

Abstract
The small heat-shock protein Hsp20 (heat-shock protein 20), also known as HspB6, has been shown to protect against a number of pathophysiological cardiac processes, including hypertrophy and apoptosis. Following β-adrenergic stimulation and local increases in cAMP, Hsp20 is phosphorylated on Ser16 by PKA (protein kinase A). This covalent modification is required for many of its cardioprotective effects. Both Hsp20 expression levels and its phosphorylation on Ser16 are increased in ischaemic myocardium. Transgenic mouse models with cardiac-specific overexpression of Hsp20 that are subject to ischaemia/reperfusion show smaller myocardial infarcts, and improved recovery of contractile performance during the reperfusion phase, compared with wild-type mice. This has been attributed to Hsp20’s ability to protect against cardiomyocyte necrosis and apoptosis. Phosphomimics of Hsp20 (S16D mutants) confer improved protection from β-agonist-induced apoptosis in the heart, whereas phospho-null mutants (S16A) provide no protection. Naturally occurring mutants of Hsp20 at position 20 (P20L substitution) are associated with markedly reduced Hsp20 phosphorylation at Ser16, and this lack of phosphorylation correlates with abrogation of Hsp20’s cardioprotective effects. Therefore phosphorylation of Hsp20 at Ser16 by PKA is vital for the cardioprotective actions of this small heat-shock protein. Selective targeting of signalling elements that can enhance this modification represents an exciting new therapeutic avenue for the prevention and treatment of myocardial remodelling and ischaemic injury.
Keywords: cardioprotection, heat-shock protein 20 (Hsp20), phosphodiesterase type 4 (PDE4), phosphorylation, protein kinase A (PKA), small heat-shock protein
Introduction
Hsps (heat-shock proteins) are a diverse group of molecular chaperones that are up-regulated in response to cellular stress. Originally described in Drosophila melanogaster as a group of proteins whose expression was increased at elevated temperatures [1], Hsps have been extensively characterized, and a variety of cellular triggers for their induction have now been identified, including I/R (ischaemia/reperfusion) [2], oxidative stress [3] and glucose deprivation [4]. The chaperone activities of Hsps include prevention of protein misfolding, refolding of denatured proteins and their targeting for proteolytic degradation [5]. Hsps may be broadly classified according to their molecular mass as either high-molecular-mass (e.g. Hsp90, Hsp70) or low-molecular-mass sHsps (small Hsps). The sHsps, which include Hsp20, are less frequently induced by heat stress, and several family members, such as Hsp27 and αB-crystallin, are known to be abundant in cardiac and skeletal muscle, where they increase in response to stress to protect against muscle ischaemia [5,6].
Many Hsps are now known to play essential protective roles in the cardiovascular system. The present mini-review focuses on the protective actions of the sHsp Hsp20 in the heart, with particular emphasis on cardiac hypertrophy and I/R injury, and how phosphorylation of Hsp20 by PKA (protein kinase A) enhances these cardioprotective effects.
Small heat-shock proteins
sHsps are a diverse group of proteins with different subcellular localization and tissue distribution [7]. To date, ten sHsp isoforms have been identified, and these are formally classified as HspB1–HspB10 [8]. Hsp20, also known as HspB6, is expressed at high levels in cardiac, skeletal and vascular smooth muscle, where it represents up to 1% of total protein [7,9]. sHsps range from 12 to 43 kDa, and are characterized by a stretch of amino acids in their C termini known as the α-crystallin domain, which facilitates their chaperone activities [8] (Figure 1). In addition to the α-crystallin domain, Hsp20 also possesses an N-terminal domain which is involved in inhibiting platelet aggregation, and a region similar to the minimal inhibitory region of troponin I, which is thought to be involved in actin binding [6] (Figure 1). Hsp20 has long been a focus of interest in the field of cardiovascular research, as it is the only sHsp to contain a PKA/PKG (protein kinase G) consensus phosphorylation sequence, of the form R13RAS16 (the bold residue is the phosphorylation site) within its N-terminus [7,10]. Thus Hsp20 may be regulated by the β-adrenergic/cAMP/PKA signalling pathway, which is known to be chronically activated in heart failure. PKG phosphorylation of Hsp20, in response to nitric oxide stimulation of guanylate cyclases and generation of cGMP, has been shown to modulate smooth muscle relaxation, and is discussed in detail elsewhere [10-12].
Figure 1.
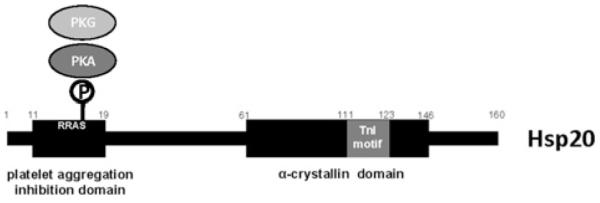
Schematic diagram of the domain structure of Hsp20 Hsp20 is a 160-amino-acid protein. A nine-amino-acid motif (WLRRAS-APL) at its N-terminus has been shown to inhibit thrombin-induced platelet aggregation [6]. This region encompasses a PKA/PKG consensus phosphorylation sequence, RRAS16, which allows the function of the protein to be regulated by β-adrenergic signalling. The C-terminal α-crystallin domain is common to all sHSPs and aids in their chaperone activities [2]. In Hsp20, this domain also encompasses a region similar to the minimal inhibitory region of the thin filament-associated protein troponin I (TnI) (GFVAREFHRRYRL), which may facilitate its interaction with actin [6]. Hsp20 has been shown to translocate to the myofilament under ischaemic conditions [27], and is able to bind actin [28], therefore it may also play a role in stabilizing the cytoskeleton during ischaemic stress [29].
β-Adrenergic signalling and Hsp20 phosphorylation
In cardiac cells, stress results in a rise in circulating catecholamines, and β-adrenergic stimulation triggers increased intracellular generation of the second messenger cAMP. The concomitant activation of PKA permits the phosphorylation of key downstream targets involved in excitation–contraction coupling. These include the ryanodine receptor and phospholamban, which regulate calcium handling at the sarcoplasmic reticulum, and myofilament proteins directly involved in contraction, such as troponin I and myosin-binding protein C. The net result is to increase the rate and efficiency of myocyte contraction, and improve cardiac output [13]. Initially, this acts to compensate for the provoking stress. However, upon chronic up-regulation of cAMP synthesis, these effects become detrimental, leading to cardiomyocyte hypertrophy, apoptosis and further deterioration in cardiac function [14].
In cells, PKA activity is tightly regulated in amplitude, space and time by the compartmentalization of cAMP [15]. A freely diffusible entity, cAMP can potentially flood the interior of the cell, causing inappropriate phosphorylation and activation of downstream PKA targets. This situation is avoided by the opposing action of cyclic nucleotide PDEs (phosphodiesterases). Members of the PDE superfamily provide the only means of hydrolysing cAMP within the cell, and intracellular targeting of PDEs enables the creation of subcellular compartments with high levels of cAMP relative to the surrounding environment [16]. Thus PDEs have the capacity to influence activation of PKA, and the phosphorylation status of PKA target proteins. Recently, we have shown that isoforms of the PDE4 family form a complex with Hsp20 [17]. Under basal conditions, this interaction maintains Hsp20 in a hypophosphorylated state. Upon prolonged β-adrenergic stimulation, as is seen in the failing heart, the pool of PDE4 tethered to Hsp20 becomes swamped, leading to a local increase in cAMP, PKA activation and phosphorylation of Hsp20 on Ser16 [17].
A further level of regulation is provided by AKAPs (A-kinase-anchoring proteins). AKAPs are a diverse group of scaffold proteins that organize cellular signalling pathways by tethering PKA and other signalling enzymes to specific locations within the cell [18]. Previously, no AKAP had been identified for the pool of PKA that phosphorylates Hsp20; however, Hsp20 has now been shown to interact with AKAP–Lbc, an anchoring protein that is enriched in cardiomyocytes (H.V. Edwards, J.D. Scott and G.S. Baillie, unpublished work). This is an interesting observation, as AKAP–Lbc has been implicated in the development of cardiac hypertrophy (discussed further below) [19].
Cardioprotective effects of Hsp20
I/R injury and cardiomyocyte apoptosis
One of the commonest causes of heart failure is myocardial ischaemia, with subsequent progression to infarction [20]. Even transient ischaemia can lead to myocardial necrosis and apoptosis. Myocardial reperfusion strategies aim to reduce morbidity and mortality by restoring tissue oxygenation, but may, in fact, result in further cellular damage, due to mitochondrial generation of reactive oxygen species. This phenomenon is known as I/R injury [21]. It is now recognized that Hsp20 plays an important role in protecting against I/R injury. Major evidence in support of this role has come from the use of TG (transgenic) mouse models. TG mice with cardiac-specific overexpression of Hsp20 subject to I/R displayed a 6-fold reduction in myocardial infarct size, and improved recovery of contractile performance during reperfusion, compared with wt (wild-type) mice [22]. The molecular mechanisms underlying this dramatic reduction in infarct size have been investigated by studies on isolated TG heart preparations. Hsp20-overexpressing TG hearts exhibited reduced release of lactate dehydrogenase, consistent with reduced necrosis, and a significant reduction in DNA fragmentation and TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP nickend labelling) staining, indicating attenuation of apoptosis. The observed effects on apoptosis may be explained by evidence that Hsp20 can interact with the pro-apoptotic protein Bax, thus preventing its translocation from the cytosol to mitochondria, and repression of caspase 3 activity [22].
Levels of phospho-Ser16 Hsp20 were more than doubled following 24 h of reperfusion in the TG hearts, suggesting that this modification may be necessary to protect against I/R injury. Indeed, the importance of Hsp20 phosphorylation by PKA in mediating its cardioprotective effects has been comprehensively illustrated by studies employing Hsp20 phosphosite mutations. Here, Ser16 is replaced by either non-phosphorylatable alanine (S16A) or aspartate (S16D) to mimic constitutive phosphorylation. Fan et al. [23] employed recombinant adenoviral transfer of wt-Hsp20, S16D-Hsp20 and S16A-Hsp20 into cultured adult cardiomyocytes. AdHsp20-wt (adenovirally expressed wt-Hsp20) protected against β-agonist-induced apoptosis, as determined by a reduction in pyknotic nuclei, and this Hsp20 was shown to be significantly phosphorylated [23]. S16D overexpression provided even greater protection against apoptosis than the wt protein, whereas the S16A mutant conferred no such protection. Consistent with these observations, caspase 3 activity was reduced by 10% in wt-infected cells and by 25% in S16D-treated cells, but was unaffected in S16A cells [23].
When S16A was exchanged for overexpression of wt Hsp20 in the TG mouse, this led to impaired functional recovery of hearts ex vivo during I/R compared with non-TG hearts. S16A TG hearts also exhibited increased necrosis and apoptosis [2]. The increased cardioprotective abilities of phosphorylated Hsp20 may be in part associated with its ability to activate autophagy pathways. Autophagy, a physiological catabolic pathway enabling the cell to degrade and recycle damaged organelles, is known to be up-regulated in myocardial ischaemia [24]. S16A TG hearts exhibited an impaired ability to activate autophagy post-I/R, compared with wt, and pre-treatment of S16A hearts with rapamycin (an activator of autophagy pathways) improved their functional recovery in response to I/R injury [2].
The role of Ser16 phosphorylation of Hsp20 in cardio-protection is exemplified further by genetic screening studies performed in patients with dilated cardiomyopathy. Screening identified a C59T base substitution in the first exon of the Hsp20 gene in a subset of screened individuals both with and without heart disease. This single nucleotide polymorphism results in a proline-to-leucine change at position 20 (P20L), just downstream of the PKA site, and is associated with changes in secondary structure [25]. Adenoviral transfer of P20L-Hsp20 into adult rat cardio-myocytes was associated with significantly diminished Ser16 phosphorylation following I/R, and complete abrogation of the anti-apoptotic effects of Hsp20 [25], again illustrating the fundamental importance of this modification. It is clear from the results presented above that modulation of Hsp20 Ser16 phosphorylation represents a key therapeutic target in the treatment of ischaemic heart disease.
Cardiac hypertrophy
Ser16 phosphorylation of Hsp20 has also been shown to protect against cardiac hypertrophy. Hypertrophy may be a beneficial physiological response, for example in the case of elite athletes, to improve cardiac output or a pathological process that occurs secondary to pressure/volume overload associated with hypertension and valvular heart disease [5]. Hsp20 expression is increased in response to hypertrophic stimuli, such as chronic β-adrenergic stimulation [2,23]. TG mice with cardiac-specific overexpression of Hsp20 and subject to chronic β-adrenergic stimulation with the synthetic agonist isoproterenol, are protected from the increases in heart weight/body weight ratio and cardiomyocyte size that are the hallmarks of cardiac hypertrophy [26]. In addition, TG hearts show reduced induction of the fetal gene response that accompanies hypertrophy. These effects have been linked to suppression of ASK-1 (apoptosis signal-regulating kinase 1) signalling, with reduced activation of the downstream JNK (c-Jun N-terminal kinase)/p38 signalling pathway [26].
The importance of phosphorylation of Hsp20 at Ser16 in protection against cardiac hypertrophy has recently been highlighted by a study on PDEs [17]. Members of the PDE4 family have been shown to form a complex with Hsp20, which maintains it in a dephosphorylated state until chronic β-adrenergic stimulation and a consequent sustained rise in the cAMP level saturates the PDE, allowing phosphorylation of Hsp20 [17]. Using peptide array technology and in vitro binding assays, the docking site for Hsp20 on PDE4 was mapped to the conserved catalytic region of the PDE. This sequence information was then used to design a cell-permeant disruptor peptide which specifically inhibited the interaction between Hsp20 and PDE4. Treatment of neonatal rat cardiomyocytes with this peptide disruptor led to highly phosphorylated endogenous Hsp20, and attenuation of β-agonist-induced hypertrophy, as determined by a reduction in cardiomyocyte size and measurement of fetal gene expression [17]. This study provides further direct evidence that alteration of the phosphorylation status of Hsp20, and not just the expression level of the protein, is required to fully realize its cardioprotective effects.
Recent work in our laboratory indicates an interaction between Hsp20 and the multi-purpose scaffold protein AKAP–Lbc to facilitate its phosphorylation by PKA (H.V. Edwards, J.D. Scott and G.S. Baillie, unpublished work). In the heart, AKAP–Lbc anchors PKA and PKC (protein kinase C) to mediate activation of a third anchored kinase, PKD (protein kinase D) 1 [19]. Interestingly, expression of AKAP–Lbc is increased in hypertrophic cardiomyocytes, and it has been proposed that this augments PKD1 activation, which favours phosphorylation and nuclear export of HDAC5 (histone deacetylase 5), and the consequent depression of hypertrophic genes [19]. Taken together, these results suggest that induction of cardiac hypertrophy may involve a complex signalling interplay centring on AKAP–Lbc, where PKD1 activation favours a hypertrophic phenotype, and PKA activation, with subsequent phosphorylation of Hsp20, opposes these effects (Figure 2). Further studies are required to understand how Hsp20 and the AKAP interact, and whether this interaction is deregulated in cardiac disease.
Figure 2.
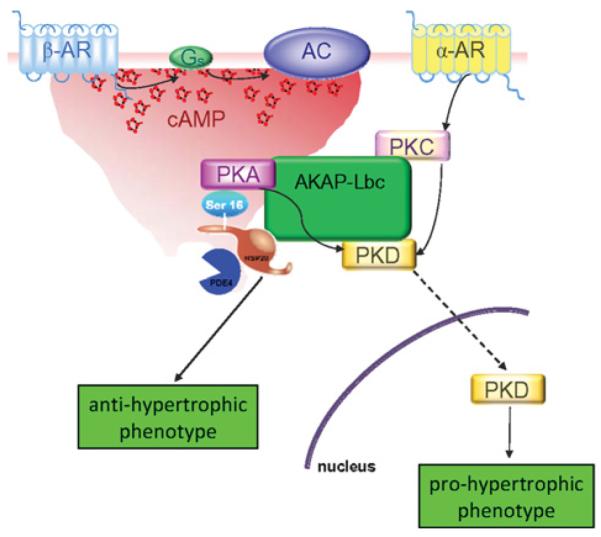
Proposed role of Hsp20 in protecting against β-agonist-induced hypertrophy Following β-adrenergic stimulation, a local cAMP gradient is generated via the action of PDE4 isoforms associated directly with Hsp20. With sustained production of cAMP, the local PDE4 complement becomes saturated, leading to the activation of PKA, which may be scaffolded by AKAP–Lbc. PKA then phosphorylates Hsp20 on Ser16, leading to induction of its cardioprotective actions [17]. Following α-adrenergic stimulation, PKC isoforms also scaffolded by AKAP–Lbc can phosphorylate anchored PKD. Combined PKA/PKC phosphorylation leads to the activation and translocation of PKD to the nucleus, where it contributes to the depression of transcription factors involved in the fetal gene response, a key mediator of the hypertrophic phenotype [19]. AC, adenylate cyclase; α-AR, α-adrenergic receptor; β-AR, β-adrenergic receptor.
Conclusions
Heart failure, resulting from ischaemic heart disease and cardiomyopathy, presents an increasing financial and medical burden. Strategies which selectively modulate components of the β-adrenergic signalling pathway have long been of interest in the search for more effective treatments for heart failure [13]. Phosphorylation of Hsp20 at Ser16 is significantly increased under ischaemic conditions in TG mice hearts [2]. The ratio of phospho-Ser16 to total Hsp20 has also been found to increase in failing human hearts compared with donor hearts [2]. Therefore, as a PKA effector with significant cardioprotective abilities, Hsp20 is of considerable interest as a potential therapeutic target.
Acknowledgements
We thank members of the Scott laboratory at the University of Washington, Seattle, for helpful discussions regarding AKAP–Lbc.
Funding
G.S.B. is supported by the Medical Research Council [grant number G0600765] and Fondation Leducq [grant number 06CVD02]. H.V.E. is supported by a Biotechnology and Biological Sciences Research Council grant. J.D.S. was supported in part by the National Institutes of Health [grant number HL088366].
Abbreviations used
- AKAP
A-kinase-anchoring protein
- Hsp
heat-shock protein
- I/R
ischaemia/reperfusion
- PDE
phosphodiesterase
- PKA
protein kinase A
- PKC
protein kinase C
- PKD
protein kinase D
- PKG
protein kinase G
- sHsp
small Hsp
- TG
transgenic
- wt
wild-type
References
- 1.Tissieres A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J. Mol. Biol. 1974;85:389–398. doi: 10.1016/0022-2836(74)90447-1. [DOI] [PubMed] [Google Scholar]
- 2.Qian J, Ren XP, Wang XH, Zhang PY, Jones WK, Molkentin JD, Fan GC, Kranias EG. Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death. Circ. Res. 2009;105:1223–1231. doi: 10.1161/CIRCRESAHA.109.200378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 4.Sciandra JJ, Subjeck JR. The effects of glucose on protein synthesis and thermostability in Chinese hamster ovary cells. J. Biol. Chem. 1983;258:12091–12093. [PubMed] [Google Scholar]
- 5.Willis MS, Patterson C. Hold me tight: role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan GC, Kranias EG. Small heat shock protein 20 (HspB6) in cardiac hypertrophy and failure. J. Mol. Cell. Cardiol. 2011;51:574–577. doi: 10.1016/j.yjmcc.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan GC, Chu GX, Kranias EG. Hsp20 and its cardioprotection. Trends Cardiovasc. Med. 2005;15:138–141. doi: 10.1016/j.tcm.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JAM, De Jong WW. The human genome encodes 10 α-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones. 2003;8:53–61. doi: 10.1379/1466-1268(2003)8<53:thgecs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dreiza CM, Komalavilas P, Furnish EJ, Flynn CR, Sheller MR, Smoke CC, Lopes LB, Brophy CM. The small heat shock protein, HSPB6, in muscle function and disease. Cell Stress Chaperones. 2010;15:1–11. doi: 10.1007/s12192-009-0127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beall A, Bagwell D, Woodrum D, Stoming TA, Kato K, Suzuki A, Rasmussen H, Brophy CM. The small heat shock-related protein, HSP20, is phosphorylated on serine 16 during cyclic nucleotide-dependent relaxation. J. Biol. Chem. 1999;274:11344–11351. doi: 10.1074/jbc.274.16.11344. [DOI] [PubMed] [Google Scholar]
- 11.Rembold CM, Foster DB, Strauss JD, Wingard CJ, Van Eyk JE. cGMP-mediated phosphorylation of heat shock protein 20 may cause smooth muscle relaxation without myosin light chain dephosphorylation in swine carotid artery. J. Physiol. 2000;524:865–878. doi: 10.1111/j.1469-7793.2000.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flynn CR, Komalavilas P, Tessier D, Thresher J, Niederkofler EE, Dreiza CM, Nelson RW, Panitch A, Joshi L, Brophy CM. Transduction of biologically active motifs of the small heat shock-related protein, HSP20, leads to relaxation of vascular smooth muscle. FASEB J. 2003;17:1358–1360. doi: 10.1096/fj.02-1028fje. [DOI] [PubMed] [Google Scholar]
- 13.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 14.El-Armouche E, Eschenhagen T. β-Adrenergic stimulation and myocardial function in the failing heart. Heart Failure Rev. 2009;14:225–241. doi: 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- 15.Houslay MD, Baillie GS, Maurice DM. cAMP-specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ. Res. 2007;100:950–966. doi: 10.1161/01.RES.0000261934.56938.38. [DOI] [PubMed] [Google Scholar]
- 16.Baillie GS. Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009;276:1790–1799. doi: 10.1111/j.1742-4658.2009.06926.x. [DOI] [PubMed] [Google Scholar]
- 17.Sin YY, Edwards HV, Li X, Day JP, Christian F, Dunlop AJ, Adams DR, Zaccolo M, Houslay MD, Baillie GS. Disruption of the cyclic AMP phosphodiesterase-4 (PDE4)-HSP20 complex attenuates the β-agonist induced hypertrophic response in cardiac myocytes. J. Mol. Cell. Cardiol. 2011;50:872–883. doi: 10.1016/j.yjmcc.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interventions. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carnegie GK, Soughayer J, Smith FD, Pedroja BS, Zhang F, Diviani D, Bristow MR, Kunkel MT, Newton AC, Langeberg LK, Scott JD. AKAP–Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell. 2008;32:169–179. doi: 10.1016/j.molcel.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005;14:170–175. doi: 10.1016/j.carpath.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Murphy E, Steenburgen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan GC, Ren XP, Qian J, Yuan QY, Nicolaou P, Wang Y, Jones WK, Chu GX, Kranias EG. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–179923. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 23.Fan GC, Chu GX, Mitton B, Song QJ, Yuan QY, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ. Res. 2004;94:1474–1482. doi: 10.1161/01.RES.0000129179.66631.00. [DOI] [PubMed] [Google Scholar]
- 24.Gustaffson AB, Gottlieb R. Autophagy in ischemic heart disease. Circ. Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicolaou P, Knoll R, Haghighi K, Fan GC, Dorn GW, Hasenfuss G, Kranias EG. Human mutation in the anti-apoptotic heat shock protein 20 abrogates its cardioprotective effects. J. Biol. Chem. 2008;283:33465–33471. doi: 10.1074/jbc.M802307200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan GC, Yuan QY, Song GJ, Wang YG, Chen GL, Qian J, Zhou XY, Lee YJ, Ashraf M, Kranias EG. Small heat-shock protein Hsp20 attenuates β-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ. Res. 2006;99:1233–1242. doi: 10.1161/01.RES.0000251074.19348.af. [DOI] [PubMed] [Google Scholar]
- 27.Golenhofen N, Perng MD, Quinlan RA, Drenckhahn D. Comparison of the small heat shock proteins αB-crystallin, MKBP, HSP25, HSP20, and cvHSP in heart and skeletal muscle. Histochem. Cell Biol. 2004;122:415–425. doi: 10.1007/s00418-004-0711-z. [DOI] [PubMed] [Google Scholar]
- 28.Brophy CM, Lamb S, Graham A. The small heat shock-related protein-20 is an actin-associated protein. J. Vasc. Surg. 1999;29:326–333. doi: 10.1016/s0741-5214(99)70385-x. [DOI] [PubMed] [Google Scholar]
- 29.Dreiza CM, Brophy CM, Komalavilas P, Furnish EJ, Joshi L, Pallero MA, Murphy-Ullrich JE, von Rechenberg M, Ho Y-SJ, Richardson B, et al. Transducible heat shock protein 20 (HSP20) phosphopeptide alters cytoskeletal dynamics. FASEB J. 2005;19:261–263. doi: 10.1096/fj.04-2911fje. [DOI] [PubMed] [Google Scholar]