

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) contribute to gastrointestinal ulcer formation by inhibiting epithelial cell migration and mucosal restitution; however, the drug-affected signaling pathways are poorly defined. We investigated whether NSAID inhibition of intestinal epithelial migration is associated with depletion of intracellular polyamines, depolarization of membrane potential (Em) and altered surface expression of K+ channels. Epithelial cell migration in response to the wounding of confluent IEC-6 and IEC-Cdx2 monolayers was reduced by indomethacin (100 μM), phenylbutazone (100 μM) and NS-398 (100 μM) but not by SC-560 (1 μM). NSAID-inhibition of intestinal cell migration was not associated with depletion of intracellular polyamines. Treatment of IEC-6 and IEC-Cdx2 cells with indomethacin, phenylbutazone and NS-398 induced significant depolarization of Em, whereas treatment with SC-560 had no effect on Em. The Em of IEC-Cdx2 cells was: −38.5±1.8 mV under control conditions; −35.9±1.6 mV after treatment with SC-560; −18.8±1.2 mV after treatment with indomethacin; and −23.7±1.4 mV after treatment with NS-398. Whereas SC-560 had no significant effects on the total cellular expression of Kv1.4 channel protein, indomethacin and NS-398 decreased not only the total cellular expression of Kv1.4, but also the cell surface expression of both Kv1.4 and Kv1.6 channel subunits in IEC-Cdx2. Both Kv1.4 and Kv1.6 channel proteins were immunoprecipitated by Kv1.4 antibody from IEC-Cdx2 lysates, indicating that these subunits co-assemble to form heteromeric Kv channels. These results suggest that NSAID inhibition of epithelial cell migration is independent of polyamine-depletion, and is associated with depolarization of Em and decreased surface expression of heteromeric Kv1 channels.
1. Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most commonly used drugs worldwide despite their well-documented gastrointestinal (GI) toxicity. Adverse GI effects of NSAIDs including GI ulceration [1–4] result in 100,000 emergency admissions, high medical costs and 17,500 deaths per year in the US alone [5, 6]. Despite extensive investigation, the mechanisms responsible for NSAID-associated GI damage are incompletely understood. A growing body of data suggests that inhibition of mucosal cyclooxygenase (COX) and loss of protective prostaglandins (PG) are insufficient to explain NSAID enteropathies [3]. Moreover, the recently recognized untoward cardiac effects of COX-2 inhibitors suggest that the development of less ulcerogenic NSAIDs through manipulation of COX-selectivity is not a viable therapeutic strategy [7, 8].
To date, NSAIDs have been hypothesized to diminish normal mucosal defense mechanisms and to promote ulcer formation not only by inhibiting cyclooxygenase (COX), but also by diminishing nitric oxide synthesis, uncoupling oxidative phosphorylation, decreasing intracellular pH, reducing surface hydrophobicity, and inducing apoptosis [3, 9–13]. It has also been shown that NSAIDs contribute to ulcer formation and delay ulcer healing by inhibiting intestinal epithelial cell migration and mucosal restitution [14–16].
With respect to the latter mechanism, the epithelial cells of the GI mucosa normally form a protective barrier to pathogens and toxins that may be present in the gut lumen. These surface epithelial cells are continuously exposed to noxious agents that may cause mucosal injury. Superficial mucosal defects typically are repaired rapidly by migration of epithelial cells into the defect from the periphery of the wound. This process, called mucosal restitution, is a primary repair modality in the GI tract and is critical to preserving mucosal integrity and to suppressing ulceration [17–20]. Restitution occurs within minutes to hours following injury in vivo or in vitro, because this process requires cell migration but not cell proliferation or differentiation [17–20]. While it is established that NSAIDs promote ulceration by interfering with restitution [14–16], the signaling pathways mediating this effect have not been fully elucidated.
In contrast to the dearth of information regarding the effects of NSAIDs on intestinal restitution, extensive knowledge of the molecular basis for polyamine-modulation of this process exists [21–25]. Depletion of intracellular polyamines inhibits intestinal cell migration by decreasing the expression of delayed rectifier potassium channels, thereby depolarizing membrane potential (Em), decreasing the driving force for calcium-influx, and inhibiting downstream [Ca2+]i-dependent signaling events [21–25]. NSAIDs have been shown to deplete the polyamine content of immortalized colon cancer cells by enhancing polyamine catabolism [26]. However, to our knowledge, there have been no experimental efforts to link NSAID inhibition of intestinal epithelial migration to depletion of intracellular polyamines, depolarization of membrane potential or surface expression of K+ channels. The present investigation was designed to identify potential mechanisms for the GI toxicity of NSAIDs by defining those relationships.
2. Materials and Methods
2.1 Reagents
Cell culture media was obtained from American Type Culture Collection (ATCC, Manassas, VA) or JRH Biosciences. Serum was obtained from Invitrogen (Calsbad, CA) and chemicals were obtained from Sigma (St. Louis, MO) unless stated otherwise. Matrigel was purchased from BD Biosciences (Bedford, MA). DiBAC4(3) was obtained from Molecular Probes (Invitrogen, Carlsbad CA). SC-560 (5-(4-chlorphenyl)-1-(4-methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole) and NS-398 (N-[2-(cyclohexyloxy)-4-nitrophenyl]-methanesulfonamide) were obtained from Cayman Chemical (Ann Arbor, MI). AVE0118, (12′(benzyloxycarbonylaminomethyl) biphenyl-2-carboxylic acid 2-(2-pyridyl)ethylamide) was obtained from Aventis Pharma (Frankfort, Germany).
2.2 Cell Culture
The IEC-6 cell line was purchased from ATCC at passage 13 (Manassas, VA). These cells, developed by Quaroni et al. [27], are non-tumorigenic and retain the character of undifferentiated intestinal crypt cells. Cell culture conditions were similar to those described previously by others [28, 29]. The basic medium consisted of DMEM supplemented with heat-inactivated fetal bovine serum (FBS, 5%), insulin (10 μg/ml) and gentamicin (50 μg/ml). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in air. Passages 15–20 were used for the experiments described below.
A line of IEC-6 cells stably transfected with the Cdx2 transcription factor (IEC-Cdx2) was obtained from Dr. E. Suh (University of Pennsylvania, Philadelphia, PA). This cell line was developed and characterized initially by Suh and Traber [30], and then characterized further by others [23, 31, 32]. The forced expression of the Cdx2 gene in IEC-6 cells results in development of a differentiated phenotype [23, 31]. The LacSwitch expression vector system (Stratgene, La Jolla, CA) is used to direct conditional expression of the Cdx2 gene, with isopropyl-β-D-thiogalactopyranoside (IPTG) serving as the inducer for gene expression. IEC-Cdx2 cell stocks were maintained in the same basic culture medium used for IEC-6, and grown in media supplemented with 4 mM IPTG for 16 days to induce the conditional expression of Cdx2 before experiments.
IEC-6 or IEC-Cdx2 cells were seeded at a density of approximately 6.25 ×104 cells/cm2 on 35 or 60 mm plates thinly coated with Matrigel and were grown to a confluent monolayer prior to the introduction of media containing experimental treatments for: optical analysis of cell migration; western analysis of K+ channel expression; assay of intracellular polyamines; and sharp electrode measurement of Em. Cells were seeded in 96 well plates at a density of 65,000 cells per well for assessment of Em by DiBAC4(3) fluorescence.
Both IEC-6 and IEC-Cdx2 cells were used in our initial investigations of drug effects on cell migration and Em. Subsequent experiments focused on the elucidating the relationship between NSAID-treatment and Kv channel expression were performed using IEC-Cdx2, exclusively.
2.3 Treatment Protocols
The experiments performed utilized NSAIDs with variable effects on intestinal restitution and variable COX-selectivity, as shown in Table 1 [33–36]. Drug effects were evaluated over the following ranges of concentrations, as indicated in the text or figure legends: indomethacin (1–100uM); phenylbutazone (1–100uM); NS-398 (1–100uM), SC-560 (10 nM-1uM). DMSO (0.1% v/v) was the vehicle control used for comparison to NSAIDs. Monolayers were exposed to NSAIDs for 72 h prior to wounding, measurement of Em or performance of biochemical assays, as indicated in the figure legends. In some experiments, an inhibitor of polyamine synthesis, α-difluoromethylornithine (DFMO, 5 mM), was added to experimental media [37]. AVE0118, (10−6 M) was employed as an antagonist of voltage-gated K+ (Kv) channels [38].
Table 1.
| INHIBITOR | IC50 (μM) Cox-1 | IC50 (μM) Cox-2 | COX SELECTIVITY* | ULCEROGENIC POTENTIAL |
|---|---|---|---|---|
| Indomethacin (human) | 0.19 | 0.44 | 0.4 | +++ |
| Indomethacin (horse) | 0.036 | 0.43 | 0.08 | +++ |
| SC-560 (human) | 0.005 | 1.4 | 0.003 | 0 |
| NS-398 (rat) | 125 | 5.6 | 22 | (+) slows ulcer healing |
| Phenylbutazone (horse) | 6.15 | 3.79 | 1.6 | +++ |
2.4 Assessment of Cell Migration
Treatment-induced changes in IEC-6 and IEC-Cdx2 cell migration were assessed using a wounding assay. A razor blade was used to remove approximately one-third of the monolayer after 72 h of exposure to media containing drug. The created cell border (scratch line) was marked. Micrographs (Nikon ACT-1) were captured immediately after creation of this defect and again 4 h later. Three methods of image analysis (Nova Prime, Bioquant, Nashville TN) were used to measure cell migration. First, the linear distance (μM) from the scratch line to the point of maximum cell migration was measured. Second, the number of cells migrating into the defect was counted and expressed in terms of cells/μm2. Finally, a standardized rectangular region of interest (120,000 μm2) was created and positioned at the scratch line; the percentage of this region occupied by migrating cells was measured over time. All experiments were done in triplicate and 2 fields per plate were examined and measured, as described previously.
2.5 Determination of Polyamine Content
IEC-6 cells were washed with chilled PBS containing protease inhibitors (P-8340, Sigma), mechanically lifted using a cell scrapper in 5% deoxycholate, transferred to cryotubes and snap frozen using liquid nitrogen, then thawed and sonicated for 3 pulses of 10 s in ice. These lysates were centrifuged for 15 min at 10,000 rpm (5415F, Eppendorf, Brinkman Inst., Westbury, NY). The supernatant was collected and stored at −70°C until analysis.
Polyamine content was measured by liquid chromatography/mass spectrometry (LC/MS). Samples, standards and quality controls (QCs) were prepared for LC/MS by combining 300 μl of cell lysate with 30 μl of internal standard (10 μg/ml ethambutol), and then filtered (Whatman Mini-Uniprep, PVDF filter media, 45 μm pore). A volume of 50 μl of filtered sample was injected onto the LC/MS system. The system was comprised of a P4000 narrow-bore quaternary pump with a vacuum degasser, AS3000 auto-sampler, UV6000 photo-diode array detector and LCQDuo ion-trap mass spectrometer (ThermoFinnigan, San Jose, CA). The HPLC column (Phenomenex, Torrance, CA) was a Luna C18(2) (30× 2.0 mm, 5 μM). Mobile phase was 10/90-acetonitrile/acetic acid (10 mM); flow rate, 0.3 ml/min. The MS used electrospray ionization to detect each ion individually, 146 (spermidine), 203 (spermine) and 205 (ethambutol) m/z.
Stock solutions of putrescine, spermidine, spermine, and ethambutol were made in water. The stock solution of ethambutol was used to make a 10 μg/ml solution in water, which was then used as an internal standard. Standards were made fresh each day that samples were analyzed. The QCs were made in bulk prior to validation and aliquoted into 300 μl samples and stored at −20 °C until used. For the initial intra-day assay validation, five QCs were randomly selected from each of the three different concentrations. For assays, two QCs at each concentration were selected at random and prepared with the standard curve and unknown samples.
Peak area ratio of analyte:internal standard was used for quantitation. The standard curve was weighted 1/x, was linear, and contained the concentrations: 0.1, 0.25, 0.5, 1.0, 1.5, 2.0, and 2.5 μg/ml (0.3, 0.75, 1.5, 3.0, 4.5, 6, and 7.5 μg/ml for spermine). Concentration vs. peak area ratio was plotted and a linear regression equation obtained for quantitation. Prepared concentrations for QCs were 0.75, 1.25, and 2.25 μg/ml (2.25, 3.75, and 6.75 μg/ml for spermine).
The validation assay was considered valid if the intra-day accuracy and precision values of the QCs were within 15% of the expected values for each of the three QC concentrations. Accuracy was calculated as and precision was calculated as , where Cint was the intended concentration, Cx̄ was the average measured concentration, and CSD was the standard deviation of the measured concentrations. An assay run was rejected if more than two of the six QCs for a given standard curve were >±15% from their intended value or if both QCs at a single concentration were >±15% from their intended concentration. A rejected run was re-assayed with freshly prepared samples.
2.6 Determination of Membrane Potential
Treatment-induced changes in Em of IEC-6 and IEC-Cdx2 cells were assessed initially by spectrophotometric determination of DiBAC4(3) fluorescence (excitation= 485 nM; emission= 527 nM; Fluoroskan Ascent FL, LabSystems Inc., Helsinki, Finland). Cells were equilibrated with 2 μM DiBAC4(3) for 30 min at 37°C prior to fluorescence measurement.
In addition, the Em of individual IEC-Cdx2 cells was measured using microelectrodes (35–55 MΩ) filled with 3M KCl. The extracellular solution contained 132 mM NaCl, 4 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM Dextrose, 20 mM Hepes (pH 7.4). Data acquisition was performed using an Axon Instruments 700B amplifier and Multiclamp Commander (Molecular Devices, Sunnyvale, CA). Recordings were made at room temperature.
2.7 Immunoblotting
Western blot analysis was used to determine the effects of NSAIDs and DFMO on K+ 17, and 18 were resolved by SDS-PAGE, transferred to nitrocellulose membranes and probed with polyclonal primary antibodies (Alomone Laboratories, Jerusalem, Israel) directed against Kv1.1 (APC-009), Kv 1.4 (APC-007), Kv 1.5 (APC-004), Kv1.6 (APC-003) and KV10.1 (APC-104).
Bound primary antibodies were visualized using an enhanced chemiluminescence detection system (SuperSignal, Pierce, Rockford, IL), and recorded on radiographic film. Densitometric analysis was performed using AlphaEaseFC (Alpha Innotech Co, San Leandro, CA). Equal loading of protein was confirmed by immunoblotting for actin. The detection system yielded a linear response over a ~1:20 concentration range when immunoblotting was performed using cell lysates that had been serially diluted. Actin was considered an appropriate loading control because its level of expression in IEC-6 cells is unaffected by interventions that disrupt cell migration, such as polyamine depletion [22, 39]. Preliminary immunoblotting experiments (not shown) confirmed that the ratio of actin to Na+/K+ATPase in IEC-6 cell lines was not significantly affected by NSAID treatments, thereby supporting the choice of actin as the loading control in experiments designed to investigate the expression of membrane proteins.
2.8 Immunoprecipitation
To preclear non-specific binding, protein G-agarose beads (Immunopure, Pierce, Rockford, IL) were incubated with samples for 2 h at 4°C with continuous gentle agitation, then pelleted by centrifugation (700 X g). A monoclonal antibody directed against Kv1.4 (Sigma K1264) was added subsequently to the supernatant and mixed overnight at 4 °C. Immune complexes were then immobilized onto protein G-agarose beads, washed three times with cold RIPA buffer and eluted from the beads with SDS sample buffer containing 5% β-mercaptoethanol. Immunoprecipitated proteins and supernatant fractions were analyzed by immunoblotting, as described above. The ability of the Sigma monoclonal antibody to pull down Kv1.4 was confirmed in preliminary experiments by immunoblotting the precipitated fraction with the Alomone polyclonal antibody Kv1.4; a protein band with the expected mobility of monomeric Kv1.4 (~96 kDa) was detected (data not shown). Attempts to pull-down Kv1.6 using commercially available polyclonal antibodies directed against that target were unsuccessful.
2.9 Biotinylation
IEC-Cdx2 cells were incubated for 45 min at room temperature with EZ-Link sulfo-NHS-biotin (Pierce, Rockford IL) in PBS, lysed in a buffer containing 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 50 mM Tris-HCl, 0.5% sodium deoxycholate, 0.1% SDS, and 1% protease inhibitor cocktail, combined with 200 μl ImmunoPure Immobilized streptavidin (Pierce) and rocked overnight at 4°C. Biotinylated proteins were pelleted by centrifugation, washed and eluted using Laemmli Sample Buffer. The starting material, supernatant, and biotinylated fractions were subsequently subjected to SDS-PAGE and immunoblot analysis as described above. Actin was used to normalize for loading. Relative cell surface expression was calculated by dividing the normalized surface protein (biotinylated fraction) by the normalized total protein (biotinylated + non-biotinylated).
2.10 Statistical Analysis
Data are expressed as mean ± SEM. Significant differences between treatment groups were identified by ANOVA and multiple comparisons were made using LSD procedure (Statistix, Analytical Software, Tallahassee, FL). Differences were considered to be significant when P≤0.05. The numbers of replicates per treatment group and independent experiments associated with specific studies are provided in the figures or accompanying legends.
3. Results
3.1 NSAIDs and Epithelial Cell Migration
Exposure to NSAIDs significantly reduced cell migration in response to the wounding of confluent IEC-6 and IEC-Cdx2 monolayers (Figures 1 and 2). Maximum cell migration, measured as linear distance from the created defect 4 h after wounding, was significantly reduced in monolayers exposed to DFMO (5 mM), indomethacin (100 μM), phenylbutazone (100 μM), NS-398 (100μM) but not SC-560 (1 μM). At these concentrations, the NSAIDs lack cytotoxic effects [40–43]. Similar results were obtained when cell migration was assessed in terms of either the number of cells in a defined (120,000 μm2) region of interest adjacent to the created wound or the percentage of this defined region covered by cells (data not shown). As expected, results obtained using these three assessment methods were highly correlated (r2=0.84).
Figure 1.
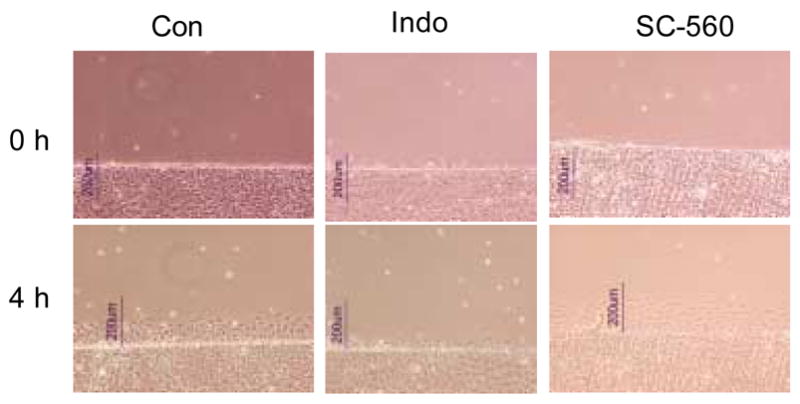
IEC-6 migration in monolayers grown in the absence (Con) or presence of NSAIDs. Drug-treated cells were exposed to either indomethacin (Indo, 100 μM) or SC-560 (1 μM) for 72 h before a defect was created in the monolayer (wounding). Images were captured immediately (0 h) or 4 h after wounding.
Figure 2.
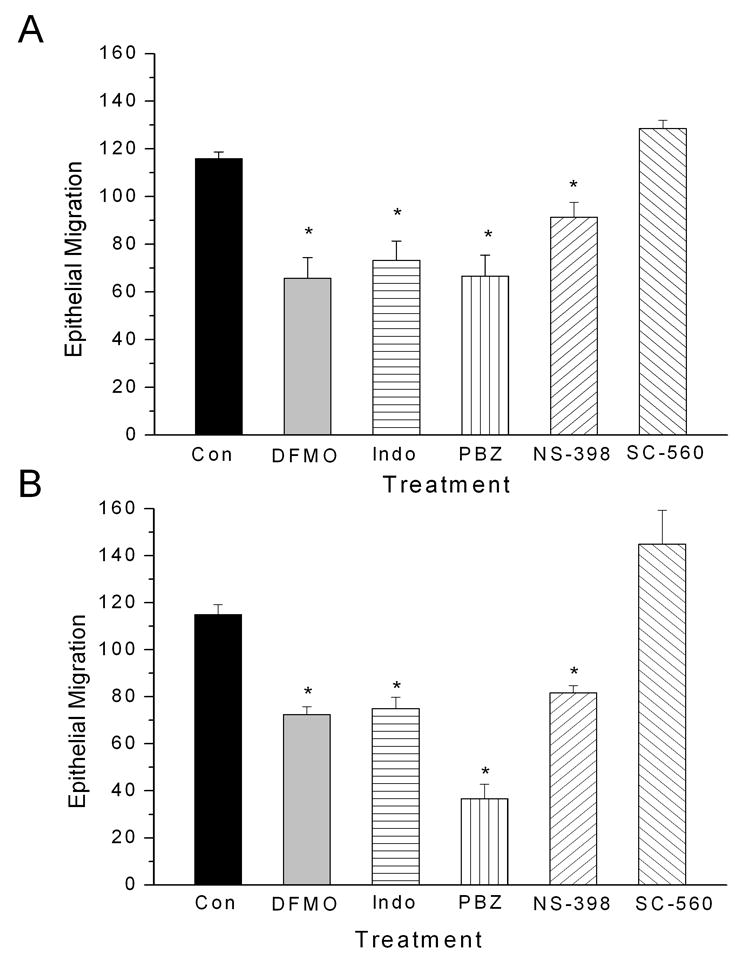
Epithelial cell migration in IEC-6 (panel A) and IEC-Cdx2 (panel B) monolayers (n=6 per group) under control conditions (Con), after depletion of polyamines (DFMO, 5.0 mM), or after treatment with the NSAIDs indomethacin (Indo, 100 μM), phenylbutazone (PBZ, 100 μM), SC-560 (1 μM) and NS-398 (100 μM1). Monolayer cultures were exposed to drug for 72 h before wounding. Migration was measured 4 h post-wounding as the maximum linear distance from the created defect. Asterisks indicate significant difference from control (p < 0.05).
3.2 NSAIDs and Depletion of Intracellular Polyamines
The determination of intracellular polyamine content yielded expected results for IEC-6 treated with DFMO with and without spermadine [29, 31]. As shown in Figure 3A, exposure to DFMO significantly decreased the intracellular polyamine content of IEC-6 cells. Polyamine depletion could be counteracted by the addition of exogenous spermidine. These data support the integrity of our method for assaying cellular polyamine content.
Figure 3.
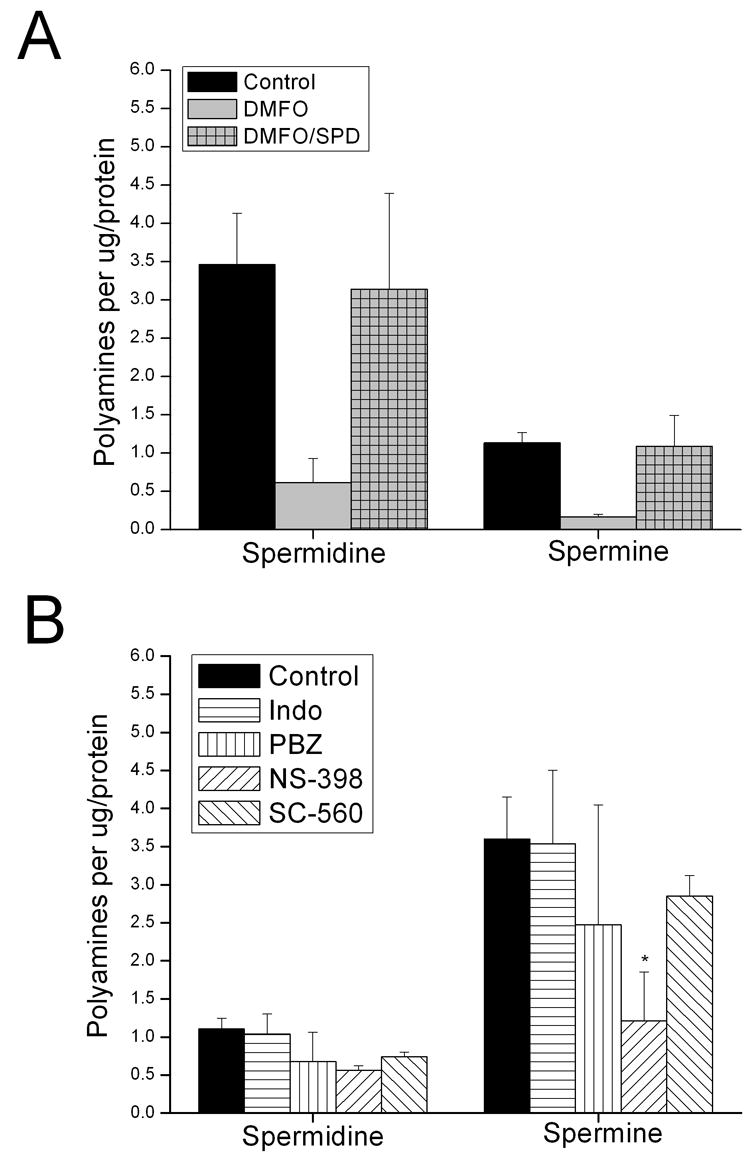
Cellular polyamine concentrations of IEC-6 exposed to NSAIDs. Panel A: Polyamine content of IEC-6 cells treated with DFMO (5 mM) in the absence and presence of exogenous spermadine (SPD, 5 μM). Panel B: IEC-6 treated with indomethacin (Indo, 100 μM), phenylbutazone (PBZ, 100 μM), SC-560 (1 μM) and NS-398 (100 μM). Cellular polyamine concentrations were measured by LC-MS at 78 h post-treatment (n= 3). Asterisks indicate significant difference from control (p < 0.05).
Treatment of IEC-6 monolayers with the NSAIDs indomethacin, phenylbutazone and SC-560 failed to significantly decrease intracellular polyamine content, while NS-398 decreased spermine but not spermidine content (Figure 3B). These data suggest NSAID-inhibition of intestinal cell migration cannot be attributed to depletion of intracellular polyamines.
3.3 NSAIDs and Epithelial Cell Resting Potential (Em)
Treatment of IEC-6 and IEC-Cdx2 cells with DFMO to induce depletion of intracellular polyamines increased the DiBAC4(3) fluorescence of treated cells, indicating depolarization of Em (Figure 4). These data are consistent with published data showing that depletion of polyamines by a 96 h exposure to DFMO induces a ~+10 mV change in the Em in IEC-6 cells measured by microelectrode techniques [25]. Thus, fluorescence imaging of DIBAC4(3)-loaded cells is a useful method for detecting treatment-induced changes in the Em of intestinal epithelial cells.
Figure 4.
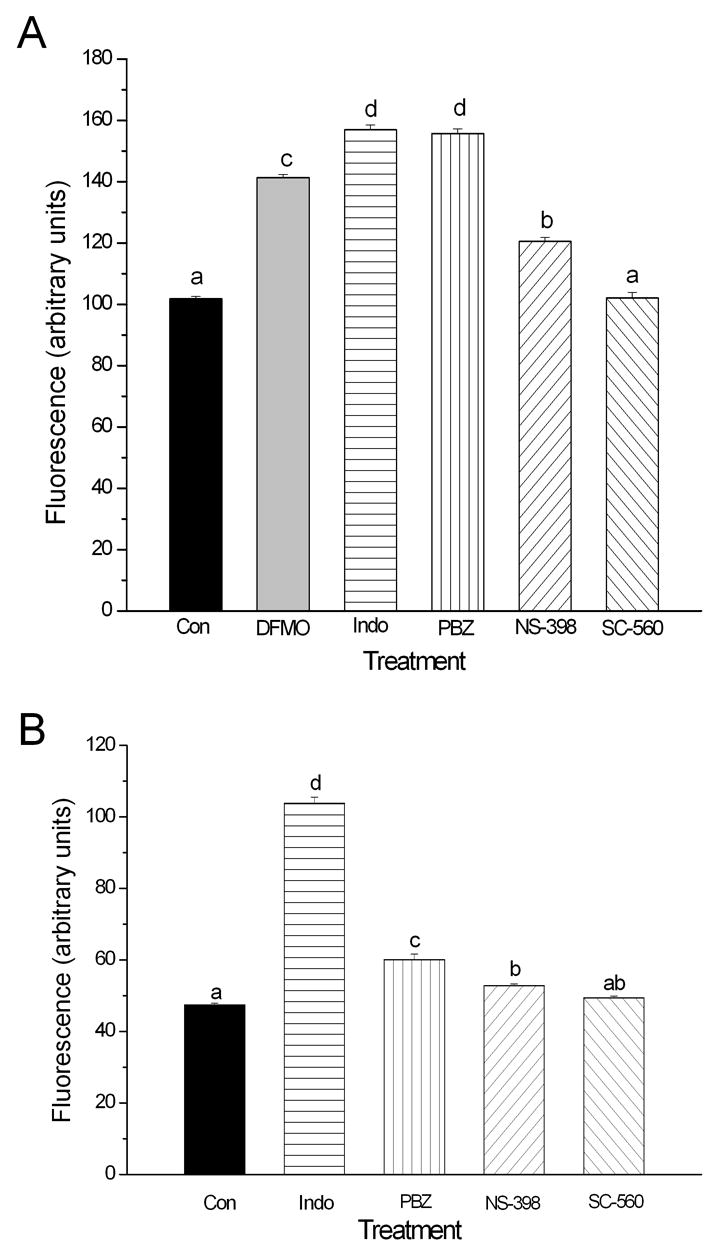
Relative Em of IEC-6 (panel A) and IEC-Cdx2 (panel B) cells under control conditions (Con, n=86), after depletion of polyamines (DFMO, 5.0 mM, n=22 ), or after 72 h treatment with the NSAIDs indomethacin (Indo, 100 μM, n=23), phenylbutazone (PBZ, 100 μM, n=39), SC-560 (1 μM, n=44) and NS-398 (100 μM, n=22). Treatment-induced changes in Em were detected by monitoring the fluorescence of cells loaded with DiBAC4(3). Increased fluorescence indicates depolarization of Em. Different superscripts indicate significant difference between groups (p < 0.05).
Interestingly, treatment of IEC-6 and IEC-Cdx2 monolayers with indomethacin, phenylbutazone and NS-398, the NSAIDs previously shown to inhibit cell migration after wounding, also induced a significant increase in DiBAC4(3) fluorescence consistent with depolarization of Em. Moreover, exposure to SC-560, an NSAID with no significant effects on epithelial cell migration, caused no significant change in DiBAC4(3) fluorescence. Microelectrode measurements of Em in IEC-Cdx2 confirmed that NSAID-inhibition of epithelial cell migration is correlated with drug-induced membrane depolarization. The Em of cells treated with SC-560 (−35.9±1.6 mV, n=8) was not significantly different from that of untreated IEC-Cdx2 cells (−38.5±1.8 mV, n=19). In contrast, the Em of indomethacin-treated cells (−18.8±1.2 mV, n=6) and the Em of NS-398-treated cells (−23.7±1.4 mV, n=15) were significantly depolarized.
3.4 NSAIDs and Expression of Kv Channels
The Em of IEC-6 and IEC-Cdx2 cells is related directly to the activity and number of K+ channels expressed on the plasma membrane. Depolarization of Em and inhibition of cell migration can be induced by antagonizing membrane K+ channels [25]. Conversely, increased K+ channel activity induces hyperpolarization and enhances the rate of epithelial cell migration [23].
It has been suggested that the Em and migration response of IEC-6 and IEC-Cdx2 cells are determined primarily by the activity of delayed rectifier K+ channels formed by Kv1 (KCNA) gene products [21, 23, 25]. Consistent with that hypothesis, the Kv channel antagonist AVE0118 (1 μM) impaired IEC-Cdx2 cell migration post-wounding (Figure 5). The inhibition of epithelial restitution by AVE0118 was comparable to that seen with the NSAIDs indomethacin, phenylbutazone and NS-398. On the basis of these findings, we hypothesized that NSAIDs depolarize Em and inhibit intestinal epithelial cell migration by attenuating the activity of Kv1 channels. Accordingly, we used immunoblotting in combination with biotinylation to determine the effects of NSAID treatment on the total expression and the cell surface expression of the Kv1 channel proteins: Kv1.1, Kv1.4, Kv1.5 and Kv1.6; mRNA for these K+ channel pore-forming subunits has been reported to be expressed by either IEC-6, IEC-Cdx2 cells or both [21, 23, 25].
Figure 5.
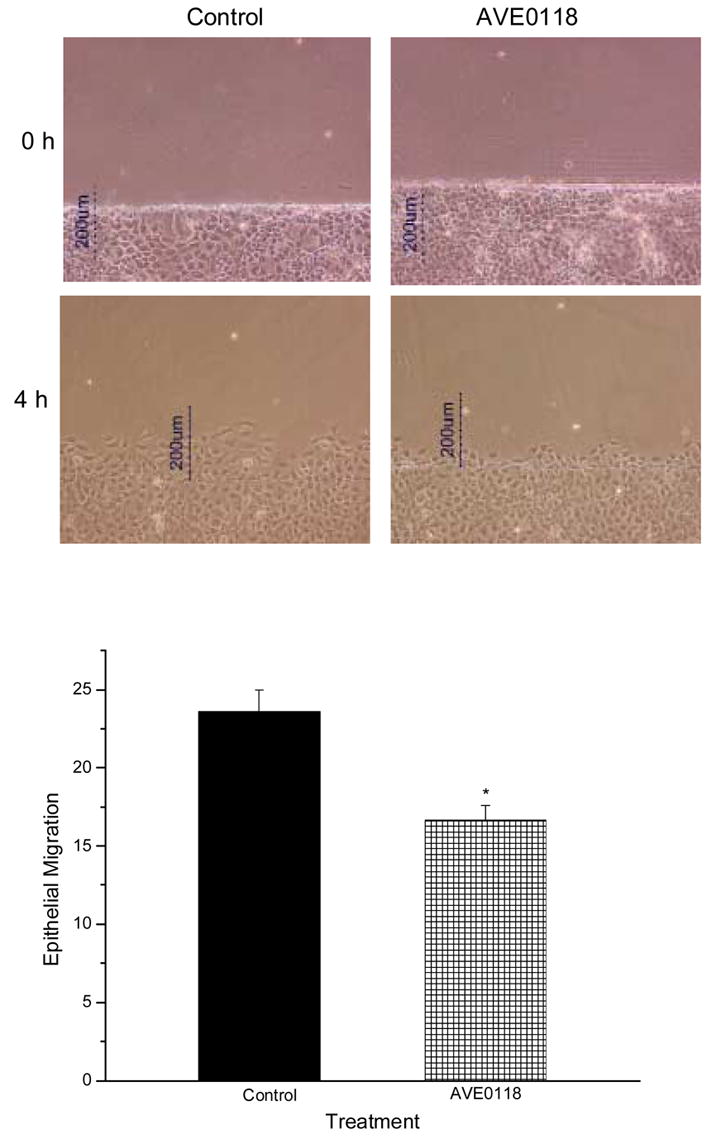
Effect of AVE0018 on epithelial cell migration in IEC-Cdx2 cells. Photomicrographs show control (left column) and drug-treated (right column) monolayers immediately (top row) and 4 h (lower row) after wounding. In the graph, epithelial migration is expressed as the percentage of a defined region adjacent to the defect that was occupied by migrating cells 4 h after monolayer wounding in the absence (Con, n=6) and presence of the Kv channel antagonist AVE0118 (1 μM, n=6). Asterisk indicates significant difference (p < 0.05).
Kv1.4, Kv1.5 and Kv1.6 channel proteins were detected routinely by immunoblotting of IEC-Cdx2 cell lysates, whereas Kv1.1 was not found consistently. Kv1.4 and Kv1.6 but not Kv1.5 were detected in the biotinylated fraction after surface biotinylation of IEC-Cdx2 cells (Figure 6). These data indicate that even though IEC-Cdx2 cells express Kv1.4, Kv1.5, and Kv1.6 channel proteins, only Kv1.4 and Kv1.6 are present on the cell surface membrane to form functional channels.
Figure 6.
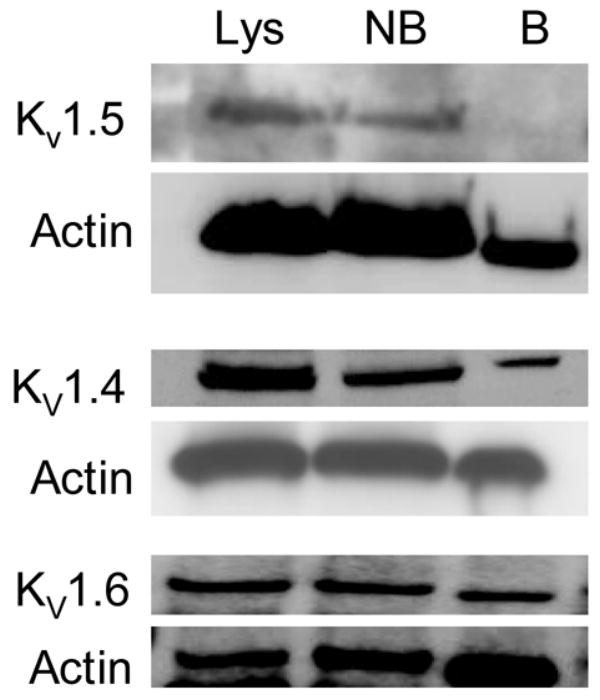
Kv1 channel expression by IEC-Cdx2 cells. Immunblots of Kv1.5, Kv1.4 and Kv1.6 channel protein in total cell lysates (Lys), non-biotinylated (NB) and biotinylated (B) fractions obtained from IEC-Cdx2 cells subjected to cell surface biotinylation. After detection of Kv channel protein (upper panel of each set), each blot was stripped and reprobed with antibody against actin to assess loading (lower panel of each set). The apparent molecular masses of Kv1.5, Kv1.4 and Kv1.6 were ~75 kDa, ~96 kDa and ~56 kDa, respectively. The immunoblots shown are representative of n ≥ 3.
The detection of Kv1.6 in the fraction of proteins precipitated by Kv1.4 antibody (Figure 7, top panel) suggests that these two subunits co-assemble to form heteromeric K+ channels. The specificity of the interaction between Kv1.4 and Kv1.6 was confirmed in two ways. First, the protein precipitated by Kv1.4 was immunoblotted with Kv1.6 antibody incubated with an excess of the fusion protein used to generate the antibody (Figure 7, middle panel). In addition, the protein pulled down by an unrelated K+ channel antibody was probed for Kv1.6 (Figure 7, lower panel). Kv1.6 protein was not detected under either experimental condition.
Figure 7.
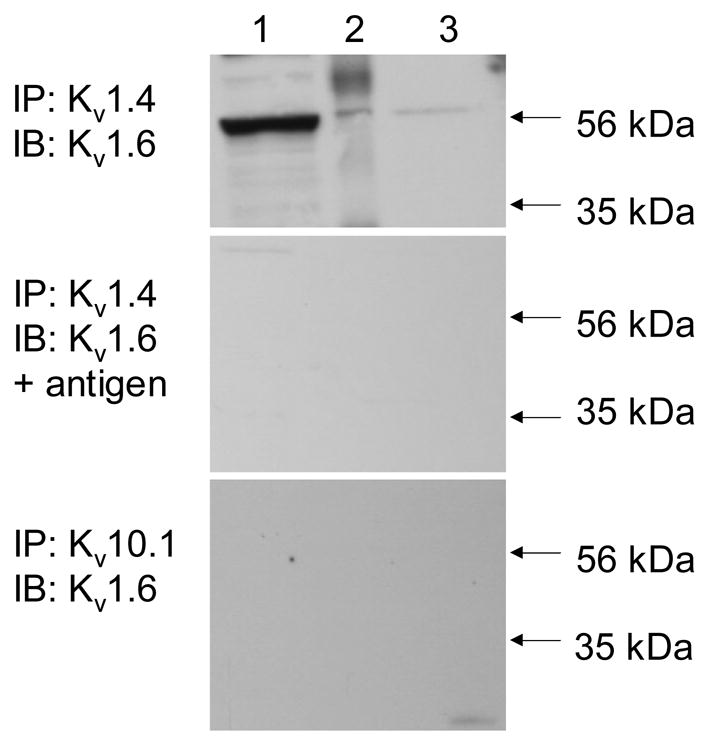
Co-association of Kv1.6 and Kv1.4 in IEC-Cdx2. Top: Immunoprecipitation (IP) was performed with antibody against Kv1.4, then the starting material (lane 1), IP (lane 2) and supernatant (lane 3) were immunoblotted (IB) directly with antibody against Kv1.6. Middle: IP was performed with antibody against Kv1.4, then the starting material (lane 1), IP (lane 2) and supernatant (lane 3) were IB with antibody against Kv1.6 pre-incubated with an excess of the fusion protein encoding the epitope used to generate the Kv1.6 antibody. Bottom: IP was performed with antibody against Kv10.1, then the starting material (lane 1), IP (lane 2) and supernatant (lane 3) were IB directly with antibody against Kv1.6. The immunoblots shown are representative of n ≥ 3.
The total cellular expression of Kv1.4 was decreased significantly by indomethacin, phenylbutazone and NS-398, the NSAIDs that impair epithelial cell migration, but not by SC-560, an NSAID with no inhibitory effects on cell migration (Figure 8A). Cell surface expression of Kv1.4 was similarly affected (Figure 8B). Interestingly, the NSAIDs that impair epithelial cell migration, indomethacin and NS-398, had no effect on the total expression of Kv1.6 (Figure 9A), although the cell surface expression of this channel subunit was decreased significantly (Figure 9B).
Figure 8.
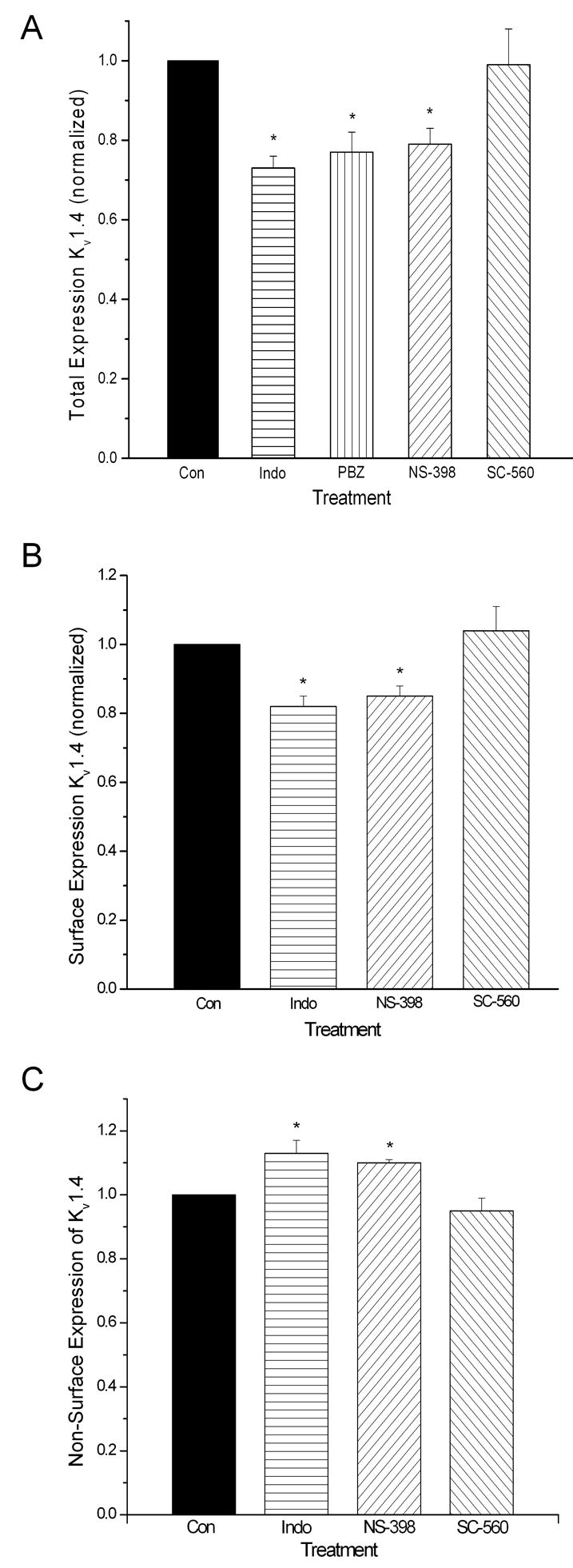
NSAIDs influence the total (panel A), cell surface (panel B) and non-surface (panel C) expression of Kv1.4 by IEC-Cdx2. Monolayer cultures were exposed to drug for 72 h before surface biotinylation and lysis. Drug concentrations were identical to those used in Figures 1–4. Asterisks indicate significant differences (p < 0.05).
Figure 9.
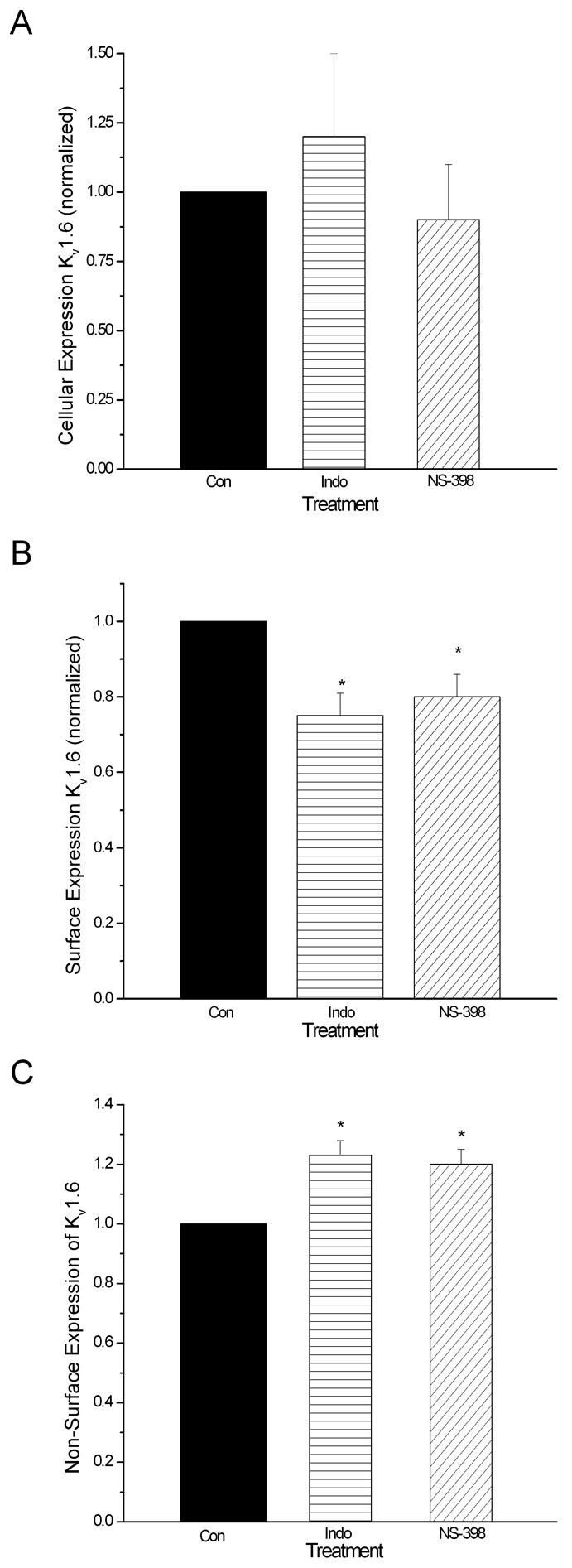
The NSAIDs that impair cell migration do not affect the total expression (panel A) of Kv1.6 by IEC-Cdx2; however, these drugs do diminish the cell surface expression (panel B) and increase the non-surface expression (panel C) of Kv1.6. Monolayer cultures were exposed to drug for 72 h before surface biotinylation and lysis. Drug concentrations were identical to those used in Figures 1–4. Asterisks indicate significant differences (p < 0.05).
4. Discussion
Early mucosal restitution, as manifest by the rapid resealing of superficial wounds secondary to cell migration is an important primary repair modality, critical to preserving GI mucosal integrity [17–20]. NSAIDs contribute to ulcer formation and delay ulcer healing by inhibiting intestinal epithelial cell migration and mucosal restitution [14–16]. The present study provides a potential mechanism for these ulcerogenic effects of NSAIDs.
Our data are consistent with published work [3] that suggests inhibition of mucosal cyclooxygenase (COX) and loss of protective prostaglandins (PG) are insufficient to explain NSAID effects on epithelial restitution. The COX-2 antagonist NS-398 and the non-specific COX anatagonist indomethacin impeded intestinal epithelial cell migration, whereas the COX-1 antagonist SC-560 did not. In our investigations, NSAID-inhibition of restitution was associated with depolarization of epithelial cell Em and altered surface expression of heteromeric K+ channels, but not with depletion of intracellular polyamines. To our knowledge, this is the first demonstration of an association between the GI toxicity of NSAIDs and their effects on Em and Kv channel expression.
4.1 NSAIDs and Depletion of Intracellular Polyamines
Previous studies have shown that depletion of intracellular polyamines by DFMO inhibits epithelial migration [22, 25, 29, 31, 37] and that NSAIDs accelerate polyamine catabolism in colonic epithelial cancer cells [26]. Based on these observations, we hypothesized that NSAIDs inhibit intestinal cell migration by depleting intracellular polyamines. In the course of testing this idea, we first confirmed that NS-398 and indomethacin inhibit epithelial cell migration in wounded IEC-6 and IEC-Cdx2 monolayers, and then validated the findings of previous investigators that depletion of intracellular polyamines inhibits migration (Figure 2). However, as shown in Figure 3B, the NSAIDs that inhibited cell migration either had no significant effect on IEC-6 polyamine content (indomethacin and phenylbutazone) or decreased spermine but not spermidine content (NS-398). Together, these results indicate that polyamine-depletion is not a major mechanism underlying NSAID-mediated inhibition of intestinal cell migration. In light of these findings, we next considered the possibility that NSAIDs with ulcerogenic potential inhibit restitution by altering Em independent of poyamine depletion, as discussed below.
4.2 NSAIDs and Epithelial Cell Resting Potential (Em)
The migration of IEC-6 and IEC-Cdx2 cells is influenced significantly by Em because of the Nernstian relationship between Em and the driving force for calcium influx, where the latter is an important determinant of downstream signaling events that are critical for epithelial restitution [21–23, 25]. Consequently, depolarization of Em reduces Ca2+ influx and thereby inhibits cell migration [23, 25]. In the present study, NSAIDs with inhibitory effects on cell migration induced a significant increase in DiBAC4(3) fluorescence consistent with depolarization of Em whereas an NSAID without negative effects on intestinal cell migration, caused no significant change in DiBAC4(3) fluorescence (Figure 4).
The global qualitative results provided by the fluorescence measurements subsequently were confirmed by direct measurements of Em in individual cells using microelectrode techniques. Those studies indicated that the resting Em of IEC-Cdx2 cells measured in this study (−38.5±1.8 mV) was similar to that reported previously (−43±2.5 mV) by Rao et al. [23] and that the NSAIDs with inhibitory effects on epithelial cell migration depolarized the Em of IEC-Cdx2 by ~15 to 20 mV. This magnitude of depolarization was associated with differential rates of intestinal cell migration in previous investigations of epithelial restitution [23, 25], supporting the contention that depolarization of Em is sufficient to account for the inhibitory effects of NSAIDs on the migration of IEC-Cdx2 cells.
4.3 NSAIDs and Expression of Kv Channels
The Em of non-excitable cells reflects the K+ gradient established by the Na+/K+-ATPase, a selective permeability to K+ and an inward “leak” driving the measured Em from the Nernst potential for K+. The membrane permeability to K+ is the most important determinant of Em, and it can be mediated through non-gated permeation pathways and/or voltage-gated potassium channels.
Previous investigations of IEC-6 and IEC-Cdx-2 cells have shown not only that these cells express multiple Kv channel subunits at the mRNA and protein levels, but also that delayed rectifier currents conducted by 4-aminopyridine-sensitive Kv channels play a major role in controlling Em and epithelial cell migration [21, 23, 25]. Our work extends those findings. We document for the first time the cell surface expression (Figure 6) and the co-association (Figure 7) of Kv1.4 and Kv1.6 channel proteins in IEC-Cdx-2 cells. Previous investigations had shown expression of mRNA encoding these two K+ channel subunits in intestinal epithelial cells using RT-PCR [21, 23]. We also demonstrate that the experimental atrial anti-arrhythmic drug AVE0118 inhibits IEC-Cdx2 migration (Figure 5). AVE0118 and 4-aminopyridine are structurally distinct K+ channel antagonists; however, both drugs inhibit the delayed rectifier currents conducted by channels composed of Kv1.x subunits [38, 44].
In one previous investigation, the pharmacological inhibition of K+ channels in intestinal epithelial cells was shown to accelerate rather than inhibit intestinal epithelial cell migration [45]. Although this result may appear to be at odds with our data, it is actually consistent with the hypothesis that Kv channels influence epithelial restitution by modulating Em and intracellular calcium. Lotz et al. [45] studied the effects of K+ channel antagonism on epithelial restitution using monolayers of the human colon carcinoma cell line T84. Colon cancer is associated with increased expression of L-type calcium channels, and surface expression of these channels has been documented in cultured T84 cells [46]. In contrast, it is well established that IEC-6 and IEC-Cdx-2 cells lack voltage-gated calcium channels [21, 23, 25]. On this basis, antagonism of Kv channels would be expected to have opposite effects on intracellular calcium and calcium-dependent cellular events in T84 cells compared to IEC-6 and IEC-Cdx-2 cells. As described above, depolarization of Em in the IEC-6 cell lines that lack voltage-gated calcium channels decreases the driving force for calcium entry thereby inhibiting calcium-dependent cell migration processes [21–23, 25]. Conversely, in T84 cells the depolarization of Em associated with Kv channel inhibition would be expected to increase the influx of calcium through voltage-gated L-type calcium channels and thus enhance epithelial cell migration.
The co-association of Kv1.4 and Kv1.6 channel subunits in IEC-Cdx-2 cells is a significant finding. The inactivation kinetics of heteromeric K+ channels containing Kv1.4 and Kv1.6 subunits differ significantly from those of homomeric Kv1.4 channels, such that channels formed by co-assembly of Kv1.4 and Kv1.6 conduct delayed rectifier K+ currents which exhibit slow, partial inactivation, while channels comprised of Kv1.4 conduct rapidly inactivating transient outward currents [47]. The expression in non-excitable cells of Kv1.4 channels does not confer sufficient K+ permeability to influence the membrane potential because of the rapid, complete inactivation of the currents [48]. In contrast, the expression in non-excitable cells of Kv channels that conduct delayed rectifier currents is sufficient to generate a resting Em that corresponds to the base of the curve relating voltage to current activation [48]. Therefore, the heteromeric channels containing Kv1.4 and Kv1.6 would be expected to contribute to the Em of IEC-Cdx2.
The NSAIDs that inhibit cell migration and depolarize the Em of IEC-6 and IEC-Cdx2 cells decrease significantly the total expression of Kv1.4 (Figure 8A) but not Kv1.6 (Figure 9A) protein in whole cell lysates. In contrast, these drugs decrease the cell surface expression of both Kv1.4 (Figure 8B) and Kv1.6 (Figure 9B) in IEC-Cdx2. These data suggest that the availability of Kv1.4 can influence the surface expression of the heteromeric channel complexes containing Kv1.4 and Kv1.6 in intestinal epithelial cells. In fact, an extensive body of work indicates not only that Kv1 channel pore-forming subunits possess distinct trafficking and surface expression properties, but also that the subunit composition of heteromeric channels determines their surface expression [49–52]. The ER export and cell-surface expression of Kv1.6 is inherently less efficient than that of Kv1.4 [50]. However, Kv1.4 enhances intracellular trafficking and surface expression of co-associated Kv1 subunits in epithelial cells [49–52]. Thus, it is highly plausible that NSAIDs inhibit intestinal cell migration and promote ulcer formation by reducing the expression of functional delayed rectifier channels on the surface membrane of intestinal epithelial cells by decreasing the total expression of Kv1.4.
Additional experiments will be needed to understand the mechanistic basis for the newly documented effects of NSAIDs on the abundance of voltage gated K+ channels on the intestinal epithelial cell surface, and to translate those findings into new strategies for mitigation of NSAID toxicity. Key questions remain to be answered: Do NSAIDs impact the cell surface abundance of Kv channels via drug effects on forward trafficking, internalization and recycling, or some combination thereof? What are the downstream effectors that link K+ channel expression and Em to epithelial cell? Do gastroprotective prostaglandins modulate NSAID effects on Em and Kv channel expression? Are the effects of NSAIDs on Kv channel expression completely independent of the drugs’ therapeutic effects?
4.4 Concluding Remarks
In summary, NS-398 and indomethacin inhibit intestinal epithelial cell migration through the depolarization of Em and the biogenic modulation of Kv channels. The data from this study suggest that these NSAIDs decrease the trafficking and cell surface expression of heteromeric Kv channel complexes by decreasing the total cellular expression of the Kv1.4 channel subunit. The mechanisms responsible for drug-induced changes in the expression of Kv1.4 remain to be elucidated. However, it is clear that depletion of intracellular polyamines is not a contributing factor. This work has identified a novel mechanism for NSAID-induced GI toxicity.
Acknowledgments
This work was supported by NIH grants P20-RR017686, T32RR017497 and T35RR007064 and the College of Veterinary Medicine at Kansas State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Israel LH, Koea JB, Stewart ID, Wright CL, Frankish PD. Nonsteroidal anti-inflammatory drug-induced strictures of the colon: report of a case and review of the literature. Dis Colon Rectum. 2001;44:1362–1364. doi: 10.1007/BF02234797. [DOI] [PubMed] [Google Scholar]
- 2.Karcher LF, Dill SG, Anderson WI, King JM. Right dorsal colitis. J Vet Intern Med. 1990;4:247–253. doi: 10.1111/j.1939-1676.1990.tb03117.x. [DOI] [PubMed] [Google Scholar]
- 3.Lichtenberger LM. Where is the evidence that cyclooxygenase inhibition is the primary cause of nonsteroidal anti-inflammatory drug (NSAID)-induced gastrointestinal injury? Topical injury revisited Biochem Pharmacol. 2001;61:631–637. doi: 10.1016/s0006-2952(00)00576-1. [DOI] [PubMed] [Google Scholar]
- 4.MacAllister CG, Morgan SJ, Borne AT, Pollet RA. Comparison of adverse effects of phenylbutazone, flunixin meglumine, and ketoprofen in horses. J Am Vet Med Assoc. 1993;202:71–77. [PubMed] [Google Scholar]
- 5.Whittle BJ. Gastrointestinal effects of nonsteroidal anti-inflammatory drugs. Fundam Clin Pharmacol. 2003;17:301–313. doi: 10.1046/j.1472-8206.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 6.Delco F, Michetti P, Beglinger C, Fried M, Szucs TD. Health care resource utilization and costs of NSAID-induced gastrointestinal toxicity. A population-based study in Switzerland. Digestion. 2004;69:10–19. doi: 10.1159/000076542. [DOI] [PubMed] [Google Scholar]
- 7.Choi HK, Seeger JD, Kuntz KM. Effects of rofecoxib and naproxen on life expectancy among patients with rheumatoid arthritis: a decision analysis. Am J Med. 2004;116:621–629. doi: 10.1016/j.amjmed.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 9.Ashton M, Hanson PJ. Disparate effects of non-steroidal anti-inflammatory drugs on apoptosis in guinea-pig gastric mucous cells: inhibition of basal apoptosis by diclofenac. Br J Pharmacol. 2002;135:407–416. doi: 10.1038/sj.bjp.0704497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahmud T, Rafi SS, Scott DL, Wrigglesworth JM, Bjarnason I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum. 1996;39:1998–2003. doi: 10.1002/art.1780391208. [DOI] [PubMed] [Google Scholar]
- 11.Sigthorsson G, Jacob M, Wrigglesworth J, Somasundaram S, Tavares I, Foster R, Roseth A, Rafi S, Mahmud T, Simpson R, Bjarnason I. Comparison of indomethacin and nimesulide, a selective cyclooxygenase-2 inhibitor, on key pathophysiologic steps in the pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in the rat. Scand J Gastroenterol. 1998;33:728–735. doi: 10.1080/00365529850171675. [DOI] [PubMed] [Google Scholar]
- 12.Somasundaram S, Sigthorsson G, Simpson RJ, Watts J, Jacob M, Tavares IA, Rafi S, Roseth A, Foster R, Price AB, Wrigglesworth JM, Bjarnason I. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther. 2000;14:639–650. doi: 10.1046/j.1365-2036.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 13.Tibble JA, Sigthorsson G, Foster R, Bjarnason I. Comparison of the intestinal toxicity of celecoxib, a selective COX-2 inhibitor, and indomethacin in the experimental rat. Scand J Gastroenterol. 2000;35:802–807. doi: 10.1080/003655200750023156. [DOI] [PubMed] [Google Scholar]
- 14.Pai R, Szabo IL, Giap AQ, Kawanaka H, Tarnawski AS. Nonsteroidal anti-inflammatory drugs inhibit re-epithelialization of wounded gastric monolayers by interfering with actin, Src, FAK, and tensin signaling. Life Sci. 2001;69:3055–3071. doi: 10.1016/s0024-3205(01)01412-6. [DOI] [PubMed] [Google Scholar]
- 15.Penney AG, Malcontenti-Wilson C, O’Brien PE, Andrews FJ. NSAID-induced delay in gastric ulcer healing is not associated with decreased epithelial cell proliferation in rats. Dig Dis Sci. 1995;40:2684–2693. doi: 10.1007/BF02220461. [DOI] [PubMed] [Google Scholar]
- 16.Rahgozar M, Pazokitoroudi H, Bakhtiarian A, Djahanguiri B. Diazoxide, a K(ATP) opener, accelerates restitution of ethanol or indomethacin-induced gastric ulceration in rats independent of polyamines. J Gastroenterol Hepatol. 2001;16:290–296. doi: 10.1046/j.1440-1746.2001.02433.x. [DOI] [PubMed] [Google Scholar]
- 17.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 18.Heath JP. Epithelial cell migration in the intestine. Cell Biol Int. 1996;20:139–146. doi: 10.1006/cbir.1996.0018. [DOI] [PubMed] [Google Scholar]
- 19.Terano A, Sakata-Horie K, Shimada T, Hiraishi H, Yoshiura K, Yoneda M, Takahashi M, Fujimori T. The role of cellular migration in the repair process of gastric epithelial cells. Life Sci. 2001;69:3083–3089. doi: 10.1016/s0024-3205(01)01414-x. [DOI] [PubMed] [Google Scholar]
- 20.Wilson AJ, Gibson PR. Epithelial migration in the colon: filling in the gaps. Clin Sci (Lond) 1997;93:97–108. doi: 10.1042/cs0930097. [DOI] [PubMed] [Google Scholar]
- 21.McDaniel SS, Platoshyn O, Yu Y, Sweeney M, Miriel VA, Golovina VA, Krick S, Lapp BR, Wang JY, Yuan JX. Anorexic effect of K+ channel blockade in mesenteric arterial smooth muscle and intestinal epithelial cells. J Appl Physiol. 2001;91:2322–2333. doi: 10.1152/jappl.2001.91.5.2322. [DOI] [PubMed] [Google Scholar]
- 22.Rao JN, Li L, Golovina VA, Platoshyn O, Strauch ED, Yuan JX, Wang JY. Ca2+-RhoA signaling pathway required for polyamine-dependent intestinal epithelial cell migration. Am J Physiol Cell Physiol. 2001;280:C993–1007. doi: 10.1152/ajpcell.2001.280.4.C993. [DOI] [PubMed] [Google Scholar]
- 23.Rao JN, Platoshyn O, Li L, Guo X, Golovina VA, Yuan JX, Wang JY. Activation of K(+) channels and increased migration of differentiated intestinal epithelial cells after wounding. Am J Physiol Cell Physiol. 2002;282:C885–C898. doi: 10.1152/ajpcell.00361.2001. [DOI] [PubMed] [Google Scholar]
- 24.Rao JN, Guo X, Liu L, Zou T, Murthy KS, Yuan JX, Wang JY. Polyamines regulate Rho-kinase and myosin phosphorylation during intestinal epithelial restitution. Am J Physiol Cell Physiol. 2003;284:C848–C859. doi: 10.1152/ajpcell.00371.2002. [DOI] [PubMed] [Google Scholar]
- 25.Wang JY, Wang J, Golovina VA, Li L, Platoshyn O, Yuan JX. Role of K(+) channel expression in polyamine-dependent intestinal epithelial cell migration. Am J Physiol Cell Physiol. 2000;278:C303–C314. doi: 10.1152/ajpcell.2000.278.2.C303. [DOI] [PubMed] [Google Scholar]
- 26.Turchanowa L, Dauletbaev N, Milovic V, Stein J. Nonsteroidal anti-inflammatory drugs stimulate spermidine/spermine acetyltransferase and deplete polyamine content in colon cancer cells. Eur J Clin Invest. 2001;31:887–893. doi: 10.1046/j.1365-2362.2001.00901.x. [DOI] [PubMed] [Google Scholar]
- 27.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang JY, Johnson LR. Polyamines and ornithine decarboxylase during repair of duodenal mucosa after stress in rats. Gastroenterology. 1991;100:333–343. doi: 10.1016/0016-5085(91)90200-5. [DOI] [PubMed] [Google Scholar]
- 29.McCormack SA, Viar MJ, Johnson LR. Polyamines are necessary for cell migration by a small intestinal crypt cell line. Am J Physiol. 1993;264:G367–G374. doi: 10.1152/ajpgi.1993.264.2.G367. [DOI] [PubMed] [Google Scholar]
- 30.Suh E, Traber PG. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol Cell Biol. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao JN, Li J, Li L, Bass BL, Wang JY. Differentiated intestinal epithelial cells exhibit increased migration through polyamines and myosin II. Am J Physiol. 1999;277:G1149–G1158. doi: 10.1152/ajpgi.1999.277.6.G1149. [DOI] [PubMed] [Google Scholar]
- 32.Rao JN, Platoshyn O, Golovina VA, Liu L, Zou T, Marasa BS, Turner DJ, Yuan JX, Wang JY. TRPC1 functions as a store-operated Ca2+ channel in intestinal epithelial cells and regulates early mucosal restitution after wounding. Am J Physiol Gastrointest Liver Physiol. 2006;290:G782–G792. doi: 10.1152/ajpgi.00441.2005. [DOI] [PubMed] [Google Scholar]
- 33.Brideau C, Kargman S, Liu S, Dallob AL, Ehrich EW, Rodger IW, Chan CC. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm Res. 1996;45:68–74. doi: 10.1007/BF02265118. [DOI] [PubMed] [Google Scholar]
- 34.Brideau C, Van Staden C, Chan CC. In vitro effects of cyclooxygenase inhibitors in whole blood of horses, dogs, and cats. Am J Vet Res. 2001;62:1755–1760. doi: 10.2460/ajvr.2001.62.1755. [DOI] [PubMed] [Google Scholar]
- 35.Kato M, Nishida S, Kitasato H, Sakata N, Kawai S. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs: investigation using human peripheral monocytes. J Pharm Pharmacol. 2001;53:1679–1685. doi: 10.1211/0022357011778070. [DOI] [PubMed] [Google Scholar]
- 36.Riendeau D, Percival MD, Brideau C, Charleson S, Dube D, Ethier D, Falgueyret JP, Friesen RW, Gordon R, Greig G, Guay J, Mancini J, Ouellet M, Wong E, Xu L, Boyce S, Visco D, Girard Y, Prasit P, Zamboni R, Rodger IW, Gresser M, Ford-Hutchinson AW, Young RN, Chan CC. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J Pharmacol Exp Ther. 2001;296:558–566. [PubMed] [Google Scholar]
- 37.McCormack SA, Johnson LR. Polyamines and cell migration. J Physiol Pharmacol. 2001;52:327–349. [PubMed] [Google Scholar]
- 38.Gogelein H, Brendel J, Steinmeyer K, Strubing C, Picard N, Rampe D, Kopp K, Busch AE, Bleich M. Effects of the atrial antiarrhythmic drug AVE0118 on cardiac ion channels. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:183–192. doi: 10.1007/s00210-004-0957-y. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Rao JN, Guo X, Liu L, Santora R, Bass BL, Wang JY. Polyamine depletion stabilizes p53 resulting in inhibition of normal intestinal epithelial cell proliferation. Am J Physiol Cell Physiol. 2001;281:C941–C953. doi: 10.1152/ajpcell.2001.281.3.C941. [DOI] [PubMed] [Google Scholar]
- 40.Kokoska ER, Smith GS, Deshpande Y, Wolff AB, Miller TA. Indomethacin increases susceptibility to injury in human gastric cells independent of PG synthesis inhibition. Am J Physiol. 1998;275:G620–G628. doi: 10.1152/ajpgi.1998.275.4.G620. [DOI] [PubMed] [Google Scholar]
- 41.Tomisato W, Tsutsumi S, Rokutan K, Tsuchiya T, Mizushima T. NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1092–G1100. doi: 10.1152/ajpgi.2001.281.4.G1092. [DOI] [PubMed] [Google Scholar]
- 42.Tomisato W, Tsutsumi S, Hoshino T, Hwang HJ, Mio M, Tsuchiya T, Mizushima T. Role of direct cytotoxic effects of NSAIDs in the induction of gastric lesions. Biochem Pharmacol. 2004;67:575–585. doi: 10.1016/j.bcp.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 43.Weiss H, Amberger A, Widschwendter M, Margreiter R, Ofner D, Dietl P. Inhibition of store-operated calcium entry contributes to the anti-proliferative effect of non-steroidal anti-inflammatory drugs in human colon cancer cells. Int J Cancer. 2001;92:877–882. doi: 10.1002/ijc.1280. [DOI] [PubMed] [Google Scholar]
- 44.Decher N, Kumar P, Gonzalez T, Pirard B, Sanguinetti MC. Binding site of a novel Kv1.5 blocker: a “foot in the door” against atrial fibrillation. Mol Pharmacol. 2006;70:1204–1211. doi: 10.1124/mol.106.026203. [DOI] [PubMed] [Google Scholar]
- 45.Lotz MM, Wang H, Song JC, Pories SE, Matthews JB. K+ channel inhibition accelerates intestinal epithelial cell wound healing. Wound Repair Regen. 2004;12:565–574. doi: 10.1111/j.1067-1927.2004.012509.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang XT, Nagaba Y, Cross HS, Wrba F, Zhang L, Guggino SE. The mRNA of L-type calcium channel elevated in colon cancer: protein distribution in normal and cancerous colon. Am J Pathol. 2000;157:1549–1562. doi: 10.1016/S0002-9440(10)64792-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roeper J, Sewing S, Zhang Y, Sommer T, Wanner SG, Pongs O. NIP domain prevents N-type inactivation in voltage-gated potassium channels. Nature. 1998;391:390–393. doi: 10.1038/34916. [DOI] [PubMed] [Google Scholar]
- 48.Felipe A, Snyders DJ, Deal KK, Tamkun MM. Influence of cloned voltage-gated K+ channel expression on alanine transport, Rb+ uptake, and cell volume. Am J Physiol. 1993;265:C1230–C1238. doi: 10.1152/ajpcell.1993.265.5.C1230. [DOI] [PubMed] [Google Scholar]
- 49.Manganas LN, Trimmer JS. Subunit composition determines Kv1 potassium channel surface expression. J Biol Chem. 2000;275:29685–29693. doi: 10.1074/jbc.M005010200. [DOI] [PubMed] [Google Scholar]
- 50.Manganas LN, Wang Q, Scannevin RH, Antonucci DE, Rhodes KJ, Trimmer JS. Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc Natl Acad Sci U S A. 2001;98:14055–14059. doi: 10.1073/pnas.241403898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu J, Watanabe I, Gomez B, Thornhill WB. Heteromeric Kv1 potassium channel expression: amino acid determinants involved in processing and trafficking to the cell surface. J Biol Chem. 2003;278:25558–25567. doi: 10.1074/jbc.M207984200. [DOI] [PubMed] [Google Scholar]
- 52.Zhu J, Gomez B, Watanabe I, Thornhill WB. Amino acids in the pore region of Kv1 potassium channels dictate cell surface protein levels: a possible trafficking code in the Kv1 subfamily. Biochem J. 2005;388:355–362. doi: 10.1042/BJ20041447. [DOI] [PMC free article] [PubMed] [Google Scholar]