

Abstract
Molecular scaffolds containing alkylfluorine substituents are desired in many areas of chemical research from materials to pharmaceuticals. Herein, we report the invention of a new reagent (Zn(SO2CF2H)2, DFMS) for the innate difluoromethylation of organic substrates via a radical process. This mild, operationally simple, chemoselective, and scalable difluoromethylation method is compatible with a range of nitrogen-containing heteroarene substrates of varying complexity as well as select classes of conjugated π-systems and thiols. Regiochemical comparisons suggest that the CF2H radical generated from the new reagent possesses nucleophilic character.
The difluoromethyl group (CF2H) is an intriguing structural motif that has great potential in the areas of pharmaceuticals, agrochemicals, and materials.1–5 In the area of medicinal chemistry, the CF2H unit is of special interest for its use in isostere-based drug design.6 As a lipophilic hydrogen bond donor, CF2H substitution offers an alternative to more traditional hydrogen bond donors while improving membrane permeability. For example, the difluoromethyl group has been utilized as a thiol mimic in the context of HCV NS3 protease inhibitors, successfully mimicking the cysteine CH2SH P1 element in the parent compounds.7 It has also been used as a hydroxamic acid hydroxyl isostere in a series of COX-2 and 5-LOX dual inhibitors, likely diminishing the metabolic toxicity expressed by some hydroxamic acids.8 While several methods exist for the fluorination and alkylfluorination of organic substrates, strategies for direct difluoromethylation are less common, particularly in the context of heteroarene substrates. In this communication, the invention of a reagent to accomplish the mild, direct, scalable, and predictably selective difluoromethylation of heteroarenes and related structures is reported.
Currently, all known methods for heteroarene difluoromethylation rely on prefunctionalization through a programmed approach (Figure 1A). Perhaps the most widely known strategy involves difluorination of aldehydes using (diethylamino)sulfur trifluoride (DAST).9,10 Heteroarene-CF2H compounds may also be accessed through a copper-catalyzed cross-coupling/decarboxylation sequence,11 or via radical debromination from a heteroarene-CF2Br precursor.12 Although these methods boast impressive levels of reactivity and site selectivity, the need for methods achieving the direct transfer of the CF2H unit has been explicitly voiced in a recent review.13
Figure 1.

Invention of a new difluoromethylation reagent.
The pioneering work reported by Minisci on the decarboxylative generation of alkyl radicals and subsequent addition to heteroarenes (a form of innate14 C–H functionalization) inspired initial forays into this area.15 Thus, difluoroacetic acid was the first CF2H source that was tested on caffeine (1) under a variety of oxidative conditions (Figure 1B). However, this strategy was unsuccessful, necessitating the exploration of alternative methods for generating CF2H-radicals. The only previously reported example of a direct radical transfer of a CF2H unit was reported by Chen and co-workers in the context of additions to olefins and alkynes (no other substrate classes reported) using non-commercial and operationally problematic HCF2I gas as a precursor.16
In contrast, we have previously demonstrated the ease and practicality associated with the use of stable fluoroalkyl metal sulfinate complexes for the generation of trifluoromethyl radicals and subsequent addition to heteroarenes.17 The seemingly simple extension of this approach to difluoromethylation was without precedent and required extensive experimentation.18 Ultimately, after several strategies were evaluated, difluoromethanesulfonyl chloride was recognized as a convenient, commercially available starting point for the synthesis of a variety of different difluoromethylsulfinate reagents (Figure 1C). Using caffeine (1) as a model substrate, systematic variation of the metal counterion led to the identification of Zn(SO2CF2H)2 (3, DFMS) as the optimum precursor for the CF2H-radical. DFMS is an easily prepared, air stable, free flowing white powder whose structure was confirmed by X-ray crystallographic analysis and shown to exist as polymer in the solid state (see SI for more details).
With this new reagent in-hand, the scope of heteroarene difluoromethylation was evaluated on a broad cross-section of heterocyclic space. Many substrates showed good reactivity towards DFMS under the standard reaction conditions using tBuOOH in a CH2Cl2/H2O solvent system, although select substrates suffered from poor conversion. In these cases, a second addition of DFMS and tBuOOH was added after 12–24 hours to drive the reaction towards completion. It was also found that TFA showed improved rate and conversion for selected nitrogen heteroarene substrates, but was not essential to achieve the desired reactivity for most cases. Pyridines (5–10, 12, 22), pyrroles (14, 15), pyrimidines (18), quinoxalines (16), pyrazines (17), xanthines (2, 19, 20), purines (21), quinoline (23), thiadiazoles(13) and pyridinones (11) are all competent participants in the described method, which is tolerant of several potentially sensitive functional groups. In most cases, substrates with multiple potential reaction sites exhibit high levels of regioselectivity, commonly producing only one observable regioisomer with C–H functionalization occurring at electron-deficient positions.17 It should be noted that all of the compounds illustrated in Table 1 are chemical entities with heretofore unreported synthetic procedures. In addition to the heteroarene substrates listed in Table 1, other organic substrates are also reactive towards difluoromethylation by the described process (Table 2). Aromatic thiols exhibit unexpected reactivity towards the CF2H-radical, leading to the generation of difluoromethyl thioethers. Radical difluoromethylation was also successful in the context of other electron-deficient π-systems such as α, β-unsaturated enones.
Table 1.
Scope of C–H difluoromethylation of heteroarene substrates.
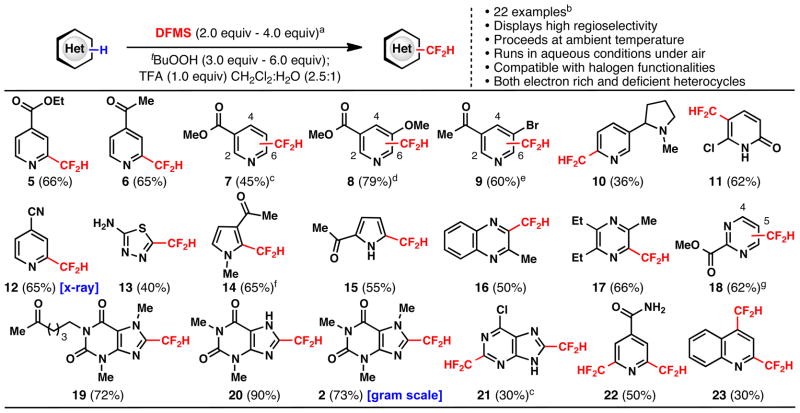
|
Heterocycle (1.0 equiv), DFMS (2.0 equiv), tert-butyl hydroperoxide (3.0 equiv), TFA (1.0 equiv), 23 °C; isolated yields of chromatographically pure products are displayed, unless otherwise noted.
Both indole and 1-(2,4-dimethylfuran-3-yl)ethanone were unreactive, giving recovered starting material.
C2:C4:C6; 1.5:1.5:2.
C2:C4:C6; 1:1:2.
C2:C4:C6; 3:1:2.
Reaction showed incomplete conversion after 12 hours, and a second addition of DFMS (2.0 equiv) and tert-butyl hydroperoxide (3.0 equiv) was added.
Heterocycle (1.0 equiv), DFMS (3.0 equiv), tert-butyl hydroperoxide (4.0 equiv), 23 °C.
C4:C5; 4:1.
Table 2.
Difluoromethylation of thiols and enones.
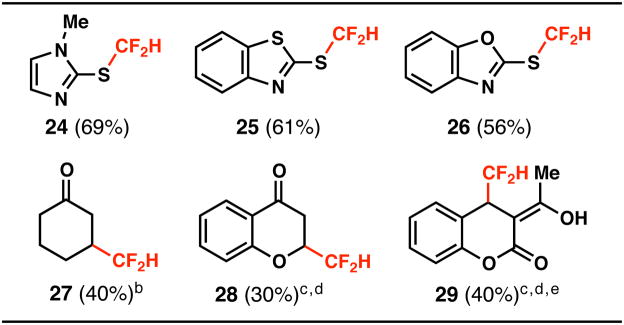
|
Standard conditions were followed and yields were obtained after silica gel chromatography.
DFMS (1.0 equiv) and tert-butyl hydroperoxide (1.5 equiv) was used.
Second addition of DFMS (2.0 equiv) and tert-butyl hydroperoxide (3.0 equiv) was added.
α,α,α-trifluorotoluene was used instead of dichloromethane.
keto:enol (1:5).
In addition to the predictable site-selectivity observed in the difluoromethylation of simple small-molecule substrates, innate C–H difluoromethylation of complex reacting partners with many potential reactive sites is also predictable. As shown in Figure 2, the site-selectivity of difluoromethylation depends on the combined electronic properties of the reacting π-system and incoming radical species. This point is illustrated through a comparison between CF3- and CF2H-radical additions to dihydroquinine and Chantix. In both cases, high levels of selectivity are observed for CF3- and CF2H-radical addition, in spite of the multiple, potentially reactive sites. For both dihydroquinine and Chantix, innate radical C–H trifluoromethylation takes place at the most electron-rich position within the arene rings (C7 and C5, 32 and 33, respectively).17 Conversely, difluoromethylation occurs exclusively at electron-poor sites adjacent to heteroatoms within the heteroarene rings (both at C2, 30 and 31). The orthogonality of these two approaches is a powerful example of the ability to fine-tune an approach to alkylfluorination when considering the innate reactivity and electronic nature of the two reacting components.
Figure 2.

Regiochemical comparison of innate difluoro- and trifluoromethylations. Isolated yields after preparative HPLC purification.
Although many substrates listed above in Table 1 produce a single observable regioisomer, it is possible to access alternative substitution in some cases by judicious choice of organic solvent media. As shown in Figure 3, 4-acetylpyridine shows exceptional regioselectivity in CH2Cl2/H2O, leading to exclusive formation of the C2-CF2H product (6-C2). However, substituting DMSO for CH2Cl2 leads to significant reversal of regiochemistry, favoring the C3-CF2H isomer (6-C3) in 1.5:1.0 ratio. Although C3-selectivity is modest under these conditions, the change in selectivity is significant considering that the C3-CF2H product is inaccessible under standard reaction conditions.
Figure 3.

Solvent effects on regioselectivity. aStandard conditions were followed. bSecond addition of DFMS (2.0 equiv) and tert-butyl hydroperoxide (3.0 equiv) was added.
In summary, a new reagent (DFMS) for direct difluoromethylation has been invented to access compounds of high value that, in almost all cases, were hitherto unknown. DFMS (3) is effective for the direct transfer of a CF2H unit to various organic substrates including heteroarenes, α, β-unsaturated enones, and aromatic thiols via user-friendly, scalable, open-flask conditions.19,20 Changes in regioselectivity may be promoted by varying the organic co-solvent in the context of 4-substituted pyridines. Further examination of this and other mechanistic features are ongoing and will be reported in due course. DFMS has recently been commercialized by Sigma-Aldrich (product number L510084).
Supplementary Material
Acknowledgments
We are grateful to Professor Arnold Rheingold and Dr. Curtis E. Moore for X-ray crystallographic analysis. Financial support for this work was provided by the NIH/NIGMS (GM-073949), the Uehara Memorial Foundation (post-doctoral fellowship for Y.F.) the NSF (predoctoral fellowship for R.A.R) and Pfizer Inc. (postdoctoral fellowship for R.D.B.).
Footnotes
ASSOCIATED CONTENT
Experimental procedures and analytical data for all new compounds, including 1H, 13C, and 19F-NMR. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smart BE. Chem Rev. 1996;96:1555–1556. doi: 10.1021/cr960075e. [DOI] [PubMed] [Google Scholar]
- 2.Filler R, Kobayashi Y, Yagupolskii LM. Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications. Elsevier; Amsterdam: 1993. [Google Scholar]
- 3.Welch JT. Tetrahedron. 1987;43:3123–3197. [Google Scholar]
- 4.Rewcastle GW, Gamage SA, Flanagan JU, Frederick R, Denny WA, Baguley BC, Kestell P, Singh R, Kendall JD, Marshall ES, Lill CL, Lee W-J, Kolekar S, Buchanan CM, Jamieson SMF, Sheperd PR. J Med Chem. 2011;54:7105–7126. doi: 10.1021/jm200688y. [DOI] [PubMed] [Google Scholar]
- 5.Furuya T, Kuttruff C, Ritter T. Curr Opin Drug Disc Dev. 2008;11:803–819. [PubMed] [Google Scholar]
- 6.Meanwell NA. J Med Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. and references therin. [DOI] [PubMed] [Google Scholar]
- 7.Narjes F, Koehler KF, Koch U, Gerlach B, Colarusso S, Steinkuhler C, Brunetti M, Altamura S, De Francesco R, Matassa VG. Bioorg Med Chem Lett. 2002;12:701–704. doi: 10.1016/s0960-894x(01)00842-3. [DOI] [PubMed] [Google Scholar]
- 8.Chowdhury MA, Abdellatif KRA, Dong Y, Das D, Suresh MR, Knaus EE. J Med Chem. 2009;52:1525–1529. doi: 10.1021/jm8015188. [DOI] [PubMed] [Google Scholar]
- 9.Markovski LN, Pahinnik VE, Kirsanov AV. Synthesis. 1973:787–789. [Google Scholar]
- 10.(a) Kuduk SD, Schlegel K-A, Yang Z-Q. Quinoline Amide M1 Receptor Positive Allosteric Modulators. PCT/US2010/060007 International Patent. ; (b) Rodgers JD, Shepard S, Arvanitis AG, Wang H, Storace L, Folmer B, Shao L, Zhu W, Glenn JP. N-(Hetero)aryl-pyrrolidine Derivatives of Pyrazol-4-yl-pyrrolo[2, 3-d]pyrimidines and Pyrrol-3-yl-pyrrolo[2, 3-d]pyrimidines as Janus Kinase Inhibitors. PCT/US2010/035783 International Patent.
- 11.Fujikawa K, Fujioka Y, Kobayashi A, Amii H. Org Lett. 2011;13:5560–5563. doi: 10.1021/ol202289z. [DOI] [PubMed] [Google Scholar]
- 12.(a) Dolbier WR, Medebielle MA–M. Tetrahedron Lett. 2001;42:4811–4814. [Google Scholar]; (b) Burkholder CR, Dolbier WR, Medebielle M. J Fluorine Chem. 2001;109:39–48. [Google Scholar]
- 13.Hu J, Zhang W, Wang F. Chem Commun. 2009:7465–7478. doi: 10.1039/b916463d. [DOI] [PubMed] [Google Scholar]
- 14.Bruckel T, Baxter RD, Ishihara Y, Baran PS. Acc Chem Res. 2011 doi: 10.1021/ar200194b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Minisci F, Fontana F, Vismara E. J Heterocycl Chem. 1990;27:79–96. [Google Scholar]; (b) Minisci F, Vismara E, Fontana F. Heterocycles. 1989;28:489–519. [Google Scholar]; (c) Duncton AJ. Med Chem Commun. 2011;2:1135–1161. [Google Scholar]
- 16.Cao P, Duan JX, Chen QY. J Chem Soc, Chem Commun. 1994:737–738. [Google Scholar]
- 17.Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Proc Natl Acad Sci. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attempts at synthesis of NaSO2CF2H based on Chen Q, Long Z, Faming F. CN 1221735 A 19990707 Shenquing Gongkai Shuomingshu. 1999 were fruitless. Synthesis of various [M]SO2CF2H reagents from fluoroalkanesulfinic acid hydrazinium salts based on Nanmyo T, Kashiwaba T, Morinaka T, Kawamoto H, Inoue S, Kume T. 2010013687 PCT Int Appl. 2010 was ineffective. Transferring CF2H via reaction of fluorocarboxylic acids of the general structure [E.W.G.]–CF2–CO2H analogous to Forat G, Mas J-M, Saint-James L. 20020042542 US Pat Appl. 2002 was unsuccessful.
- 19.A sample of DFMS left open to air on the bench-top retained its original purity and reactivity after two months.
- 20.A provisional patent on this work has been filed, application number 61565756.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.