

Abstract
We conducted a study to evaluate the protective efficacy in mice of vaccination with novel adenovirus vectors expressing an influenza A nucleoprotein (AdFluA-NP) based on isolates from non-human primates. In a previous study, we had observed that AdFluA-NP vectors can induce similar T cell responses in mice yet differ in ability to protect animals from lethal challenge with influenza A virus. To better define correlates of protection, we extended our study design to include additional novel AdFluA-NP vectors, and to evaluate cytotoxic T lymphocyte (CTL) responses in the spleens and lungs of immunized mice prior to virus challenge. As in our previous study, all vectors induced similar numbers of antigen-specific interferon gamma (IFN-γ) secreting T cells and memory T cells in the spleen four weeks post-immunization, but differed in their ability to protect the animals from lethal infection. However, cytokine-secreting NP antigen-specific CTLs in the lungs of mice from immunization groups that survived lethal challenge showed greater proliferative ability and higher CD27 expression. In addition, NP antigen-specific peripheral blood lymphocytes from protected mice showed greater proliferative ability after ex vivo stimulation. Our results provide additional correlates of protection that should be considered when developing anti-influenza vaccines.
Keywords: Influenza, vaccine, CTL
1. Introduction
Recent public health concerns over the emergence of new H1N1 and H5N1 influenza A (Flu A) viruses highlight the need for vaccines that afford cross-protection against multiple virus strains. Current vaccine modalities protect by inducing antibodies against viral proteins, which requires close antigenic matching between vaccine and circulating virus strains [1]. Such vaccines are only effective for a few years since the targets of such antibodies, the haemaglutinin (HA) and neuraminidase (NA) proteins, vary considerably across strains [2, 3] and can change rapidly through genetic drift and reassortment of viral genome segments in hosts infected with multiple viruses [4]. Strain-specific vaccines must be reformulated and re-administered every year, and are unlikely to protect against a pandemic outbreak caused by a novel virus strain.
In contrast, the cellular immune response to more conserved viral components, including the nucleoprotein (NP) and matrix protein (MP), has been shown to cross-protect against virus types [5, 6]. CTLs were identified as being involved in the protection afforded by immunization of mice with purified NP from a H3N2 virus, which effected substantial cross-protection against infection with a H1N1 virus as well [7]. The cellular immune response induced by immunization of mice with a H1N1-specific NP DNA vaccine protected mice against lethal challenge with H3N2 virus [8]. However, NP specific antibodies are not able to clear the infection as passive transfer of such Abs did not protect naive immunodeficient scid recipient mice[9]. Due to these, cellular mediated vaccines based on one or both of these NP proteins are now being investigated actively in the effort to develop pan-influenza A vaccines.
Vaccination with purified protein or DNA relies on passive uptake of the inoculums by antigen-presenting dendritic cells before an immune response can be initiated [10]. A more robust response can be achieved using viral vectors that actively deliver the DNA or immunogen into dendritic cells. We and other investigators have shown that immunization of mice with adenovirus-based vectors induces a potent CTL response against encoded transgenes through efficient transduction of target cells [11, 12]. Vectors based on HAdV5, a subgroup C adenovirus, were used primarily in these studies. Unfortunately, HAdV5 is a common human pathogen; NAbs against it are quite common in the human population, interfering with its effectiveness as a gene delivery vector for human therapeutics [13, 14]. Efforts to surmount this problem with HAdV5 and other common human adenoviruses have focused on modification of virus capsid components, masking viral antigens with polyethylene glycol, development of chimeric viruses, and identification of novel adenovirus isolates to which the human population is naive.
To increase the repertoire of vectors available for vaccine development, we recently isolated a series of novel adenoviruses from stools of higher order primates (manuscript in preparation). We have now engineered three of these viruses to express the Flu A NP, and used them to vaccinate Balb/c mice, challenging one month after vaccination with a lethal dose of influenza A virus A/Puerto Rico/8/34/Mount Sinai (PR8-MTS). The novel vectors differed in their ability to protect mice after challenge. To understand the basis for protection we analyzed several aspects of the cellular immune response engendered by each vector prior to virus challenge. Surprisingly, all immunization groups had similar numbers of cytokine secreting antigen-specific T cells in their spleens. However, cytokine-secreting NP antigen-specific CTLs in the lungs of mice from immunization groups that survived lethal challenge showed greater proliferative ability and higher CD27 expression. In addition, NP antigen-specific peripheral blood lymphocytes from protected mice showed greater proliferative ability after ex vivo stimulation. Our study raises important concerns about judgments of vaccine efficacy predicated upon limited analysis of the CTL response.
2. Materials and methods
2.1. Viruses and vectors
The novel adenoviruses used in this study were described previously [15]. Influenza A NP-expressing adenovirus vectors (AdFluA-NP) were constructed from selected viruses as described [16]. Briefly, an expression cassette, composed of a synthetic, codon-optimized H1N1 Flu A NP gene (from PR8-MTS, GenBank accession number AF389119.1) under the control the human cytomegalovirus early promoter and followed by the bovine growth hormone polyadenylation signal, was inserted in place of an E1deletion in human adenovirus 5 (HAdV5), and simian adenoviruses 28 (SAdV28), 30 (SAdV30) and 39 (SAdV39), by the construction of plasmid molecular clones, as described [17]. HAdV5 vectors also carried an additional deletion of the E3 region. The recombinant adenoviruses (HAdV5-NP, SAdV28-NP, SAdV30-NP and SAdV39-NP) were rescued by transfection of plasmids into HEK 293 cells. Adenoviruses and adenovirus-based vectors were purified by cesium chloride density gradient centrifugation (PennVector, University of Pennsylvania, Philadelphia, PA). Particle counts in purified virus stocks were determined by measuring absorbance at 260 nm.
Influenza A PR8-MTS virus was a gift from Dr. Jan Erikson at Wistar institute, Philadelphia. The virus was preparation by propagating in the allantoic cavity of embryonated hen's eggs as previously described [18] and titered using a limiting dilution assay [19]. Titers are expressed as the 50% tissue culture infectious dose (TCID50) of Madin-Darby canine kidney (MDCK) cells.
2.2. Vaccination and challenge of Balb/c mice
Protocols for all animal studies were approved by institutional animal care and use committees of the University of Pennsylvania. Female Balb/c mice (6–8 weeks old) were obtained from Jackson Laboratories (Bar Harbor, Maine). For vaccination, mice from each group (n=11) were immunized by intramuscular injection of 50 μl of PBS containing the specified number of virus particles into the hind limb. Four mice from each group were sacrificed one month post immunization and the isolated T cells were phenotyped and characterized using functional assays. The remaining mice (n=7) were infected two months post immunization by intranasal inoculation with a lethal dose (10 LD50) of PR8-MTS virus, as previously described [16]. Animals were monitored daily for clinical signs of influenza infection and body weight. Mice that lost greater than 30% of body weight were euthanized.
2.3. Peptides
A peptide corresponding to the H2-Kd-restricted immunodominant CTL epitope of the influenza A NP protein (amino acids 197–205: TYQRTRALV) [20] was synthesized by Mimotopes (Clayton Victoria, Australia) and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mg/ml. DMSO content in assay reactions was kept below 0.1% (v/v). A pool of overlapping peptides spanning the protein sequence of the influenza A NP was obtained from Dr. Gary kobinger at University of Manitoba. All peptides were 15 amino acids in length, overlapping by 10 amino acids, and present in the pool at equimolar concentration.
2.4. Isolation of lymphocytes from lungs and spleen
Methods used have been described previously [21]. Briefly, lung tissue was diced to about 1 mm3 fragments in RPMI 1640 containing 1 mg/ml collagenase IV (Sigma), then incubated at 37°C for 1 h. The digested tissue suspension was then passed through a 100 μm nylon mesh filter to remove cell clumps and non-dissociated tissue. Cells were collected from the clarified suspension, washed three times with PBS, then layered onto a 56–64% Percoll density gradient, which was centrifuged for 30 min at 2,000 rpm. The lymphocyte band was recovered and the lymphocytes were washed three times in PBS prior to use.
Lymphocytes were isolated from spleens following mechanical disruption to generate single cell suspensions.
2.5. MHC class I tetramer staining and memory cell phenotype analysis
Phycoerythrin (PE)-conjugated major histocompatibility complex class I (MHC-I) H2-Kd:TYQRTRALV tetramer complex was obtained from the NIH Tetramer Core Facility. Cells collected from heparinized whole blood or lymphocytes isolated from spleen or lungs were stained by incubation for 30 min at room temperature with PE-conjugated tetramers, FITC-conjugated anti-CD8a antibody (Ly-2; BD Biosciences Pharmingen, San Diego, CA), PE Cy5-conjugated anti-CD127 antibody (eBioscience, CA) and PE Cy7-conjugated anti-CD62L antibody (eBioscience, CA). Red blood cells were then lysed, and cells were fixed with iTAg MHC tetramer fix solution (Beckman Coulter) for 10 minutes at room temperature. The fixed cells were washed three times with PBS, fixed again with BD CytoFix (BD Biosciences) for 20 min at 4°C, and examined by flow cytometry using a FC500 flow cytometer (Beckman Coulter). Data were analyzed using FlowJo software (Tree Star, San Carlos, CA). Lymphocytes were selected on the basis of characteristic forward and side scatter. CD8+/Tetramer+ lymphocytes were gated from the total population and evaluated further for CD127 and CD62L staining.
2.6. Analysis of cytokine production and expression of T-cell functional markers
Cytokine production and levels of CD27, CD43 and PD-1 proteins on CD8+/ IFN-γ+ T cells were evaluated by flow cytometry following combined surface and intracellular immunostaining with fluorochrome-labeled monoclonal antibodies using the BD Cytofix/CytoPerm™ Plus kit (PharMingen, CA), following protocols recommended by the manufacturer. For evaluation, 106 lymphocytes isolated from spleen or lung tissue were stimulated by incubation with Flu NP peptide at 2 μg/ml in DMEM supplemented with 2% FBS, 50 μM 2-mercaptoethanol, and 1 μg/ml of GolgiPlug™ protein transport inhibitor (BD PharMingen, San Diego, CA) for 5 h at 37°C. For evaluation of cytokine production, stimulated cells were washed and stained with FITC-conjugated antibody to mouse CD8a (Ly-2), then fixed and permeabilized, and stained with PE-conjugated anti-mouse IFN-γ (BD Biosciences Pharmingen, CA), PE-Cy7-conjugated anti-tumor necrosis factor alpha (TNF-α) (BD Biosciences Pharmingen, CA), and allophycocyanin (APC)-conjugated anti-interleukin-2 (IL-2) (BD Biosciences Pharmingen, CA). To evaluate expression of T cell functional markers stimulated cells were stained with FITC- or PE-Cy5 conjugated anti-mouse CD8 antibody, APC-conjugated anti-mouse CD27 antibody (eBioscience, CA), FITC-conjugated anti-mouse CD43 antibody (eBioscience, CA) or FITC-conjugated anti-mouse PD-1 (eBioscience, CA), then permeabilized and stained with PE-conjugated anti-mouse IFN-γ. Cells were analyzed by multi-color flow cytometry using a FC500 flow cytometer (Beckman Coulter). Data were analyzed with FlowJo software (Tree Star, San Carlos, CA).
2.7. CFSE-dye dilution assay for lymphocyte proliferation
Freshly isolated PBMCs were stained with CFSE as previously described [22]. Briefly, 5 million cells in one ml of PBS were mixed with an equal volume of 5 μM CFSE. Following staining, cells were stimulated by incubation with the NP peptide pool in alpha MEM for 6 days. Equivalent reactions were set up with no peptide and with an irrelevant peptide from HIV-gag to serve as controls. Following stimulation, cells were stained with antibodies to APC-conjugated anti-mouse CD4 and PE-Cy5-conjugated anti-mouse CD8 to identify T cell populations. Proliferating cells were defined as the CFSElow/CD4+ and CFSElow /CD8+ populations gated from total CD4+ and CD8+ cells respectively. Samples with numbers of proliferating cells four times higher than in control wells, including more than 1% of total CD4+ or CD8+ cells were considered positive.
3. Results
3.1. Protection from lethal virus challenge following immunization with novel vectors
In a recent survey of adenoviruses in non-human primates, we isolated more than 30 novel strains from the stools of wild macaques, chimpanzees, bonobos, orangutans, and gorillas [15]. Three chimpanzee isolates, SAdV28 (adenovirus subgroup B), SAdV30 (adenovirus subgroup E) and SAdV39 (adenovirus subgroup E) were chosen for AdFluA-NP vaccine development based on the yield and genetic stability of E1 deleted version of the recombinant virus rescued after transfection of molecular clones in 293 cells. Mice were immunized with 1011 particles of SAdV28-NP, SAdV30-NP, SAdV39-NP, HAdV5-NP, or a E1/E3-deleted HAdV5 vector expressing the E. coli lacZ gene (HAdV5 lacZ), included as a negative control. One month after immunization, spleen and lung tissue samples and peripheral blood were collected from a subset of the mice for analysis of the cell mediated immune response to vaccination. The remaining mice were challenged with a lethal dose of live PR8-MTS virus (10 LD50) by intranasal inoculation and followed for signs of illness for three weeks.
Very clear differences in protection following vaccination were seen among the vectors used for immunization. All mice in the HAdV5 lacZ control group either succumbed to infection within eight days of challenge or were euthanized within 12 days due to loss of >30% of body weight (Fig. 1A). All mice immunized with HAdV5-NP and on average >95% mice that received SAdV30-NP were protected from lethal infection and survived until the end of the study (~3 weeks) (Fig. 1A and 1C). Animals in these groups showed signs of illness, losing ~20–30% of their body weight after the challenge, but recovered most of the lost weight over the observation period (Fig. 1B). In contrast, 70% of the mice immunized with SAdV28-NP had to be euthanized between eight and twelve days of challenge infection due to body weight losses exceeding 30% (Fig. 1B). Interestingly, immunization with SAdV39-NP, an adenovirus which shares the same subgroup as SAdV30-NP, was also protective (Fig. 1A). These findings raise the possibility that vaccine induced T cell responses were similar within adenovirus subgroups than without.
Figure 1.

Protection effect mediated by new Ad vaccines after a lethal dose of PR8-MTS virus challenge. Mice in different groups were vaccinated with 4 serotypes of AdFluANP vaccines (1011 particles/mouse) and HAdV5 LacZ as negative control, followed by challenged with a lethal dose (10LD50) of PR8-MTS virus 1 month post immunization. Their survival (A) and average percentage of normal body weight (B) were monitored for the following 3 weeks. For further assessing the possible dose-dependent protection of vaccines, we immunized mice with 109, 1010 and 1011 particles of SAdV28 and 30 Flu NP vaccines. Survival (C) and average percentage of normal body weight (D) were followed post challenge for 3 weeks.
A second study was completed comparing protection following immunization with SAdV28-NP and SAdV30-NP at several doses. Mice immunized with 109, 1010 or 1011 vector particles were inoculated with 10 LD50 of PR8-MTS one month after vaccination and monitored for survival and body weight for three weeks post challenge, as in the initial study. As before, all mice immunized with HAdV5 lacZ succumbed to the virus challenge. Dose-dependent protection was seen with SAdV30-NP, with 95% survival at the highest dose, 70% survival at the intermediate dose, and 30% survival at the lowest dose (Fig. 1C). In contrast, only mice immunized with the highest dose of SAdV28-NP were protected, with a survival rate of 43%. All mice immunized with either of the lesser doses succumbed to the challenge infection. In addition, surviving animals vaccinated with SAdV28-NP lost more body weight compared to mice vaccinated with SAdV30-NP (Fig 1D).
This suggests the subgroup E virus is close to 100-fold more potent than the subgroup B virus, a difference unlikely due to vector specific effect on transgene expression. In fact, in vitro, transduction studies with these vectors indicated comparable level of transgene expression (data not shown).
3.2. Analysis of antigen-specific CD8+ T lymphocytes from spleen and lungs of immunized mice
Lymphocytes isolated from lung and spleen tissue of mice one month after vaccination with SAdV30-NP, SAdV28-NP, SAdV39-NP, and HAdV5-NP were processed and stained to evaluate various aspects of the NP-specific cell mediated immune response engendered by each vector.
CTLs specific for the dominant NP epitope were enumerated in lymphocytes isolated from spleens of immunized mice by simultaneous staining with anti-CD8 antibody and fluorescented H2-Kd: TYQRTRALV tetramer complexes. Numbers of CD8+/tetramer+ lymphocytes were similar in spleens from all treatment groups (Fig 2A). Further staining of this population with antibodies to cell surface markers CD62L, a lymph node homing receptor, and CD127, the IL-7 receptor, was performed to evaluate memory cell compartmentalization. CD127+/CD62L− cells are effector memory cells found in peripheral circulation, distinct from CD127+/CD62L+ central memory cells that predominantly home to lymphoid organs and CD127−/CD62L− double negative effector cells that have yet to convert to a memory phenotype. Surprisingly, no clear differences in numbers or phenotypes of NP-specific memory cells were observed among mice immunized with the different vectors (Fig. 2B).
Figure 2.
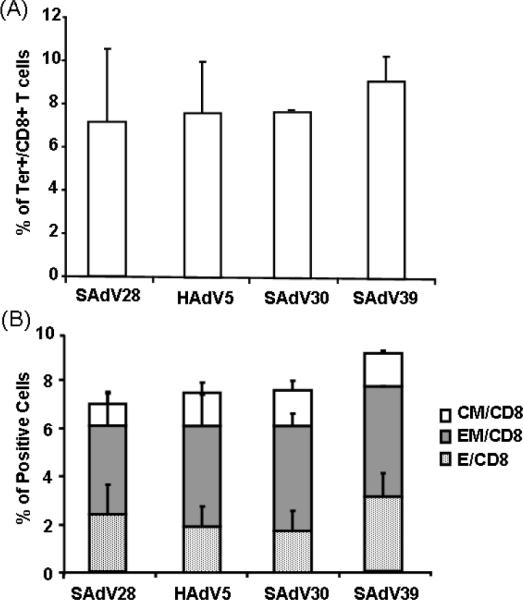
NP specific CD8+ T cell responses and memory phenotype analyses in PBMC. Mice were vaccinated with 1011 particles of Ad Flu NP vaccines. 4 weeks later before Flu virus challenge, splenocytes were isolated and the NP specific CD8 T cells responses were measured by NP tetramer staining (A). Memory phenotypes of NP tetramer specific CD8+ T cells were also determined by the expression of CD127 and CD62L on NP specific T cells (B). Data are shown as mean with SD, n=4.
Epitope-specific cytokine production by CTLs isolated from spleen and lung was evaluated following ex vivo stimulation for six hrs with the immunodominant NP epitope peptide. In spleen cytokine production (IFNγ, TNFα, and IL2) by CD8+ T cells was similar across immunization groups (Fig 3A). All vectors induced similar numbers of polyfunctional double (IFNγ+TNFα+IL2−) and triple (IFNγ+TNFα+IL2+) cytokine expressing cells. In contrast, the numbers of cytokine expressing NP-specific CTLs in the lung were three to four fold lower in mice immunized with SAdV28-NP and which did not survive the lethal challenge, than in the lungs of mice immunized with vectors that afforded protected (Fig. 3B). These assays were performed on cells harvested at the time of challenge.
Figure 3.

NP specific polyfunctional CD8 T-cell responses in tissue-derived cells from BALB/c mice. Lymphocytes were isolated from the spleens and lungs and were evaluated by ICCS to measure the production of cytokines including IFN-γ, TNF-α and IL-2 combined with anti-CD8 antibody surface staining in the context of stimulation with NP dominant peptide. Data are expressed as the frequencies of multiple-cytokine-secreting antigen-specific CD8 T cells isolated from spleen (A) and lung (B) 4 weeks after i.m. Ad immunization. Data are presented as means and standard deviations for four mice per group. Lung samples were analyzed as pools due to the relatively low yields.
Epitope-specific cytokine-producing CTLs (CD8+IFNγ+) from the lung were further characterized for expression of several T cell functional markers, PD-1, CD27 and CD43. PD-1 is a negative regulator of CTL responses; CD43 is a T cell activation marker and CD27, a member of the TNF receptor family, has been shown to be essential for the survival and accumulation of virus-specific T cells in the lung [23]. No differences were observed among immunization groups in either PD-1 or CD43 expression in CD8+IFNγ+ lymphocytes isolated from the lung (Fig 4). However, significantly fewer CD8+IFNγ+ CTLs from the lungs of mice immunized with SAdV28-NP showed evidence of CD27 up-regulation. Lower numbers of CD27 expressing CTLs may be the basis for the inability of SAdV28-NP's to engender a protective response.
Figure 4.

Activation characteristics of NP specific CD8+ T cells isolated from lung. Fresh lung-isolated lymphocytes were evaluated by ICCS to measure intracellular IFNγ production in combination with the expression of activation and apoptosis markers including PD-1, CD27 and CD43 with anti-CD8 antibody in the context of stimulation with NP dominant peptide. IFNγ+CD8+ T cell population are gated and analyzed the expression of PD-1, CD27 and CD43 by histogram. The numbers in the figure indicate the high expression populations for PD-1 and CD27 and positive populations (black line) compared to naïve CD8+ population (gray line) for CD43 expression. The data is the representation of two independent assays.
3.3. Epitope-specific proliferation of peripheral blood T cells from immunized mice
The ability of epitope-specific T cells to proliferate in response to antigen stimulation has been identified as an important correlate of vaccine protection; impaired proliferative ability has been associated with an inability to clear viral infections [24]. An ex vivo CFSE dye-dilution assay was used to evaluate proliferation of circulating NP-specific T cells in peripheral blood samples from the vaccinated mice prior to challenge. PBMCs were stained with CSFE, stimulated by incubation with a NP peptide pool for six days, and finally immunostained with antibodies to CD4 and CD8. CD4+ and CD8+ populations were evaluated for CFSE levels. Proliferating T cells were defined as the CFSElow/CD4+ and CFSElow/CD8+ populations among total CFSE-labeled cells.
Antigen-induced proliferation of both CD4+ and CD8+ populations was lowest in SAdV28-NP-immunized mice (Fig 5A and 5B), with three to four times fewer CFSElow cells than were observed for mice immunized with HAdV5-NP and SAdV30-NP. These results support antigen-specific T cell proliferative capacity as a marker for vaccine efficacy, providing another possible explanation for SAdV28-NP's failure to protect from lethal infection.
Figure 5.
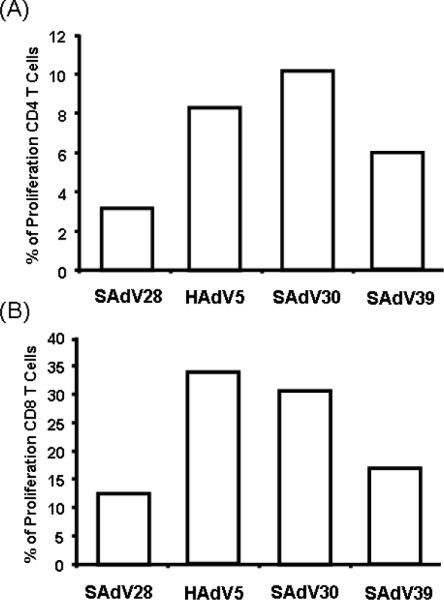
NP specific CD4 and CD8 T cells proliferative potential. 4 weeks post immunization, the levels of CD4+ and CD8+ T cell proliferation was assessed by a CFSE dye-dilution assay. CFSE labeled CD4+ and CD8+ T cells were stimulated ex vivo for 6 days with Flu NP peptide library. Proliferating cells were defined as the CFSElow population among the total CFSE labeled CD4+ (A) or CD8+ (B) populations. The levels of background measured by naïve CD4+ and CD8+ population in the stimulation of NP library and control population in the stimulation of HIV gag (un-specific peptide) are less than 0.03%.
4. Discussion
With the present study, we build on previous work in our laboratory [16] and by others [25] to identify immune correlates of vaccine efficacy in a rodent model of influenza A virus infection. We generated FluA NP expression vectors using three recently isolated chimpanzee adenoviruses, SAdV28, SAdV30 and SAdV39, belonging to subgroups B, E and E respectively, and used them to vaccinate mice against influenza infection. We extended our previous study protocol to include analysis of antigen-specific T cell function one month after vaccination, just prior to challenge with a lethal dose of virus, increased the number of T cell functional assays performed, and analyzed CTLs isolated from both lung and spleen.
Vaccination with two of the novel vectors, SAdV30-NP and SAdV39-NP, and the control HAdV5-NP vector engendered a strong immune response, protecting immunized mice against challenge with a lethal dose of PR8-MTS virus. However, the third novel vector tested, SAdV28-NP, clearly differed from the others, providing little protection against challenge even at the highest immunizing dose. This difference correlated with impairment in three aspects of the cell mediated immune response to the NP transgene - fewer antigen-specific cytokine-secreting CTLs in the lungs, decreased expression of CD27 on antigen-specific cytokine-secreting CTLs in the lung, and decreased ability of circulating antigen-specific T cells to proliferate upon ex vivo stimulation. No differences were observed in numbers or types of memory or cytokine secreting CTLs in spleen.
Hendricks et al observed that the expression of CD27, a member of the TNF receptor superfamily, strongly correlates with the ability of T cells to home to the lung [26]. In their study, CD27−/− mice inoculated intranasally with influenza showed a decrease in the absolute numbers of T cells that infiltrated the lungs; the effect was more pronounced following a secondary challenge with the virus. Their results support the conclusion that lack of protection following immunization with SAdV28-NP results from insufficient numbers of influenza-specific CTLs in the lungs due to low expression of CD27.
Diminished antigen-specific proliferation was observed in both CD8+ and CD4+ T cell subsets in the mice immunized with SAdV28-NP relative to mice immunized with other vectors. At present it is not clear what could cause this difference in response. It is known that the lack of T cell proliferation observed in CD27−/− mice is not due to a block in entry into cell division cycle, but rather due to a lack of survival of activated T cells following successive rounds of division [23]. It should be noted that we observed no difference in expression of PD-1, which is up-regulated in exhausted T cells [27].
Why should influenza specific T cells in mice immunized with SAdV28 show reduced CD27 expression? This could arise from differences in targeting and activation of antigen presenting cells (APCs) by the different adenoviruses. The interaction of CD27 with its ligand CD70, which is expressed on APCs, is required for an optimal T cell response [28, 29]. However, APCs only express CD70 upon maturation. It is possible that differential targeting and/or activation of APCs by adenoviruses could result in sub-optimal APC-T cell interactions that result in reduced CD27 expression. On the other hand, variation in biodistribution of vectors, including the specific lymph nodes in which interaction of T cells with APCs occurs, could create vector-specific functional differences [30].
The presence of neutralizing antibodies to influenza A virus has been unambiguously established as an immune correlate of protection against infection [31, 32]. However, the primary targets of such antibodies are highly variable proteins displayed on the surface of the virus, so this humoral immunity offers only limited protection against heterotypic strains. Understanding of immune correlates of protection following administration of a T cell vaccine are more limited, although studies have documented a role for NP-specific CD8 T cells in protection against influenza infection in animal models [8, 16]. Christensen et al. demonstrated that a large pool of CD8+ memory T cells may be only partially utilized to deal with a potentially lethal influenza infection [7]. McMichael et al. conclude that cytotoxic T cells play a part in recovery from influenza virus infection and high levels of cytotoxic T cells reduced the amount and period of viral shedding [33]. Another group found that influenza NP-specific CD8+ CTL can play a direct role in clearance of influenza virus [34]. In the study reported here, we provide additional data to further the understanding of this important topic. Our results indicate that the proliferative capacity of NP-specific CTL and their ability to home to the lung are significant correlates of adenovirus vaccine-mediated protection against influenza virus infection. A caveat of the present study is that immune responses were noted following immunization via i.m. administrations and may differ when vectors are administered via other routes of administration.
The adenovirus vector platform established in our laboratory allows us not only to screen many vectors for their utility as vaccines, but also to define possible immune correlates of protection through comparison of efficacy across an expanding variety of constructs. Additional studies are ongoing in our group to study the cell-mediated immune response against influenza virus induced by vaccines based on additional novel non-human primate adenoviruses.
Acknowledgement
The authors appreciate the NIH Tetramer Core Facility at Emory University for providing quality tetramer reagents, and the support from the Vector Core and Animal Model Group of the Gene Therapy Program at the University of Pennsylvania. The authors thank Dr. Jan Erikson at Wistar institute for providing the Influenza A PR8-M strain of virus and Dr. Gary kobinger at University of Manitoba for supplying the NP peptide library. This research was supported by P30-DK-47757 (JMW), P01-HL-059407 (JMW), AI-30034 (DCM) and a grant from GlaxoSmithKline Pharmaceuticals, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure J.M.W. is a consultant to ReGenX Holdings, and is a founder of, holds equity in, and receives a grant from affiliates of ReGenX Holdings; in addition, he is an inventor on patents licensed to various biopharmaceutical companies, including affiliates of ReGenX Holdings.
References
- [1].Fiore AE, Bridges CB, Cox NJ. Seasonal influenza vaccines. Curr Top Microbiol Immunol. 2009;333:43–82. doi: 10.1007/978-3-540-92165-3_3. [DOI] [PubMed] [Google Scholar]
- [2].Kilbourne ED, Johansson BE, Grajower B. Independent and disparate evolution in nature of influenza A virus hemagglutinin and neuraminidase glycoproteins. Proc Natl Acad Sci U S A. 1990;87(2):786–90. doi: 10.1073/pnas.87.2.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoskins TW, Davies JR, Smith AJ, Miller CL, Allchin A. Assessment of inactivated influenza-A vaccine after three outbreaks of influenza A at Christ's Hospital. Lancet. 1979;1(8106):33–5. doi: 10.1016/s0140-6736(79)90468-9. [DOI] [PubMed] [Google Scholar]
- [4].Furuse Y, Suzuki A, Shimizu M, Kishi M, Sawayama R, Saito M, et al. Reassortment between amantadine-resistant and -sensitive H1N1 influenza A viruses generated an amantadine-sensitive virus during the 2007–2008 season. J Infect Dis. 2009;200(11):1766–73. doi: 10.1086/647989. [DOI] [PubMed] [Google Scholar]
- [5].Saha S, Yoshida S, Ohba K, Matsui K, Matsuda T, Takeshita F, et al. A fused gene of nucleoprotein (NP) and herpes simplex virus genes (VP22) induces highly protective immunity against different subtypes of influenza virus. Virology. 2006;354(1):48–57. doi: 10.1016/j.virol.2006.04.015. [DOI] [PubMed] [Google Scholar]
- [6].Altstein AD, Gitelman AK, Smirnov YA, Piskareva LM, Zakharova LG, Pashvykina GV, et al. Immunization with influenza A NP-expressing vaccinia virus recombinant protects mice against experimental infection with human and avian influenza viruses. Arch Virol. 2006;151(5):921–31. doi: 10.1007/s00705-005-0676-9. [DOI] [PubMed] [Google Scholar]
- [7].Christensen JP, Doherty PC, Branum KC, Riberdy JM. Profound protection against respiratory challenge with a lethal H7N7 influenza A virus by increasing the magnitude of CD8(+) T-cell memory. J Virol. 2000;74(24):11690–6. doi: 10.1128/jvi.74.24.11690-11696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259(5102):1745–9. doi: 10.1126/science.8456302. [DOI] [PubMed] [Google Scholar]
- [9].Gerhard W, Mozdzanowska K, Furchner M, Washko G, Maiese K. Role of the B-cell response in recovery of mice from primary influenza virus infection. Immunol Rev. 1997;159:95–103. doi: 10.1111/j.1600-065x.1997.tb01009.x. [DOI] [PubMed] [Google Scholar]
- [10].Akbari O, Panjwani N, Garcia S, Tascon R, Lowrie D, Stockinger B. DNA vaccination: transfection and activation of dendritic cells as key events for immunity. J Exp Med. 1999;189(1):169–78. doi: 10.1084/jem.189.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hosoya N, Miura T, Kawana-Tachikawa A, Koibuchi T, Shioda T, Odawara T, et al. Comparison between Sendai virus and adenovirus vectors to transduce HIV-1 genes into human dendritic cells. J Med Virol. 2008;80(3):373–82. doi: 10.1002/jmv.21052. [DOI] [PubMed] [Google Scholar]
- [12].Varnavski AN, Schlienger K, Bergelson JM, Gao GP, Wilson JM. Efficient transduction of human monocyte-derived dendritic cells by chimpanzee-derived adenoviral vector. Hum Gene Ther. 2003;14(6):533–44. doi: 10.1089/104303403764539323. [DOI] [PubMed] [Google Scholar]
- [13].Mast TC, Kierstead L, Gupta SB, Nikas AA, Kallas EG, Novitsky V, et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine. 2009 doi: 10.1016/j.vaccine.2009.10.145. [DOI] [PubMed] [Google Scholar]
- [14].Sumida SM, Truitt DM, Lemckert AA, Vogels R, Custers JH, Addo MM, et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174(11):7179–85. doi: 10.4049/jimmunol.174.11.7179. [DOI] [PubMed] [Google Scholar]
- [15].Roy S, Vandenberghe LH, Kryazhimskiy S, Grant R, Calcedo R, Yuan X, et al. Isolation and characterization of adenoviruses persistently shed from the gastrointestinal tract of non-human primates. PLoS Pathog. 2009;5(7):e1000503. doi: 10.1371/journal.ppat.1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Roy S, Kobinger GP, Lin J, Figueredo J, Calcedo R, Kobasa D, et al. Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine. 2007;25(39–40):6845–51. doi: 10.1016/j.vaccine.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roy S, Gao G, Lu Y, Zhou X, Lock M, Calcedo R, et al. Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Hum Gene Ther. 2004;15(5):519–30. doi: 10.1089/10430340460745838. [DOI] [PubMed] [Google Scholar]
- [18].Mozdzanowska K, Feng J, Gerhard W. Virus-neutralizing activity mediated by the Fab fragment of a hemagglutinin-specific antibody is sufficient for the resolution of influenza virus infection in SCID mice. J Virol. 2003;77(15):8322–8. doi: 10.1128/JVI.77.15.8322-8328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Prince GA, Hemming VG, Horswood RL, Baron PA, Chanock RM. Effectiveness of topically administered neutralizing antibodies in experimental immunotherapy of respiratory syncytial virus infection in cotton rats. J Virol. 1987;61(6):1851–4. doi: 10.1128/jvi.61.6.1851-1854.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Andreansky SS, Stambas J, Thomas PG, Xie W, Webby RJ, Doherty PC. Consequences of immunodominant epitope deletion for minor influenza virus-specific CD8+-T-cell responses. J Virol. 2005;79(7):4329–39. doi: 10.1128/JVI.79.7.4329-4339.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin J, Zhi Y, Mays L, Wilson JM. Vaccines based on novel adeno-associated virus vectors elicit aberrant CD8+ T-cell responses in mice. J Virol. 2007;81(21):11840–9. doi: 10.1128/JVI.01253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hawkins ED, Hommel M, Turner ML, Battye FL, Markham JF, Hodgkin PD. Measuring lymphocyte proliferation, survival and differentiation using CFSE time-series data. Nat Protoc. 2007;2(9):2057–67. doi: 10.1038/nprot.2007.297. [DOI] [PubMed] [Google Scholar]
- [23].Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med. 2003;198(9):1369–80. doi: 10.1084/jem.20030916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Day CL, Kiepiela P, Leslie AJ, van der Stok M, Nair K, Ismail N, et al. Proliferative capacity of epitope-specific CD8 T-cell responses is inversely related to viral load in chronic human immunodeficiency virus type 1 infection. J Virol. 2007;81(1):434–8. doi: 10.1128/JVI.01754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Quan FS, Huang C, Compans RW, Kang SM. Virus-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J Virol. 2007;81(7):3514–24. doi: 10.1128/JVI.02052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol. 2000;1(5):433–40. doi: 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- [27].Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- [28].Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood. 2009;113(21):5167–75. doi: 10.1182/blood-2008-03-148007. [DOI] [PubMed] [Google Scholar]
- [29].Hashimoto-Okada M, Kitawaki T, Kadowaki N, Iwata S, Morimoto C, Hori T, et al. The CD70-CD27 interaction during the stimulation with dendritic cells promotes naive CD4(+) T cells to develop into T cells producing a broad array of immunostimulatory cytokines in humans. Int Immunol. 2009;21(8):891–904. doi: 10.1093/intimm/dxp056. [DOI] [PubMed] [Google Scholar]
- [30].Sheets RL, Stein J, Bailer RT, Koup RA, Andrews C, Nason M, et al. Biodistribution and toxicological safety of adenovirus type 5 and type 35 vectored vaccines against human immunodeficiency virus-1 (HIV-1), Ebola, or Marburg are similar despite differing adenovirus serotype vector, manufacturer's construct, or gene inserts. J Immunotoxicol. 2008;5(3):315–35. doi: 10.1080/15376510802312464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ito R, Ozaki YA, Yoshikawa T, Hasegawa H, Sato Y, Suzuki Y, et al. Roles of anti-hemagglutinin IgA and IgG antibodies in different sites of the respiratory tract of vaccinated mice in preventing lethal influenza pneumonia. Vaccine. 2003;21(19–20):2362–71. doi: 10.1016/s0264-410x(03)00078-1. [DOI] [PubMed] [Google Scholar]
- [32].Belshe RB, Gruber WC, Mendelman PM, Mehta HB, Mahmood K, Reisinger K, et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine. J Infect Dis. 2000;181(3):1133–7. doi: 10.1086/315323. [DOI] [PubMed] [Google Scholar]
- [33].McMichael AJ, Gotch FM, Noble GR, Beare PA. Cytotoxic T-cell immunity to influenza. N Engl J Med. 1983;309(1):13–7. doi: 10.1056/NEJM198307073090103. [DOI] [PubMed] [Google Scholar]
- [34].Mbawuike IN, Zhang Y, Couch RB. Control of mucosal virus infection by influenza nucleoprotein-specific CD8+ cytotoxic T lymphocytes. Respir Res. 2007;8:44. doi: 10.1186/1465-9921-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]