

Abstract
Background & Aims
Transgenes delivered to livers of mice via adeno-associated virus (AAV) are expressed stably, via induction of immune tolerance. However, transgene expression is lost in higher-order primates. We investigated whether inflammatory processes, which likely differ between species, affect the stability of transgene expression.
Methods
We developed a mouse model of vector-unrelated, systemic inflammation following AAV-mediated transfer of genes to liver.
Results
Inflammation eliminated previously stable expression of trangenes delivered by AAV; the limited tissue destruction and persistence of AAV genomes implicated immune responses that were not mediated by cytotoxic T cells. Tumor necrosis factor (TNF)-α downregulated transgene expression from AAV, indicating a role for inflammatory cytokines in loss of transgene expression.
Conclusions
Inflammation and inflammatory cytokines such as TNF-α can reduce AAV-mediated expression of transgenes in livers of mice. Inflammation might therefore affect expression of transgenes from viral vectors in humans.
Keywords: Hepatic gene therapy, loss of tolerance, immune response, liver inflammation
Adeno-associated virus (AAV) is a promising and widely used gene therapy vector. A parvovirus belonging to the genus dependovirus, AAV delivers therapeutic transgenes without inducing toxicity, inflammation or disease in animals and humans. Hence, vectors based on AAV have become the preferred delivery vehicle for gene transfer to multiple organs such as the eye, muscle, brain, heart and lungs. A principal target organ for AAV gene transfer has been the liver given its preponderance in metabolic disorders and access of its parenchymal cells, hepatocytes, to vector through its fenestrated endothelium. In addition, numerous rodent studies have documented that upon systemic delivery most AAV vector encoded transgenes are immunologically inert and rarely induce a humoral or cellular immune response; a prerequisite for persistent transgene expression in the treatment of recessive genetic diseases 1-4.
Unexpectedly, however, when AAV was administered to the liver of nonhuman primates, transgene expression was completely ablated with a concomitant increase in T lymphocyte (CTL) response to the foreign transgene product GFP 5. This finding that induction of tolerance to AAV encoded transgenes in mice cannot always be extrapolated to pre-clinical studies in higher mammals is a major concern and warrants the study of circumstances under which such tolerance can be broken. An important difference is that in contrast to inbred mice that live in a pathogen-free environment, non-human primates and more importantly human subjects are more likely to be exposed to pathogens, toxins or environmental stimuli that induce inflammation. Such differences could be more pronounced in gene therapy patients as many disorders under consideration for hepatic gene transfer are associated with liver pathology that may induce an inflammatory milieu in the liver 6, 7. Furthermore, one cannot exclude the possibility of the development of infectious or chemically induced liver inflammation subsequent to gene transfer by processes unrelated to the vector. It has been reported that in mice tolerant to a superantigen or virus, challenge with antigen-unrelated viral, bacterial or parasitic infections broke tolerance by restoring the effector T cell response 8, 9. We hypothesized that it was the presence of inflammatory processes that influenced the stability of AAV transgene expression following translational studies in higher order primates. In the present study, we developed a mouse model to mimic a scenario where a subject receiving hepatic AAV gene transfer subsequently develops vector-unrelated systemic inflammation. Using this model we demonstrate that immunological tolerance to AAV encoded transgene can be broken following a potent inflammatory response, and describe the likely pathways and effector molecules involved in the loss of transgene expression. To our knowledge the study presented here is the first of its kind in demonstrating the loss of previously stable AAV transgene expression in the liver following an inflammatory insult.
Materials and Methods
Vectors
All vectors were produced by Penn Vector at the University of Pennsylvania.
Animals
C57BL/6 mice and RAG-deficient mice on C57BL/6 background (6-8 weeks of age; Jackson Laboratory, Bar Harbor, ME) with three mice per group were injected via the tail vein with 1011 vector genomes (VG) of AAV serotype-8 vector, or 1010 VG of human adenovirus 5 vector (Ad). Blood was drawn by retro-orbital bleeding at the indicated time points, and serum samples were analyzed for alanine aminotransferase (ALT) and aspartate transaminase (AST) by Antech Diagnostics. Mice were housed under specific pathogen-free conditions at the University of Pennsylvania’s Translational Research Laboratories. All animal procedure protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. All experiments were performed in triplicate on at least two separate occasions.
Transgene detection
Animals were sacrificed at 28 days after Ad or AAV administration. To examine expression of nuclear β-galactosidase (β-gal), X-gal staining on snap-frozen liver cryosections was performed according to standard protocols 10. Quantification of LacZ staining was performed with ImageJ software (NIH, Bethesda, MD). Plasma α1-antitrypsin (AAT) levels were measured by an ELISA as described 11.
Splenocyte isolation
Splenocytes were isolated as previously described 12.
IFN-γ ELISPOT assay
The IFN-γ ELISPOT assay was performed according to the manufacturer’s instructions (BD Biosciences, San Jose, CA) at 9 days after Ad administration, unless indicated otherwise. Splenocytes from individual mice were added to wells at a density of 105 or 5 × 105 cells/well along with 2 μg/ml of β-gal CD8 T-cell epitope [ICPMYARV; described in 13], 2 μg/ml of AAT CD8 T-cell epitope [FALVNYIFF; described in 14] or an overlapping 15-mer peptide library spanning the full length Ad hexon divided into 4 (A, B, C and D) pools (Mimotopes, Victoria, Australia). Splenocytes were incubated at 37 °C, 5% CO2 for 18 hours. Spots were counted using the AID ELISPOT reader system (Cell Technology, Columbia, MD).
Intracellular IFN-γ staining
At 10 days after Ad administration, splenocytes (106 cells) from individual mice were stimulated with Ad hexon peptide pools (2 μg/ml) for 5 hours at 37 °C, 10% CO2 in the presence of Brefeldin A (GolgiPlug; BD Biosciences) and IL-2 (BD Biosciences). Cells were stained with antibodies from BD Biosciences and analyzed on a Cytomics FC500 flow cytometer (Beckman-Coulter, Miami, FL). Data were analyzed using FlowJo software (TreeStar, San Carlos, CA).
Quantitative PCR analysis
For real-time TaqMan PCR, a thyroid hormone binding globulin gene (TBG) promoter assay was designed against the TBG moiety present in AAV cis-plasmids. Primers and probes used were as follows: TBG Fwd, AAACTGCCAATTCCACTGCTG; TBG Rev, CCATAGGCAAAAGCACCAAGA; and TBG Probe, 6FAM-TTG GCC CAA TAG TGA GAA CTT TTT CCT GC-TAMRA.
Toll-like receptor (TLR) ligands and cytokines
TLR4 (Ultrapure E. coli LPS) and TLR9 ligand (Type C CpG oligonucleotide) were purchased from Invivogen (San Diego, CA). Recombinant mouse TNF-α and IL-6 were from R&D Systems (Minneapolis, MN). Mice were administered intraperitoneally (i.p.) with LPS (10 ng), CpG (50 μg), TNF-α (0.5 μg) or IL-6 (1.25 μg). TLR ligands and cytokines were administered for 4 consecutive days starting with the day of Ad administration, AAV administration or in some instances after an interval following AAV administration.
Statistics
Statistical analysis of the presented data was performed using a two-tailed Student’s t-test. A value of P < 0.05 was considered as statistically significant.
Results
Inflammatory signals extinguish transgene expression in a mouse model of liver-directed AAV-mediated gene transfer
We reported earlier that C57BL/6 mice administered intravenously (i.v.) with an AAV8 serotype vector expressing human AAT induced tolerance to the transgene when these animals were subsequently challenged with an Ad vector expressing the same transgene and continued to express AAT due to active suppression of the AAT-specific CTL response that occurs following AdAAT in naive mice 3. The current study used a similar model to explore the consequences of systemic inflammation subsequent to hepatic gene transfer. C57BL/6 mice were i.v. administered with 1011 VG of AAV expressing LacZ under the control of the CMV-enhanced chicken β-actin (CB) promoter, followed 2 weeks later by i.v. challenge with 1010 VG of AdLacZ plus daily i.p. LPS or CpG injections for 4 consecutive days (Figure 1A). As expected, control mice administered with AAVLacZ followed by AdLacZ showed high and stable LacZ expression along with diminished CTL responses to LacZ; elevations in liver transaminases, a surrogate for hepatocellular toxicity, were unremarkable (Figure 1A and C). Strikingly, however, in mice challenged with AdLacZ plus 4 daily injections of LPS or CpG there was a loss of LacZ expression accompanied by a LacZ-specific CTL response (Figure 1A and B).
Figure 1.

Inflammatory signals extinguish LacZ expression in a mouse model of liver-directed AAV-mediated gene transfer. (A) C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of AdLacZ plus daily i.p. injections of LPS or CpG (TLRL) for 4 days. At 28 days after AdLacZ challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (B) At 9 days after AdLacZ challenge, splenocytes were stimulated with the LacZ CD8 T cell epitope and subjected to IFN-γ ELISPOT. Background spot-forming unit (SFU) values were subtracted prior to plotting. (C) At 14 days after AdLacZ challenge, mouse blood was collected to measure AST and ALT levels. Data represent groups of three mice in at least three independent experiments. A two-tailed Student t test was used for statistical analysis. *P < 0.05. **P < 0.01. ***P < 0.001.
As expected, a CTL response to Ad hexon was detected in all animals that received the vector with or without TLR ligands; the magnitude of the CTL response was not influenced by an earlier administration of an AAV vector (Supplementary Figure 1). Mild elevations in liver transaminases were noted following Ad and TLR ligands (Figure 1C and Supplementary Figure 2).
The β-gal transgene used in these studies encodes a nuclear localization signal. It is thus possible that antigenic determinants that are extra-nuclear may enter quite distinct cellular processing pathways leading to distinct outcomes upon Ad challenge. As an example of a gene product most distinct from the nuclear targeted LacZ we tested an AAV vector expressing a secreted human AAT transgene. Upon challenge with Ad plus TLR ligands these mice exhibited dramatically reduced circulating AAT levels and a modest CTL activity (Supplementary Figure 3). These results indicate that loss of expression is less likely to be influenced by the nature of the transgene i.e. whether intracellular or secreted, and more likely due to the induction of inflammation.
Another important finding was that, in the absence of AdLacZ, 4-day injections of LPS or CpG at the time of AAVLacZ administration or at later time point had no impact on LacZ expression (Supplementary Figure 4), indicating the requirement for a combination of both low dose Ad and TLR ligands in elimination of transgene expression.
Inflammation extinguishes transgene expression in the absence of substantial CTL induction to transgene or capsid
In a similar experimental setup, C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and 2 weeks later i.v. challenged with 1010 VG of Ad expressing an irrelevant transgene, GFP, plus daily LPS or CpG injections for 4 consecutive days (Figure 2A). LacZ expression was stable with an absence of LacZ-specific CTLs and transaminitis in mice challenged with AdGFP without additional TLR signaling (Figure 2A, B and C). In stark contrast, LacZ expression was extinguished by coadministration of AdGFP and TLR ligands. Surprisingly, we noted only a weak CTL response to LacZ that was accompanied by a modest elevation in liver transaminases. We found no detectable CTLs against AAV capsid in any of the experimental groups (data not shown).
Figure 2.

Inflammation extinguishes LacZ expression in the absence of substantial CTL induction to LacZ. (A) C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of AdGFP plus daily i.p. injections of LPS or CpG (TLRL) for 4 days. At 28 days after AdGFP challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (B) At 9 days after AdGFP challenge, splenocytes were stimulated with the LacZ CD8 T cell epitope and subjected to IFN-γ ELISPOT. Background SFU values were subtracted prior to plotting. (C) At 14 days after AdGFP challenge, mouse blood was collected to measure AST and ALT levels. Data are representative of three mice per group in at least three independent experiments. A two-tailed Student t test was used for statistical analysis. *P < 0.05. **P < 0.01. ***P < 0.001.
TNF-α response extinguishes transgene expression in the absence of a CTL response
Since TNF-α and IL-6 are downstream mediators of both LPS- and CpG-driven inflammatory processes 15-18, we examined whether these cytokines are involved in the elimination of AAV transgene expression. C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and 2 weeks later i.v. challenged with 1010 VG of AdLacZ along with daily injections of TNF-α or IL-6 for 4 consecutive days (Figure 3A). LacZ expression was lost in TNF-α-treated mice, although, interestingly, we found no LacZ-specific CTL response nor liver transaminase elevations (Figure 3B, C and Supplementary Figure 2). Administration of TNF-α in the absence of Ad had no effect on AAV transgene expression (Supplementary Figure 4). Although not statistically significant, mice treated with a maximum tolerated dose of IL-6 exhibited a trend toward diminished LacZ expression.
Figure 3.

TNF-α response extinguishes LacZ expression in the absence of a CTL response. (A) C57BL/6 mice (n = 3 per group) were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of AdLacZ plus daily i.p. injections of IL-6 or TNF-α for 4 days. At 28 days after AdLacZ challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (B) At 9 days after AdLacZ challenge, splenocytes were stimulated with the LacZ CD8 T cell epitope and subjected to IFN-γ ELISPOT. Background SFU values were subtracted prior to plotting. (C) At 14 days after AdLacZ challenge, mouse blood was collected to measure AST and ALT levels. Data represent three independent experiments. A two-tailed Student t test was used for statistical analysis. ***P < 0.001.
AAV vector genomes persist despite the loss of transgene expression
To determine whether hepatocyte killing was responsible for the loss of transgene expression, we quantified AAV genome copies in the liver of AAVLacZ transduced mice that were challenged with AdLacZ or AdGFP plus 4-day injections of LPS, CpG or TNF-α. Only a 5- to 10-fold drop in the number of AAV genome copies was noted in mice receiving Ad plus TLR ligands, in comparison to mice challenged with Ad alone (Figure 4A). AAV genome copies also remained unchanged in mice challenged with Ad plus TNF-α. This persistence of AAV vector genomes occurred in groups which had extinguished LacZ expression (Figure 4B). The fact that AAV vector genomes persist, together with only modest elevations of liver transaminases in animals that lost LacZ expression, suggests that the elimination of transgene expression is predominantly the consequence of transcriptional silencing rather than due to killing of target cells. The absence of significant numbers of apoptotic cells in any groups post challenge (data not shown) is consistent with this hypothesis.
Figure 4.

AAV vector genomes persist despite the loss of LacZ expression. C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of AdLacZ or AdGFP plus daily i.p. injections of LPS, CpG or TNF-α for 4 days. (A) At 28 days after Ad challenge, total cellular DNA was extracted from the liver and AAV2/8 vector genomes were quantified by real-time PCR. (B) At 28 days after Ad challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. Groups consisted of three mice, and the data are representative of three independent experiments. A two-tailed Student t test was used for statistical analysis. *P < 0.05. **P < 0.01. ***P < 0.001.
Inflammation inhibits AAV encoded transgene expression driven by a liver-specific promoter
Since promoters vary in their interaction with transcriptional factors, we sought to examine the effect of alternative promoters, such as a tissue-specific promoter, in being a target for transcriptional inactivation. Thus, C57BL/6 mice were i.v. injected with 1011 VG of AAV8 expressing LacZ under the control of the liver-specific TBG promoter. Two weeks later, mice were i.v. challenged with 1010 VG of AdLacZ or AdGFP plus 4-day injections of LPS, CpG or TNF-α. LacZ expression was dramatically reduced, however, not completely eliminated, in all groups (Figure 5A) and the effect on expression was less pronounced when compared to that observed with the CB promoter. AAV vector genomes were virtually unchanged in all groups, when compared to mice that were challenged with Ad alone (Figure 5B). This, together with insignificant transaminase elevations (Figure 5C), suggests partial inactivation of the TBG promoter. It is possible that the kinetics of transgene elimination is slower with the TBG promoter and a more dramatic drop in LacZ expression may have been observed at a later time point or that it is less susceptible to inflammation-induced inactivation.
Figure 5.
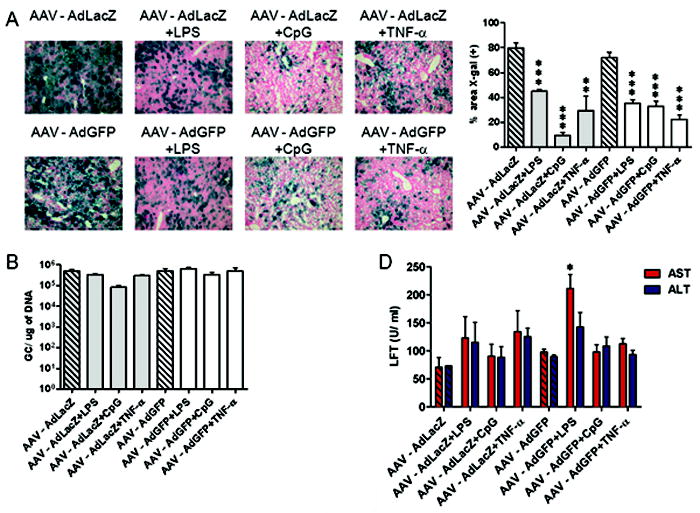
Inflammation inhibits AAVLacZ expression driven by a liver-specific promoter. C57BL/6 mice (n = 3 per group) were i.v. injected with 1011 VG of AAVTBGLacZ and i.v. challenged 2 weeks later with 1010 VG of AdLacZ or AdGFP plus daily i.p. injections of LPS, CpG or TNF-α for 4 days. (A) At 28 days after Ad challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (B) At 28 days after Ad challenge, total cellular DNA was extracted from the liver and AAV2/8 vector genomes were quantified by real-time PCR. (C) At 14 days after Ad challenge, mouse blood was collected to measure AST and ALT levels. Data are representative of three independent experiments. A two-tailed Student t test was used for statistical analysis. *P < 0.05. ***P < 0.001.
A certain level of CTL activity is required to mediate extinction of transgene expression
Studies described above indicate that both Ad and the TLR ligand are required for extinction of expression. The role of the Ad vector becomes even more ambiguous due to inherent nature of these vectors in activating both the innate and adaptive immune response. It is possible that the innate immune response to Ad complements the inflammatory response generated by TLR ligands and this inflammatory milieu is sufficient to knockdown AAV transgene expression. Alternatively, a CTL response, even though not specific to the transgene, may be required along with TLR signaling to eliminate expression. We interrogated these possibilities using an Ad vector incapable of gene expression (UV-inactivated Ad – UVAd) or an immunocompromised mouse model lacking CTLs RAG -/-). First, C57BL/6 mice were i.v. injected with 1011 VG of AAVLacZ and 2 weeks later administered with 1010 VG of either infectious AdGFP or AdGFP that has been UV-inactivated in combination with 4-day injections of CpG (Figure 6A). As anticipated, LacZ expression was unaffected by Ad challenge in the absence of the TLR ligand. However, coadministration of CpG with either infectious or UV-inactivated AdGFP resulted in extinction of AAV-mediated LacZ expression. Next, we performed a similar experiment in RAG -/- mice. Specifically, RAG mice received AAVLacZ followed by AdGFP plus CpG challenge as described above. Surprisingly, in these mice, inflammatory challenge did not lead to extinction of expression (Figure 6B), indicating the requirement of T cells. In attempting to reconcile these different but seemingly interesting findings, we noted that the inactivated Ad vectors did generate a CTL response to Ad capsids when administered to mice (Figure 6C and D); the response was similar or better than that induced by an infectious vector. The CTLs, a result of cross-presentation of Ad hexon proteins, nevertheless were sufficient to impact expression from the unrelated AAV encoded transgene. Collectively, these data suggest that a combination of inflammation and CTLs, even those unrelated to the AAV vector yet perhaps targeting the liver, may be a requirement for extinction of AAV transgene expression.
Figure 6.

A certain level of CTL activity is required to mediate extinction of LacZ expression. (A) C57BL/6 mice (n = 3 per group) were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of UVAdGFP plus daily i.p. injections of CpG for 4 days. At 28 days after UVAdGFP challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (B) Rag-/- mice (n = 3 per group) were i.v. injected with 1011 VG of AAVLacZ and i.v. challenged 2 weeks later with 1010 VG of AdGFP plus daily i.p. injection of CpG for 4 days. At 28 days after AdGFP challenge, liver tissues were evaluated for LacZ expression by X-gal histochemistry. The percentage of liver area positive for LacZ staining was quantified using ImageJ software. (C) C57BL/6 mice (n = 3 per group) were i.v. injected with 1010 VG of AdGFP or UVAdGFP. At 10 days after Ad challenge, splenocytes were stimulated with Ad5 hexon peptide pools A-D and subjected to IFN-γ ELISPOT. Background SFU values were subtracted prior to plotting. (D) C57BL/6 mice (n = 3 per group) were i.v. injected with 1010 VG of AdGFP or UVAdGFP. At 10 days after Ad challenge, splenocytes were stimulated with Ad5 hexon peptide pools A-D and stained for CD8 and IFN-γ. Representative density plots show percentage of CD8+ cells expressing IFN-γ. Data represent two independent experiments. A two-tailed Student t test was used for statistical analysis. ***P < 0.001.
Discussion
It is a well established fact that in mice, AAV delivery of non-self transgenes to the liver achieves life-long expression due to the induction of immunological tolerance 2, 3. In the present study we found that coadministration of low dose Ad vector encoding an irrelevant transgene and LPS or CpG broke this tolerance, resulting in extinction of transgene expression. This finding supports previous data showing a breach of T cell tolerance in mice challenged with infectious or proinflammatory agents. For instance, infection with the nematode parasite Nippostrongylus brasiliensis reversed staphylococcal enterotoxin B-induced anergy of CD4+ T cells 9. In another study, LCMV peptide-specific CTLs adoptively transferred into mice expressing this peptide as a transgene were expanded in the presence of pathogens or LPS and Poly:IC, and deleted in their absence 8.
While a common theme in these studies was that the T cell response was responsible for the reversal of tolerance, antigen-specific T cells in our study were measurable but not robust. An important question is whether AAV vectors induced LacZ-specific T cells that were anergic and later reactivated following administration of Ad+TLR ligand, or whether such T cells were only primed. It is possible that there were anergic T cells following AAV administration which may have been missed in our analysis due to the use of a functional assay as a surrogate for the detection of antigen-specific T cell. In keeping with this notion, AAV administered to the skeletal muscle has been shown to elicit exhausted CTLs to the transgene 19, 20. On the other hand, T cells could have been primed only following AdLacZ+TLR ligand or AdGFP+TLR ligand administration; although AdGFP does not express the homologous transgene, the combination of inflammation and tissue destruction may have enabled cross-presentation of LacZ from AAV-transduced cells. Finally, a role for CTLs also comes from the combined observations that TLR ligands alone were unable to cause loss of transgene expression and, moreover, expression was stable in RAG-deficient mice challenged with Ad+TLR ligands. The fact that the CTL activity contributes to the loss of transgene expression suggests that diseases posing the greatest risk would be those due to pathogen-induced inflammation rather than chemically induced injury.
What were the mechanisms by which CTLs mediated loss of AAV LacZ-transduced hepatocytes? An interesting observation was the complete loss of LacZ expression in some experimental groups despite only a 5- to 10-fold reduction in AAV vector genomes. The persistence of a substantial number of genomes strongly suggests that the loss of expression is mediated primarily by suppression of transcription. These findings were further corroborated by only modest elevations of liver transaminases that accompanied the loss of AAV encoded LacZ. Such noncytopathic effect of CTLs has previously been shown to suppress HBV replication 21, 22. Interestingly, this effect was attributed to the cytokine TNF-α which was also observed in our study to act in concert with Ad to downregulate AAV transgene expression, emphasizing the role of such cytokine signaling downstream of TLR activation in effecting transgene expression.
To reconcile the observations of poor CTL response, limited cell killing and loss of LacZ expression in the majority of hepatocytes, we propose the possible scenario depicted in Figure 7. Systemic administration of AAV alone at a dose of 1011 VG leads to over 80% hepatocyte transduction, while systemic Ad at a dose of 1010 VG transduces only 5-10% of hepatocytes 23, 24. Based on these observations, we expect that hepatocytes in mice administered with both vectors would contain a mixed population with those that harbor AAV, Ad or AAV+Ad. The coadministration of Ad+TLR ligands overcomes suppression mediated by AAV vectors by modulating the nexus involving CTLs, Tregs and antigen presenting cells. A potent CTL response to Ad capsids, or Ad expressed transgenes, would be expected to eliminate Ad and AAV+Ad transduced cells (Figure 7, condition I); a possible explanation for the partial loss of AAV vector genomes. On the other hand, a weak AAV transgene-specific CTL response observed in our study would be expected to eliminate a fraction of cells transduced with AAV (Figure 7, condition II). Nevertheless, such CTL responses may non-specifically inactivate AAV genomes (i.e. transcriptional silencing) through secreted factors such as TNF-α (Figure 7, condition III). CTLs are known to produce proinflammatory cytokines such as TNF-α, type I IFN and IL-6, which, in turn, have been shown to suppress transcription of various genes 25-28. The mechanisms by which cytokines induce such transcriptional silencing are largely unknown, and contribution of promoter methylation, chromatin remodeling or acetylation of transcription factors needs to be explored. Further supporting the role of transcriptional silencing is the fact that the observed loss of AAV encoded GFP expression in the liver of nonhuman primates seen in the presence of CTLs was not associated with elimination of AAV vector genomes 5. It is also important to point out that if CTL killing was the main explanation for loss of transduction in 80-90% of hepatocytes, then mice did not show symptoms of such widespread hepatocellular toxicity.
Figure 7.
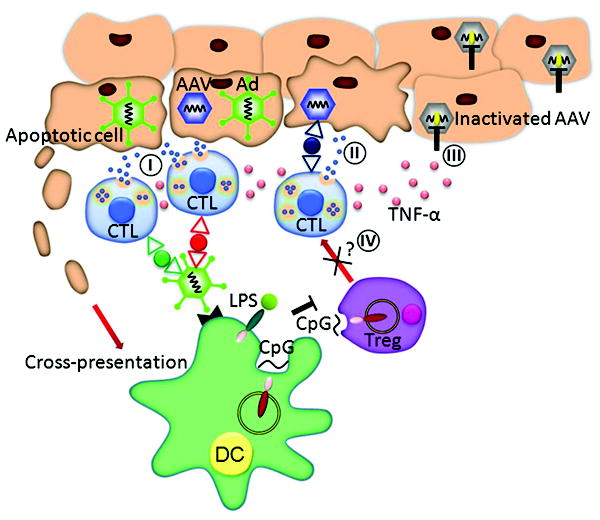
A possible mechanism of inflammation-induced extinction of AAV transgene expression. TLR stimulation leads to an enhanced CTL response to viral capsid and/or transgene that may involve engagement of cognate receptors and activation of APCs, CD8 T cells or Tregs; in the latter case TLR ligation relieves suppression. Activated CTLs kill a fraction of hepatocytes that present antigen (condition I and II), and nonspecifically silence AAV gene transcription through release of cytokine mediators such as TNF-α (condition III).
It is conceivable that the ligation of a single TLR is insufficient and the role of Ad is to merely provide additional inflammatory signaling. Indeed, Ad vectors have been recently shown to trigger the induction of multiple TLRs 29. Moreover, recent observations indicate that a combination of TLR ligands caused marked enhancement of CTL effector function 30, 31. Thus, studies analyzing various TLR ligand combinations for their ability to extinguish transgene expression in the absence of Ad will provide a more in depth understanding of the nature of the inflammatory insult.
Although we observed no changes in the number of hepatic and splenic Tregs, or in their production of IL-10 following coadministration of Ad with TLR ligands (data not shown), it is important to consider that Tregs are likely to play a role in the inflammation-induced extinction of transgene expression, contributing to the mechanisms described above (Figure 7). Indeed, ligation of TLR4 and TLR9 on DCs has been shown to overcome Treg-mediated suppression in vitro 32. Furthermore, Tregs themselves express TLRs and their triggering can lead to the loss of suppressive activity 33, 34.
In conclusion, our data suggest that inflammatory pathology, as seen for instance in some patients with liver metabolic disorders, may impact the outcome of AAV gene transfer trials to the liver. We propose that delineation of signaling pathways downstream of the inflammatory response will allow the development of therapeutic agents against the components of these pathways that can prolong hepatic transgene expression 35, 36. Additionally, elucidation of the molecular mechanism responsible for the inhibition of transgene transcription will allow identification of alternative promoters or cis-acting sequences that preclude inflammation-induced silencing.
Supplementary Material
Acknowledgments
We thank Deirdre McMenamin and Regina Munden for their assistance with animal studies, Penn Vector at the University of Pennsylvania for providing the vectors, and Levi Rupp for technical assistance.
Grant support: This work was funded by the following grants to JMW: NIH/NICHD OTC P01 [5P01 HD057247-03], NIH/NHLBI P01[5P01 HL059407-12] and NIH/NIDDK P30 [5P30 DK047757-17].
Abbreviations
- AAT
α-1 antitrypsin
- AAV
adeno-associated vector
- Ad
adenovirus vector
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- β-gal
β-galactosidase
- CB
chicken β-actin
- CTL
cytotoxic T lymphocyte
- SFU
spot-forming unit
- TBG
thyroid hormone binding globulin gene
- TLR
toll-like receptor
- Treg
regulatory T cell
- VG
vector genome
Footnotes
Author contributions: E.B. and S.S. designed and performed research and wrote the manuscript; P.B. performed research; and J.M.W. designed research and wrote the manuscript.
Disclosures: J.M.W. is a consultant to ReGenX Holdings, and is a founder of, holds equity in, and receives a grant from affiliates of ReGenX Holdings; in addition, he is an inventor on patents licensed to various biopharmaceutical companies, including affiliates of ReGenX Holdings. E. B. is currently at Department of Medicine II, University of Freiburg, Freiburg, Germany.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Snyder RO, Miao CH, Patijn GA, Spratt SK, Danos O, Nagy D, Gown AM, Winther B, Meuse L, Cohen LK, Thompson AR, Kay MA. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat Genet. 1997;16:270–6. doi: 10.1038/ng0797-270. [DOI] [PubMed] [Google Scholar]
- 2.Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, Arruda VR, High KA, Herzog RW. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111:1347–56. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612–21. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koeberl DD, Alexander IE, Halbert CL, Russell DW, Miller AD. Persistent expression of human clotting factor IX from mouse liver after intravenous injection of adeno-associated virus vectors. Proc Natl Acad Sci U S A. 1997;94:1426–31. doi: 10.1073/pnas.94.4.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao G, Wang Q, Calcedo R, Mays L, Bell P, Wang L, Vandenberghe LH, Grant R, Sanmiguel J, Furth EE, Wilson JM. Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum Gene Ther. 2009;20:930–42. doi: 10.1089/hum.2009.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer HP, Ortiz-Pallardo ME, Ko Y, Esch C, Zhou H. Chronic liver disease in heterozygous alpha1-antitrypsin deficiency PiZ. J Hepatol. 2000;33:883–92. doi: 10.1016/s0168-8278(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 7.Real JT, Martinez-Hervas S, Garcia-Garcia AB, Civera M, Pallardo FV, Ascaso JF, Vina JR, Chaves FJ, Carmena R. Circulating mononuclear cells nuclear factor-kappa B activity, plasma xanthine oxidase, and low grade inflammatory markers in adult patients with familial hypercholesterolaemia. Eur J Clin Invest. 2010;40:89–94. doi: 10.1111/j.1365-2362.2009.02218.x. [DOI] [PubMed] [Google Scholar]
- 8.Ehl S, Hombach J, Aichele P, Rulicke T, Odermatt B, Hengartner H, Zinkernagel R, Pircher H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J Exp Med. 1998;187:763–74. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rocken M, Urban JF, Shevach EM. Infection breaks T-cell tolerance. Nature. 1992;359:79–82. doi: 10.1038/359079a0. [DOI] [PubMed] [Google Scholar]
- 10.Bell P, Limberis M, Gao G, Wu D, Bove MS, Sanmiguel JC, Wilson JM. An optimized protocol for detection of E. coli beta-galactosidase in lung tissue following gene transfer. Histochem Cell Biol. 2005;124:77–85. doi: 10.1007/s00418-005-0793-2. [DOI] [PubMed] [Google Scholar]
- 11.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 12.Limberis MP, Figueredo J, Calcedo R, Wilson JM. Activation of CFTR-specific T Cells in cystic fibrosis mice following gene transfer. Mol Ther. 2007;15:1694–700. doi: 10.1038/sj.mt.6300210. [DOI] [PubMed] [Google Scholar]
- 13.Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Breous E, Somanathan S, Wilson JM. Identification of the immunodominant cytotoxic T-cell epitope of human alpha-1 antitrypsin. Gene Ther. 2009;16:1380–2. doi: 10.1038/gt.2009.100. [DOI] [PubMed] [Google Scholar]
- 15.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–76. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 16.Raetz CR. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–70. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 17.Lipford GB, Bauer M, Blank C, Reiter R, Wagner H, Heeg K. CpG-containing synthetic oligonucleotides promote B and cytotoxic T cell responses to protein antigen: a new class of vaccine adjuvants. Eur J Immunol. 1997;27:2340–4. doi: 10.1002/eji.1830270931. [DOI] [PubMed] [Google Scholar]
- 18.Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A. 1996;93:2879–83. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin SW, Hensley SE, Tatsis N, Lasaro MO, Ertl HC. Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific CD8+ T cells in mice. J Clin Invest. 2007;117:3958–70. doi: 10.1172/JCI33138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velazquez VM, Bowen DG, Walker CM. Silencing of T lymphocytes by antigen-driven programmed death in recombinant adeno-associated virus vector-mediated gene therapy. Blood. 2009;113:538–45. doi: 10.1182/blood-2008-01-131375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavanaugh VJ, Guidotti LG, Chisari FV. Inhibition of hepatitis B virus replication during adenovirus and cytomegalovirus infections in transgenic mice. J Virol. 1998;72:2630–7. doi: 10.1128/jvi.72.4.2630-2637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 23.Tao N, Gao GP, Parr M, Johnston J, Baradet T, Wilson JM, Barsoum J, Fawell SE. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol Ther. 2001;3:28–35. doi: 10.1006/mthe.2000.0227. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Wang H, Bell P, McCarter RJ, He J, Calcedo R, Vandenberghe LH, Morizono H, Batshaw ML, Wilson JM. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther. 2010;18:118–25. doi: 10.1038/mt.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solis-Herruzo JA, Brenner DA, Chojkier M. Tumor necrosis factor alpha inhibits collagen gene transcription and collagen synthesis in cultured human fibroblasts. J Biol Chem. 1988;263:5841–5. [PubMed] [Google Scholar]
- 26.Sung RS, Qin L, Bromberg JS. TNFalpha and IFNgamma induced by innate anti-adenoviral immune responses inhibit adenovirus-mediated transgene expression. Mol Ther. 2001;3:757–67. doi: 10.1006/mthe.2001.0318. [DOI] [PubMed] [Google Scholar]
- 27.Andus T, Geiger T, Hirano T, Kishimoto T, Heinrich PC. Action of recombinant human interleukin 6, interleukin 1 beta and tumor necrosis factor alpha on the mRNA induction of acute-phase proteins. Eur J Immunol. 1988;18:739–46. doi: 10.1002/eji.1830180513. [DOI] [PubMed] [Google Scholar]
- 28.Wispe JR, Clark JC, Warner BB, Fajardo D, Hull WE, Holtzman RB, Whitsett JA. Tumor necrosis factor-alpha inhibits expression of pulmonary surfactant protein. J Clin Invest. 1990;86:1954–60. doi: 10.1172/JCI114929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhee EG, Blattman JN, Kasturi SP, Kelley RP, Kaufman DR, Lynch DM, La Porte A, Simmons NL, Clark SL, Pulendran B, Greenberg PD, Barouch DH. Multiple innate immune pathways contribute to the immunogenicity of recombinant adenovirus vaccine vectors. J Virol. 2011;85:315–23. doi: 10.1128/JVI.01597-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Q, Egelston C, Gagnon S, Sui Y, Belyakov IM, Klinman DM, Berzofsky JA. Using 3 TLR ligands as a combination adjuvant induces qualitative changes in T cell responses needed for antiviral protection in mice. J Clin Invest. 2010;120:607–16. doi: 10.1172/JCI39293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, Schmitt E, Schild H, Radsak MP. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood. 2006;108:544–50. doi: 10.1182/blood-2005-10-4015. [DOI] [PubMed] [Google Scholar]
- 32.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 33.Urry Z, Xystrakis E, Richards DF, McDonald J, Sattar Z, Cousins DJ, Corrigan CJ, Hickman E, Brown Z, Hawrylowicz CM. Ligation of TLR9 induced on human IL-10-secreting Tregs by 1alpha,25-dihydroxyvitamin D3 abrogates regulatory function. J Clin Invest. 2009;119:387–98. doi: 10.1172/JCI32354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Wang RF. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380–4. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 35.Mencin A, Kluwe J, Schwabe RF. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–20. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.