or deletion, while 7 had genomic copy number changes of <5%. Three of the four 3-marker− MSI tumors had some genomic instability (gain or loss), as did all of the MSS tumors.
FIG. 1.

Chromosomal arm losses and gains as determined by CGH analysis (see methods) is shown for 3 groups of primary colorectal cancers: true MSI cancers (16 cases) for which the 3-marker MSI test was positive, putative MSI cancers (4 cases) for which the NCI 5-marker test was positive but the 3-marker test was negative, and MSS cancers (5 cases). Chromosomal arm gain is shown in black; arm loss is shown in white
TABLE 3 Chromosomal arm loss
| Diploid (chromosomal arm losses = 0) | Aneuploid (any chromosomal arm loss) | P value | |
|---|---|---|---|
| 3-Marker+ MSI cases | 16 | 0 | n/a |
| 3-Marker− MSI cases | 1 | 3 | .000* |
| MSS cases | 3 | 2 | .012* |
P < .001, for comparisons to 3-marker− cases
Patient Survival
The majority of patients who were classified as MSI by the 3-marker assay had disease confined to the colon and/or regional lymph nodes (93%), while 32% of those with MSS tumors had distant metastatic disease. The 5-year disease specific survival was 88% for the 3-marker+ patients, 67% for 3-marker− MSI patients, and 62% for MSS patients (P = .0001; Fig. 2).
DISCUSSION
Microsatellite instability is an important marker for genetic and clinical correlative studies in colorectal cancer. The presence of MSI indicates a deficiency in the DNA mismatch repair pathway and identifies 2 distinct genetic subtypes of colorectal cancer. In hereditary nonpolyposis colorectal cancer (HNPCC), DNA mismatch repair function is lost because of bi-allelic inactivation (germline mutation plus subsequent somatic inactivation) in 1 of 5 genes in the DNA mismatch repair pathway (MSH2, MLH1, MSH6, PMS2, and PMS1). Sporadic MSI cancers (12–15% of patients) have deficiencies in DNA mismatch repair due to methylation silencing of the MLH1 gene (CIMP pathway). Detection of MSI can be used for identifying probands in families with HNPCC, for studying genetic mechanisms of colorectal carcinogenesis, and for stratifying patients in prognostic marker studies and chemotherapy trials. However, for MSI testing to be clinically useful, it is crucial that the PCR assays identify the presence of MMR deficiency with a high degree of accuracy.
Because a vast number of microsatellite loci throughout the genome can be affected and potentially informative, investigators have used a variety of microsatellite markers and scoring criteria to determine MSI status. In 1998, an NCI consensus statement recommended that: (1) the term MSI become standard in the field, (2) 5 microsatellite markers (mononucleotide markers BAT25, BAT26; dinucleotide markers D2S123, D5S346, and D17S250) be used as a reference panel for future research, and (3) tumors be stratified into MSI-H (≥2 of 5 markers with size instability), MSI-L (1 of 5 markers), and MSS groups (0 of 5 markers).9 Subsequent research has shown the NCI 5-marker test to have poor specificity for MSI.1,4,10 This stems primarily from an overabundance of dinucleotide markers in the test. The dinucleotide markers D5S346 and D17S250 have sensitivities of about 50% for detecting tumors with mismatch repair deficiency.1 Like all dinucleotide markers, they are inherently prone to slippage and loss of heterozygosity during DNA replication. Such random slippage of dinucleotide microsatellite markers during tumor development can result in false-positive MSI tests in tumors that do not have true defects in MMR. For this reason, clinical correlative studies using the traditional NCI assay have produced inconsistent conclusions about prognosis and chemosensitivity of MSI cancers.4,6–8 Furthermore, the classification of MSI-L tumors by the 5-marker assay (1 of 5 markers positive for ASI) has questionable significance, since size instability of a single dinucleotide microsatellite marker can occur randomly and be clonally preserved within the tumor. In this report we provide strong evidence that a simplified assay for MSI, which uses only 3 markers in the NCI panel: the 2 mononucleotide markers (BAT25, BAT26) and the most accurate (D2S123) of the 3 dinucleotide markers, is a more specific and equally sensitive test for identifying tumors with MMR deficiency.
The conventional NCI criteria for MSI (at least 2 of 5 markers showing allele size instability: BAT 25, BAT26, D2S123, D5S346, and D17S250) significantly overestimate the number of cases with mismatch repair deficiency. It is estimated that up to 50% of papers in the late 1990 s and early 2000 s overestimated MSI because of poor marker selection.4 In our study, the overall proportion of colorectal cancers identified as MSI by the 5-marker assay was high (27%). However, when using the modified 3-marker test (at least 2 of 3 markers showing size instability: BAT25, BAT26, and D2S123), the rate of MSI drops to 16%. When the clinical features of these patients are compared, the 3-marker MSI assay reliably identified a group of patients with proximal, poorly differentiated, low-stage tumors with a significantly better 5-year disease-specific survival than most sporadic colorectal cancers. Additionally, this group of patients exhibited a lower rate of KRAS mutations and a higher rate of BRAF mutations, findings that are characteristic of MSI tumors in the CIMP pathway. We also found a dramatically lower incidence of chromosomal arm loss and a stronger association with HNPCC in the 3-marker+ cases compared with the 3-marker− cases. It is important to recognize that the additional 11% of patients identified only by the 5-marker assay (3-marker− group) were dependent on allele size instability in 1 or both of the dinucleotide markers excluded from the 3-marker assay. These additional patients did not have the clinical, morphological, or genetic characteristics of tumors with true mismatch repair deficiency. In fact, all of these additional tumors had clinical features indistinguishable from patients in the MSS group. In our previous work, we showed that the dinucleotide markers D5S346 and D17S250 had poor sensitivity and poor specificity for MSI by testing known MMR deficient (Lynch syndrome) cancers for MSI.1 The current study provides additional clinical evidence that these 2 markers are not only unnecessary but in fact add many false-positive cases to the true MSI group.
Our 3-marker assay uses BAT25, BAT26, and D2S123 and is driven by the high accuracy rates of BAT25 and BAT26. Some advocate using just BAT25, BAT26, or both as the preferred MSI assay. However, this is problematic because neither marker alone is perfectly sensitive or specific.10 Our data show that, when used in combination, BAT25 and BAT26 are concordant in 94% of cases. To assign MSI status to the 6% of cases that show discordance, a third marker is needed. We show that the dinucleotide marker, D2S123, serves this role well.
For example, the sensitivities of BAT25, BAT26, and D2S123 in our assay are 100%, 94%, and 72%, respectively.1 So 6% of the time, the mononucleotide markers will be discordant, but adding D2S123 means that 72% of those cases (or 4.3% of all patients tested) will be successfully identified by adding D2S123. Thus, we estimate the 3-marker assay misidentifies fewer than 2% of MSI tumors. Adding more markers to increase the sensitivity makes testing both more complicated and also more vulnerable to false-positive results that lower its specificity. We show that such complexity is unnecessary and that highly accurate MSI scoring can be achieved simply by modifying or reanalyzing data collected by the traditional 5-marker test.
Our study supports the findings of 2 recent publications that advocate the use of primarily mononucleotide markers for MSI testing. One such panel, named the pentaplex panel, uses BAT25, BAT26, NR21, NR22, and NR24 in a multiplex PCR. This pentaplex panel does not require normal tissue as control because mononucleotide sequences are highly conserved in tissue, except when neoplastic.10 The sensitivity and positive predictive values of the pentaplex assay are 95.8% and 88.5%, respectively for identifying tumors with an MMR protein deficiency.11 This study reinforces our findings that the NCI 5-marker assay, by including 3 dinucleotide markers in its MSI analysis, gives a high false-positive rate. The pentaplex panel was incorporated into the revised Bethesda guidelines in 2004 for testing for MSI in suspected HNPCC patients because of its high degree of accuracy. Our results indicate similar accuracy can be achieved by using only 3 of the 5 markers within the traditional 5-marker assay.
We sought to validate the accuracy of the 3-marker MSI assay by correlating MSI status with other tests. Immunohistochemical staining for MMR proteins (MSH2, MLH1, MSH6, and PMS1) was performed on 126 tumors for which paraffin blocks were available. Among 23 tumors classified as MSI+ by the 3-marker assay, 16 (71%) showed loss of expression of 1 or more MMR proteins. On the other hand, among 19 cases identified as MSI only by the NCI 5-marker assay and 84 MSS cases, none showed loss of MMR protein expression. Thus, all tumors identified as deficient in MMR protein expression were found in the 3-marker+ MSI group. These data strongly validate the 3-marker assay as technically sound and clearly show the suboptimal performance of the 5-marker assay. These data also support the idea that MSI testing is more sensitive for MMR deficiency than immunostaining. One explanation for the lower sensitivity of immunostaining is that many missense mutations in MMR genes result in an antigenically intact protein, as often happens with MLH1.13,20 These tumors have a nonfunctional protein that is recognized by the immunostain, resulting in the failure to identify a tumor with MMR deficiency. In addition, IHC staining can give erroneous results because of heterogeneous tissue staining or inadequate tissue sampling.13
We also correlated MSI status with comparative genomic hybridization, which evaluates tumors for chromosomal gains and losses. True MSI cancers are either diploid or near diploid, showing relatively few gains or losses throughout the genome.21,22 Among 16 cancers identified as MSI+ by the 3-marker assay, all 16 were shown to lack any losses or gains of chromosomal arms. On the other hand, MSS cancers as well as the additional MSI cancers identified only by the 5-marker assay showed a mixture of diploid and aneuploid tumors. We conclude that only the 3-marker MSI assay is able to define a homogeneous, biologically concordant group.
Given the distinct biological and clinical properties of colorectal cancers with MSI, why has MSI testing not gained greater use in clinical practice and clinical trials? For screening of families for HNPCC, immunohistochemistry for MMR proteins has become more popular because of its low cost and widespread availability. However, because of difficulty in diagnosis of many HNPCC families, MSI testing still has an important role.20,23 For example, in families with HNPCC, germline mutations are found in only 70%; on the other hand, MSI is present in more than 95% of colorectal cancers that occur in these families. With regard to stratifying patients for clinical trials or selecting patients for particular chemotherapy treatments, the lack of clear standards for MSI testing has been harmful. According to some reports, therapy with 5-FU is of no benefit in colorectal cancers with MSI, while other reports state the opposite.7,8 A close study of these reports shows that MSI testing is frequently suspect because it has relied on assays that contain dinucleotide markers. The National Cancer Institute–National Surgical Adjuvant Breast and Bowel Project (NCI–NSABP) Collaborative Study published in 2007 found that MSI was not predictive of 5-FU chemotherapy response.7 In this study, MSI was assigned using a 6-marker assay, and 3 of the markers were dinucleotide repeats (D2S123, Mfd15, and D5S346). On the other hand, a 2002 Taiwanese study showed that patients with MSI tumors had better response to 5-FU chemotherapy after palliative bowel resection than those that were not MSI.8 This study used the 5-marker assay to assign MSI status. Because MSI assignment is suspect in both studies, it is impossible to conclude whether MSI has no predictive value or simply has not been properly assessed. Of note, an ongoing Eastern Cooperative Oncology Group (ECOG) trial of stage II CRC patients (E5202) is using MSI and 18q LOH testing to select patients for observation versus adjuvant chemotherapy with FOLFOX ± bevacizumab.5 The patients with tumors identified as low risk for recurrence based on presence of MSI and retention of 18q are observed. However, MSI testing in this trial is done using a modified NCI protocol that includes dinucleotide markers.
Given its strong association with a unique tumor biology and favorable prognosis, MSI testing should be useful in the management of colorectal cancer patients. However, technical pitfalls of the assay has obscured its optimal value and hindered its widespread adoption. The biggest problem is the inclusion of dinucleotide markers in the most widely used MSI assays, which leads to a high false-positive rate (42% in our study) and renders them inaccurate and ineffective for clinical management decisions. However, we have demonstrated that highly accurate MSI testing can be done using 3 (BAT25, BAT26, and D2S123) of the 5 markers contained within the traditional NCI microsatellite panel. This simplified test provides a clear division of colorectal cancer patients into biologically distinct groups and allows for clinical correlative studies that are meaningful, robust, and applicable to daily clinical practice. Prognostic scoring systems, association studies, and clinical trials must use validated MSI assays in order to maximize the likelihood of meaningful biological correlation.
REFERENCES
- 1.Nash GM, Gimbel M, Shia J, Culliford AT, Nathanson DR, Ndubuisi M, et al. Automated, multiplex assay for high-frequency microsatellite instability in colorectal cancer. J Clin Oncol. 2003;21:3105–12. doi: 10.1200/JCO.2003.11.133. [DOI] [PubMed] [Google Scholar]
- 2.Gryfe R. Clinical implications of our advancing knowledge of colorectal cancer genetics: Inherited syndromes, prognosis, prevention, screening and therapeutics. Surg Clin N Am. 2006;86:787–817. doi: 10.1016/j.suc.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Sinicrope FA, Rego RL, Halling KC, Foster N, Sargent DJ, La Plant B, et al. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131:729–37. doi: 10.1053/j.gastro.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Laghi L, Bianchi P, Malesci A. Differences and evolution of the methods for the assessment of microsatellite instability. Oncogene. 2008;27:6313–21. doi: 10.1038/onc.2008.217. [DOI] [PubMed] [Google Scholar]
- 5.O'Dwyer PJ. [Accessed 12 February 2009];Oxaliplatin, leucovorin, and fluorouracil with or without bevacizumab in treating patients who have undergone surgery for stage II colon cancer; 2008. Available: http://www. clinicaltrials.gov.
- 6.Jo W-S, Carethers JM. Chemotherapeutic implications in microsatellite unstable colorectal cancer. Cancer Biomark. 2006;2:51–60. doi: 10.3233/cbm-2006-21-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim GP, Colangelo LH, Wieand HS, Paik S, Kirsch IR, Wolmark N, et al. Prognostic and predictive roles of high-degree microsatellite instability in colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J Clin Oncol. 2007;25:767–72. doi: 10.1200/JCO.2006.05.8172. [DOI] [PubMed] [Google Scholar]
- 8.Liang JT, Huang KC, Lai HS, Lee PH, Cheng YM, Hsu HC, et al. High-frequency microsatellite instability predicts better chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV sporadic colorectal cancer after palliative bowel resection. Int J Cancer. 2002;101:519–25. doi: 10.1002/ijc.10643. [DOI] [PubMed] [Google Scholar]
- 9.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57. [PubMed] [Google Scholar]
- 10.Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123:1804–11. doi: 10.1053/gast.2002.37070. [DOI] [PubMed] [Google Scholar]
- 11.Xicola RM, Llor X, Pons E, Castells A, Alenda C, Pinol V, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst. 2007;99:244–52. doi: 10.1093/jnci/djk033. [DOI] [PubMed] [Google Scholar]
- 12.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shia JR. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:301–7. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander J. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527–35. doi: 10.1016/S0002-9440(10)63994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward R, Meagher A, Tomlinson I, O'Connor T, Norrie M, Wu R, et al. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48:821–9. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guidoboni M, Gafà R, Viel A, Doglioni C, Russo A, Santini A, et al. Microsatellite instability and high content of activated cytotoxic lymphocytes identify colon cancer patients with a favorable prognosis. Am J Pathol. 2001;159:297–304. doi: 10.1016/S0002-9440(10)61695-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterlongo P, Nafa K, Lerman GS, Glogowski E, Shia J, Ye TZ, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer. 2003;107:571–9. doi: 10.1002/ijc.11415. [DOI] [PubMed] [Google Scholar]
- 18.Khanna M, Cao W, Zirvi M, Paty P, Barany F. Ligase detection reaction for identification of low abundance mutations. Clin Biochem. 1999;32:287–90. doi: 10.1016/s0009-9120(99)00020-x. [DOI] [PubMed] [Google Scholar]
- 19.Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, Forslund A, et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008;265:258–69. doi: 10.1016/j.canlet.2008.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li LS, Kim NG, Kim SH, Park C, Kim H, Kang HJ, et al. Chromosomal Imbalances in the Colorectal Carcinomas with Microsatellite Instability. Am J Pathol. 2003;163:1429–36. doi: 10.1016/S0002-9440(10)63500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trautmann K, Terdiman JP, French AJ, Roydasgupta R, Sein N, Kakar S, et al. Chromosomal instability in microsatellite-unstable and stable colon cancer. Clin Cancer Res. 2006;12:6379–85. doi: 10.1158/1078-0432.CCR-06-1248. [DOI] [PubMed] [Google Scholar]
- 23.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. 1998;338:1481–7. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]