

Abstract
Objective: To determine if cardiopulmonary bypass (CPB), together with inhibition of the sodium–hydrogen exchanger (NHE), limits myocardial and neurological injury and improves recovery after prolonged (unwitnessed) cardiac arrest (CA), as NHE inhibition improved recovery after deep hypothermic circulatory arrest. Methods: Twenty-seven pigs (31–39 kg) underwent 15 min of prolonged (no-flow) CA followed by 10 min of cardiopulmonary resuscitation-advanced life support (CPR-ALS). Subjects with restoration of spontaneous circulation (ROSC) during CPR-ALS received either no drug (n = 6) or an inhibitor of the NHE (HOE-642; n = 5). In the 16 unsuccessfully resuscitated animals, peripheral normothermic CPB was instituted, and either no drug (n = 9) or similar HOE-642 (n = 7) therapy started. Hemodynamic data, a species-specific neurological deficit score (0 = normal to 500 = brain death), and mortality were recorded at 24 h, and biochemical variables of organ injury measured. Results: CPR-ALS restored ROSC in 41% (11/27) of animals, but was unsuccessful in 59% (16/27) that required CPB. Without CPB, HOE-642 increased cardiac index and decreased vascular resistance; with CPB, HOE-642 caused higher pump flows (3.4 ± 0.6 l min−1 m−2 vs 2.5 ± 0.7 l min−1 m−2; p ≪ 0.001) and higher post-arrest cardiac index; but animals required more vasopressors (p = 0.019) from drug-induced vasodilation. No differences between biochemical markers of oxidative and organ injury and overall 24-h mortality (20%) were found between groups. Neurological score was improved at 24 h compared with 4 h only after HOE-642 treatment with (150 ± 34 vs 220 ± 43; p = 0.003) or without CPB (162 ± 39 vs 238 ± 48; p ≤ 0.001), but failed to reach statistical difference with respect to the untreated group. Conclusions: CPB is an effective resuscitative tool to treat prolonged CA but there is limited improvement of neurological function. NHE inhibition augments cardiac and neurological function, but its effect was less pronounced than in other studies.
Keywords: Cardiac arrest, Cardiopulmonary resuscitation, Cardiopulmonary bypass, Sodium–hydrogen exchanger (NHE) inhibition
1. Introduction
Brain injury and its sequelae in post-resuscitation survivors remain an unsolved problem that exists, despite our capacity to restore heart function in a limited number of instances [1,2]. To circumvent inadequate systemic perfusion, cardiopulmonary bypass (CPB) as a resuscitative tool has been repeatedly investigated in experimental [3] and clinical studies [4], with promising results. Experimental studies demonstrate 80–100% survival using CPB after 5–10 min of cardiac arrest (CA) [3]. Clinically, up to 40% survival has been reported after in-hospital CA, when CPB was established after unsuccessful cardiopulmonary resuscitation (CPR) [4,5]. Compared with CPR-advanced life support (CPR-ALS), CPB facilitates stable restoration of cerebral and cardiac blood flow, and may allow recovery of the stunned myocardium even after prolonged ischemic periods [3–5]. We have recently reported a 64% 24-h survival using normothermic CPB after 15 min of prolonged CA that was unresponsive to CPR-ALS in pigs [6]. However, despite improved survival with CPB, hypoxic brain injury was present in all animals, and was coupled to severe neurological impairment.
CPB provides the unique opportunity to apply known principles of controlled reperfusion to improve recovery of the heart and brain by limiting the post-resuscitation reperfusion injury after prolonged CA [7,8]. Calcium is one of the primary mediators of the reperfusion injury of organs, and plays a central role in post-ischemic myocardial dysfunction and neuronal cell death [9]. Counteracting intracellular acidosis after ischemia, the sodium–hydrogen ion antiporter (NHE) activation leads to sodium overload of the cell that subsequently promotes intracellular calcium accumulation. In previous experimental studies, we have demonstrated markedly improved myocardial and neurological recovery by controlling post-ischemic intracellular calcium accumulation through NHE inhibition with HOE-642 after prolonged deep hypothermic circulatory arrest (DHCA) [10,11]. The aims of this study were to determine whether use of CPB as a resuscitation tool and NHE inhibition with HOE-642 will limit whole-body-reperfusion injury and improve myocardial and neurological recovery in an experimental model of 15 min no-flow CA.
2. Material and methods
The study protocol was approved by the Institutional Animal Research Committee of the University of California at Los Angeles, and animals received humane care in compliance with the 1996 NRC Guide for the Care and Use of Laboratory Animals. The experimental protocol used in this study has been described in detail previously [6], and is, therefore, briefly summarized.
After anesthesia and endotracheal intubation, 30 Yorkshire–Duroc pigs (31–39 kg) were mechanically ventilated with respirator settings adjusted to keep oxygen tension, carbon dioxide tension, and pH values within the normal range. The left internal thoracic artery was cannulated for arterial pressure measurements under sterile surgical techniques. In addition, a balloon-tipped pulmonary artery catheter (Baxter Healthcare Corp, Irvine, CA, USA) was inserted via the right subclavian vein to measure cardiac output (thermodilution technique). After baseline hemodynamic measurements and systemic heparinization (300 IE kg−1) ventricular fibrillation (VF) was induced by delivering a 60-Hz (4.5 A) alternating current (AC-Fibrillator, Medtronic Inc., Minneapolis, MN, USA) to the right ventricular endocardium using a catheter (Bard, Electrophysiology, Lowell, MA, USA) that was inserted through the right subclavian vein.
The resuscitation protocol was initiated at 15 min of CA, and was in accordance with the CPR-ALS algorithm of the American Heart Association. Closed-chest CPR was provided for 10 min, with manual chest compressions at a rate of 80–100 min−1, and manual ventilation with room air at a rate of 12–15 breaths per minute. Defibrillation of VF was attempted after five CPR cycles with 200 J (Lifepak 9P, Physio-Control Corp., Redmond, WA, USA), and repeated after every five CPR cycles with 300–360 J until restoration of spontaneous circulation. Intravenous epinephrine was administered every 2–3 min as needed (0.02–0.04 mg kg−1 min−1), and lidocaine (1 mg kg−1) was given and repeated until maximal dosage was reached (3 mg kg−1). Manual CPR was performed by two investigators (O.L. and N.H.) to provide maximal consistency, and coronary perfusion pressures (CPPs) of >15 mmHg were targeted, as indicated by the difference of diastolic arterial pressure and central venous pressure.
In each study, the 10-min CPR-ALS interval was used to connect the arterial (femoral artery; 12F, Medtronic, Inc., Minneapolis, MN, USA) and venous cannulas (jugular vein; 19–21F, Medtronic, Inc., Minneapolis, MN, USA) to the CPB circuit, consisting of a membrane oxygenator (Affinity NT 541, Medtronic, Inc., Minneapolis, MN, USA), an arterial roller pump (Sarns, Ann Arbor, MI, USA), and a venous centrifugal pump (Biopump BPX-80, Medtronic Inc., Minneapolis, MN, USA) for active venous suction with a standardized pump prime (800 ml Plasma-Lyte solution; 700 ml of stored porcine donor blood; and 100 IU kg−1 heparin, normocalcemic).
Normothermic, full-flow CPB (100–150 ml kg−1 min−1) was initiated, if the 10-min CPR-ALS interval did not successfully restore stable spontaneous circulatory function (ROSC), defined as either persistence of ventricular fibrillation or return of supraventricular rhythm, but inability to maintain a mean arterial pressure >50 mmHg for 1 min [6].
2.1. Experimental groups
2.1.1. Preliminary studies
In an initial series of three animals, a 5 mg kg−1 bolus of the NHE inhibitor HOE-642 (Aventis Pharma, Frankfurt, Germany) used previously in our DHCA studies [12,13] was administered intravenously after 15 min of no-flow CA with 10 min of CPR-ALS (one animal with ROSC; two without ROSC and subsequent CPB). Severe hypotension due to vasodilation with acidosis led to the death of the animal with ROSC after CPR-ALS. In the two CPB subjects, persistent vasodilation occurred despite CPB, and each subject eventually succumbed from hypotension and failure to wean from CPB after 175 ± 43 min.
This adverse hemodynamic event led to subsequent adjustment of the HOE-642 dose to 1 mg kg−1 to attenuate the aforementioned adverse drug effects. The new dose was then employed in the 27 remaining pigs, whereby HOE-642 was given as a bolus (1 mg kg−1) immediately 10 min after CPR (or put in the bypass prime in subjects placed on CPB), and followed by a continuous infusion of 1 mg kg−1 over 30 min. Thus, data of all three animals receiving the 5 mg kg−1 HOE-642 dose were excluded from this study.
2.1.2. CPR with ROSC
If ROSC occurred during the 10 min of CPR-ALS, animals were randomly assigned to receive either HOE-642 or no treatment (no Rx).
2.1.3. CPR without ROSC
In animals without ROSC after 10 min of CPR-ALS, CPB was initiated and either no treatment (CPB and no Rx) or HOE-642 was administered.
CPB management and post-resuscitation care of subjects was facilitated, as previously reported [6]. After successful cardioversion of VF on CPB following repeated countershocks, discontinuation of CPB was initiated when spontaneous hemodynamic performance maintained mean arterial pressure >50 mmHg with or without dopamine (5–10 μg kg−1 min−1) or norepinephrine (0.01–0.15 μg kg−1 min−1) support. Systemic heparinization was reversed with protamine, wounds were closed, and animals were allowed to regain consciousness and extubated, when adequate spontaneous breathing was present. They were monitored for 24 h after CPB. At the end of the protocol, the animals were euthanized using 30 mg kg−1 pentobarbital and potassium chloride.
2.2. Hemodynamic and biochemical measurements
Hemodynamic measurements were made before induction of VF (baseline), and after discontinuation of CPB. Cardiac output was determined by triplicate injections of 4 °C cold saline solution, and cardiac index (CI) and systemic and pulmonary vascular resistance indices (SVRI and PVRI) calculated. In addition, mean arterial pressure and arterial pump flow were recorded during CPB for estimation of systemic vascular resistance. The number of shocks during CPR, minutes of CPR until cardioversion, and number of subjects requiring dopamine and/or norepinephrine support and mortality are summarized.
Arterial blood was sampled at baseline, after CPB, and at 4 and 24 h. Blood samples were immediately centrifuged (3000 rpm) for 10 min and stored at −20 °C until biochemical analysis [6]. As a marker of oxidant-mediated lipid peroxidation, conjugated diene (CD) levels were determined spectrophotometrically. Cellular injury was determined by creatinine kinase (CK-MB), serum aspartate aminotransferase (AST), and alanine aminotransferase (ALT) activity using the ultraviolet (UV)-spectrophotometric method (Sigma Chemical Co., St. Louis, MO, USA). Enzyme immunometric assays were performed to measure endothelin-1 (ACE EIA kit, Cayman Chemical Company, Ann Arbor, MI, USA) for vascular- and neuron-specific enolase (NSE; SynX Pharma Inc., Toronto, ON, Canada) for brain injury.
2.3. Neurologic deficit score
Neurologic assessment was performed at 4 and 24 h after CPB using a species-specific neurological deficit score [6] that assesses five general components including cranial nerves’ function, respiration, motor and sensory function, consciousness, and behavior. Each category has a maximal score of 100: 0 is normal and 500 indicates brain death. In addition, seizure activity was recorded. Two laboratory team members graded neurological function, with mean values recorded for different evaluations.
2.4. Statistical analysis
Data were expressed as mean ± standard deviation (SD) or standard error of mean (SEM) as indicated. Two-way analysis of variance (ANOVA) for repeated measurements with the Tukey post hoc procedure was used to analyze intra- and inter-group differences. Perioperative characteristics of groups were analyzed by unpaired t-test for continuous data and χ2 or Fisher’s exact test for categorical data. Probability values ≪0.05 were considered statistically significant.
3. Results
Perioperative variables of treatment groups are summarized in Table 1 and Fig. 1 . Low-amplitude VF was present in all animals after the 15-min period of CA. Restoration of spontaneous circulation with CPR-ALS was achieved in 11 of 27 animals (41%), requiring 4.2 ± 1.1 countershocks after an average CPR duration of 6.2 ± 1.9 min. CPB was instituted in 16 animals (59%) without ROSC, following 10 min of CPR-ALS. Extracorporeal support allowed stable systemic perfusion, and 6.0 ± 1.5 shocks were needed to achieve successful conversion in all pigs with CPB.
Table 1.

Perioperative variables.
Fig. 1.
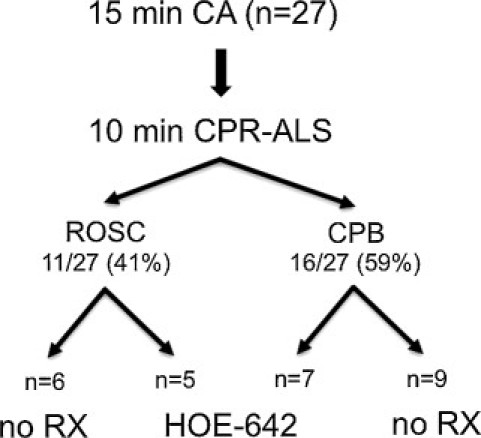
Resuscitation outcome of animals (n = 30) subjected to 15 min of cardiac arrest (CA) followed by 10 min of cardiopulmonary resuscitation advanced life support (CPR-ALS). Note: CPR-ALS restored stable circulation (ROSC) in 40% (12/30) of animals, whereas in 60% (18/30) without ROSC cardiopulmonary bypass (CPB) was established. No RX: subjects receiving no treatment; HOE-642: subjects receiving intravenous HOE-642 treatment.
3.1. Hemodynamic measurements
Hemodynamic data of groups were similar at baseline and shown in Table 2 . Post-resuscitation operative heart rate (p ≪ 0.001), mean pulmonary artery pressure (p ≪ 0.001), and PVRI (p ≪ 0.01) were significantly increased compared with baseline. Experimental groups did not differ with respect to post-resuscitation dopamine support, but use of HOE-642 resulted in prolonged duration of CPB (p = 0.046) and increased need for vasopressor therapy (p = 0.019) compared with untreated animals (Table 1). After resuscitation without CPB, HOE-642 treatment resulted in higher CI (p = 0.061) and lower SVRI (p = 0.068) compared with baseline, without reaching statistical difference compared with untreated animals. Of note, severe vasodilation existed during CPB following HOE-642 treatment, as higher pump flows were required to maintain adequate perfusion pressure compared with the untreated group (Figs. 2–4 ) at 30 min (CPB–HOE-642, CI: 3.5 ± 0.5 l min−1 m−2 vs CPB–no Rx: 2.8 ± 0.4 l min−1 m−2; p = 0.016) and 60 min (HOE-642, CI: 3.4 ± 0.6 l min−1 m−2 vs no Rx: 2.5 ± 0.7 l min−1 m−2; p = 0.001). Systemic vasodilation persisted after CPB following HOE-642 treatment, as treated animals maintained a higher CI (p = 0.014) with decreased SVRI (p = 0.003) compared with baseline values despite higher vasopressor support (Table 1), but missed statistical significance compared with the untreated group (CI: p = 0.094; SVRI: p = 0.172).
Table 2.

Hemodynamic data.
Fig. 2.
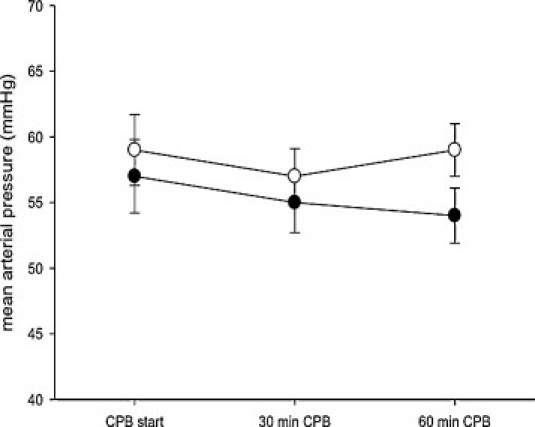
Mean arterial pressure of animals on cardiopulmonary bypass (CPB) with (HOE-642) or without HOE-642 treatment (no Rx). Data are depicted as mean ± standard error of mean (SE).
Fig. 3.
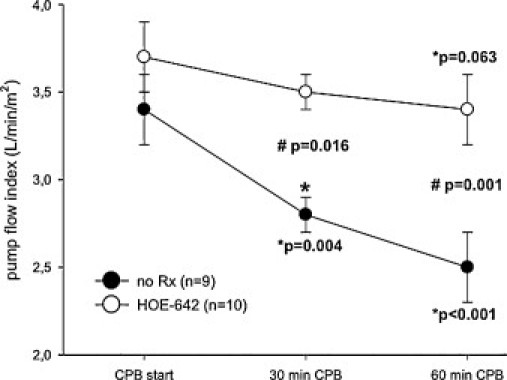
Pump flow index of animals on cardiopulmonary bypass (CPB) with (HOE-642) or without HOE-642 treatment (no Rx). Note: Higher pump flow was required to maintain adequate perfusion pressure compared to the untreated group at 30 and 60 min. Data are depicted as mean ± standard error of mean (SE); (*) compared to baseline values; (#) compared to untreated group (ANOVA for repeated measurements with the Tukey post-hoc test).
Fig. 4.
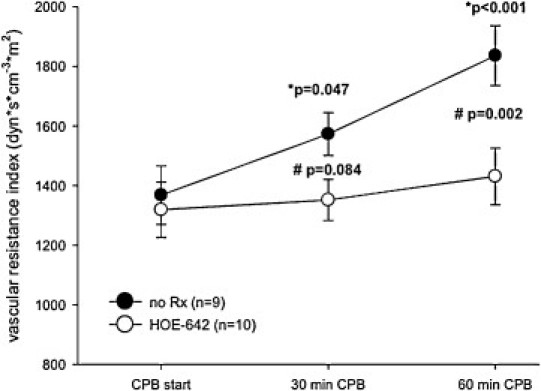
Systemic vascular resistance of animals on cardiopulmonary bypass (CPB) with (HOE-642) or without HOE-642 treatment (no Rx). Note: Systemic vascular resistance was decreased in animals treated with HOE-642. Data are depicted as mean ± standard error of mean (SE); (*) compared to baseline values; (#) compared to untreated group (ANOVA for repeated measurements with the Tukey post-hoc test).
3.2. Mortality
Overall 24-h survival was 89% (24/27), with 91% survival (10/11) for animals with ROSC during CPR and 88% survival (14/16) in animals without ROSC and CPB. Mortality did not differ between treatment groups, with no mortality occurring in both HOE-642 groups (with or without CPB) compared with 16% (1/6) and 22% (2/9) mortality in pigs with no HOE-642 treatment and ROSC during CPR (no Rx) or without ROSC and CPB (CPB and no Rx group), respectively (Table 1).
Excluded in this result overall analysis are three pigs (one with ROSC, two with CPB) receiving the high HOE-642 doses (5 mg kg−1), which succumbed to severe hypotension due to vasodilation. These observations were vital to establish the cariporoide dose that was tested in the other 27 pigs.
3.3. Biochemical measurements
Table 3 summarizes biochemical markers, as CK-MB, AST, and ALT levels were equally increased after surgery and until 24 h compared with baseline (p ≪ 0.001) in all treatment groups, indicating severe tissue damage. Similarly, oxidant injury measured by CD, and vascular injury (endothelin, ET-1) were increased in animals after resuscitation (p ≪ 0.001) without differences between treatment groups. NSE levels were significantly elevated in all groups after resuscitation, with or without CPB. However, NSE values in HOE-treated animals with CPB tended to decrease at 24 h compared with untreated subjects (p = 0.16), but without reaching statistical difference.
Table 3.

Biochemical data.
3.4. Neurologic function
Overall neurological function was impaired at 4 and 24 h, with an average score of 227 ± 43 at 24 h and 178 ± 41 at 4 h (p ≪ 0.001), respectively (Table 4 ). Although all animals regained consciousness, there was generalized clonic seizure activity in 67% (16 of 24 animals) of survivors, without inter-group differences. HOE-642 treatment, with or without CPB, caused a mild improvement of neurological recovery after 24 h when compared with 4 h (HOE-642: p = 0.003; CPB and HOE-642: p ≪ 0.001), and resulted in better consciousness (cloudy level), positioning, reflexes, and muscle tonus compared with untreated animals that, however, failed to reach statistical difference. However, no animal was able to stand, walk, eat, or drink by itself at 24 h after surgery, despite HOE-642 treatment.
Table 4.

Neurologic deficit score and seizures.
4. Discussion
This study affirmed the capacity of normothermic CPB to allow near-complete hemodynamic recovery after 15 min of no-flow CA, as the 37% (10/27) survival after CPR-ALS alone was extended to 89% (24/27) recovery in animals that would have succumbed following use of only CPR-ALS. Temporary CPB successfully salvaged the heart, but severe neurological damage occurred in survivors that did not receive other therapy. HOE-642 therapy augmented cardiac output and reduced the adverse neurological score, but this improvement was quite limited in comparison to the complete recovery observed in our previous study [10,11]. A potential culprit is the adverse effect of this agent on vascular tone, as our preliminary studies showed that profound hemodynamic vasodilation following the conventional 5 mg kg−1 HOE-642 dose allowed complete brain recovery after prolonged ischemia with DHCA [10,11]. These observations led to the dose reduction to only 1 mg kg−1 HOE-642 bolus followed by a continuous intravenous infusion of 1 mg kg−1 over 30 min that was tested in this prolonged CA sudden cardiac death model.
HOE-642-treated animals improved post-arrest CI, as there was no mortality in these animals, but it decreased vascular resistance, requiring extrinsic vasopressor support to maintain adequate perfusion pressure. Administration of the HOE-642 dose of 5 mg kg−1 used in prior studies [10,11] was not possible, as this dosage resulted in 100% mortality in a preliminary study of three pigs due to fatal hypotension. Consequently, marked reduction in the dosage was thereby required to complete this study. No deaths occurred after adjusting the HOE-642 dose, but vasodilation persisted, especially in animals undergoing CPB, where higher pump flows and prolonged CPB perfusion were needed before discontinuation of bypass was possible.
These findings demonstrate that NHE inhibition markedly limits vascular tone after prolonged CA, and earmarks the need for subsequent dose–response studies because the severe systemic no-flow state in our CPR model makes NHE-inhibitor drugs act differently than observed in our previous DHCA studies [10,11]. This supposition was emphasized from this study, as the 5 mg kg−1 HOE-642 dose produced disastrous hypotension after 15 min of normothermic sudden death. By contrast, this dose had no adverse effects on systemic vascular resistance after 90 min of DHCA [10,11], with near-complete neurological recovery as well as significant improvement of other ischemic injury markers. The current study showed that HOE-642 at 1 mg kg−1 restricted hypotension but resulted in negligible neurological improvement at 24-h compared with no-treatment groups.
The role of NHE inhibition is clear from prior information, as intracellular calcium accumulation after ischemia and reperfusion plays major role in myocardial stunning and neurological injury [7–9]. The myocardial benefits of NHE inhibition are evident from studies showing improved effectiveness of closed chest CPR, increased resuscitability, and prevention of post-resuscitation myocardial dysfunction in pigs subjected to 8 min of untreated VF followed by CPR [12]. NHE inhibition allows better neuronal energy preservation by reducing cerebral edema and infarction and improving neurological recovery, even when given upon reperfusion following experimental brain reperfusion injury [12,13]. Heart surgery patients pre-treated with HOE-642 before CPB showed lower serum levels of S-100B and NSE, suggesting better preservation of the blood–brain barrier [14]. Of note, the Sodium-Proton Exchange Inhibition to Prevent Coronary Events in Acute Cardiac Conditions (EXPEDITION) trial demonstrated a significant reduction of myocardial necrosis in patients undergoing CABG, who were treated with the NHE-1 isoform inhibitor, cariporide [15]. However, the same study revealed a marked increase in the overall incidence of cerebrovascular events from 3% in placebo-treated to 5.2% in cariporide-treated patients that was associated with a higher mortality. Although the underlying reasons for these unexpected findings of the EXPEDITION trial remain unknown, a higher susceptibility for ischemic brain injury, resulting from the unique molecular structure of the drug and the abrupt postoperative withdrawal of cariporide, was postulated as a possible cause by the investigators.
Our prior reports support the cardiac and neurological benefits of NHE inhibition [10,11], but these unequivocal salutary effects could not be reproduced in the current study, as HOE-642 treatment had no effect on NSE, and was associated only with mild neurological improvement. There are three possible explanations for why HOE-642 did not exhibit a more profound effect in this study. The first arises from the increased norepinephrine use in the treated animals, as α-adrenergic agonists increase NHE sensitivity and thereby may attenuate neuroprotective effects of HOE-642 [16]. Furthermore, the lower vascular resistance, along with the increase dose of norepinephrine, may have led to inadequate perfusion of the deep brain; hence, the reduced effect of HOE-642 may be secondary to inadequate blood flow and distribution. Second, the reduced HOE-642 dose of 1 mg kg−1 might have been insufficient to inhibit the reperfusion-related whole-body injury in this model of prolonged CA. However, we consider the most likely explanation is related to the timing of HOE-642 administration. When HOE-642 was effective in these previous studies, it was given either as a pre-treatment [11], or before initiation of reperfusion [10]. Conversely, in the present study, HOE-642 was given after 10 min of CPR, and therefore, the ischemic tissue had already been exposed to uncontrolled reperfusion with normal blood. Therefore, significant tissue reperfusion injury may have already occurred by the time HOE-642 was administered. Although we could have tested giving HOE-642 prior to starting CPR, this was not done in the present study, as this does not simulate the clinical situation of starting CPR immediately on finding the patient in sudden death. However, it raises an interesting dilemma, as if we found that giving HOE-642 before CPR improved results, this would imply that CPR should not be the initial treatment in patients with prolonged sudden death. Subsequent studies by Trummer et al. [17] tested this option of avoiding CPR in unwitnessed arrest, by employing controlled reperfusion on CPB without CPR following 15 min of sudden death, and document complete systemic and neurologic recovery. This observation implies that reconsideration of avoiding CPR as the initial treatment in patients with prolonged (unwitnessed) sudden death merits further attention.
Evidence of global reperfusion injury following prolonged CA was garnered from the marked elevation of biochemical markers of organ injury and persistent neurological damage at 24 h. We recognize that NHE inhibition was used to counteract only one component of reperfusion damage (i.e., calcium overload), and understand from prior experiments that many factors must be modified during controlled reperfusion [7,8] to offset the damage, as shown both in the heart as well as in our studies following 90 min of DHCA, where the entire pump prime reperfusate as part of a multimodal concept (leukocyte depletion, hypocalcemia, hypermagnesemia, pH-stat strategy, normoxia, mannitol, and NHE inhibition) was altered [10,18]. Consequently, the potential for similar results after prolonged CA will be tested subsequently by altering many aspects of the prime of the CPB circuit, including CPB-assisted cooling to mild hypothermia, which has been shown clinically to improve neurological recovery in post-arrest survivors. These future experiments will determine whether a whole-body (controlled) reperfusate can be useful when an extracorporeal circuit is needed to support resuscitation in the event of sudden death.
The current study has several limitations that include absence of confirming the effects of NHE inhibition by measuring calcium, sodium, and brain edema or to evaluate neurologic injury by determining apoptosis and/or necrosis. Neurological deficit score was determined to evaluate the functional hallmark of brain recovery, but post-arrest serial assessments of neurological deficit score beyond the 24-h period (3–7 days post arrest) are needed to determine the final extent of brain injury and neurological recovery. Although animals were randomly allocated to treatment groups before each study, an inherent risk of bias is still possible, as investigators were not blinded to drug treatment and a vehicle or placebo administration was not performed in control animals. We also acknowledge that inefficiency of NHE inhibition after prolonged CA in our study may have resulted from inadequate timing of HOE-642 administration and dosage, with the latter being reduced on an empirical basis, and due to significant hemodynamic adverse effects of the drug.
In conclusion, temporary CPB is a useful resuscitative tool after prolonged CA of 15 min that is unresponsive to CPR-ALS, and facilitates myocardial recovery and high survival rates, but fails to improve neurological function. NHE inhibition during reperfusion may improve neurological recovery, but adversely variance of vascular resistance will limit its’ potential usefulness. Establishment of a dose–response curve may potentially define the safe HOE-642 dosage, and thus might enhance its usefulness following prolonged CA periods.
This work was supported by a grant from the National Institutes of Health (R01-HL-71729-04).
References
- [1].Madl C., Holzer M. Brain function after resuscitation from cardiac arrest. Curr Opin Crit Care. 2004;10:213–217. doi: 10.1097/01.ccx.0000127542.32890.fa. [DOI] [PubMed] [Google Scholar]
- [2].Athanasuleas C.L., Buckberg G.D., Allen B.S., Beyersdorf F., Kirsh M.M. Sudden cardiac death: directing the scope of resuscitation towards the heart and brain. Resuscitation. 2006;70:44–51. doi: 10.1016/j.resuscitation.2005.11.017. [DOI] [PubMed] [Google Scholar]
- [3].Safar P., Abramson N.S., Angelos M., Cantadore R., Leonov Y., Levine R., Pretto E., Reich H., Sterz F., Stezoski S.W. Emergency cardiopulmonary bypass for resuscitation from prolonged cardiac arrest. Am J Emerg Med. 1990;8:55–67. doi: 10.1016/0735-6757(90)90298-e. [DOI] [PubMed] [Google Scholar]
- [4].Nichol G., Karmy-Jones R., Salerno C., Cantore L., Becker L. Systematic review of percutaneous cardiopulmonary bypass for cardiac arrest or cardiogenic shock states. Resuscitation. 2006;70:381–394. doi: 10.1016/j.resuscitation.2006.01.018. [DOI] [PubMed] [Google Scholar]
- [5].Sung K., Lee Y.T., Park P.W., Park K.H., Jun T.G., Yang J.H., Ha J.K. Improved survival after cardiac arrest using emergent autopriming percutaneous cardiopulmonary support. Ann Thorac Surg. 2006;82:651–656. doi: 10.1016/j.athoracsur.2006.03.017. [DOI] [PubMed] [Google Scholar]
- [6].Liakopoulos O.J., Allen B.S., Buckberg G.D., Hristov N., Tan Z., Villablanca J.P., Trummer G. Resuscitation after prolonged cardiac arrest: role of cardiopulmonary bypass and systemic hyperkalemia. Ann Thorac Surg. 2010;89:1972–1979. doi: 10.1016/j.athoracsur.2010.02.052. [DOI] [PubMed] [Google Scholar]
- [7].Buckberg G.D. Stroke and extra-cardiac perfusion: new vantage points in brain protection. Eur J Cardiothorac Surg. 2004;26:S62–S65. [PubMed] [Google Scholar]
- [8].Beyersdorf F., Kirsh M., Buckberg G.D., Allen B.S. Warm glutamate/aspartate-enriched blood cardioplegic solution for perioperative sudden death. J Thorac Cardiovasc Surg. 1992;104:1141–1147. [PubMed] [Google Scholar]
- [9].Kristian T., Siesjo B.K. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- [10].Allen B.S., Veluz J.S., Buckberg G.D., Aeberhard E., Ignarro L.J. Deep hypothermic circulatory arrest and global reperfusion injury: avoidance by making a pump prime reperfusate—a new concept. J Thorac Cardiovasc Surg. 2003;125:625–632. doi: 10.1067/mtc.2003.96. [DOI] [PubMed] [Google Scholar]
- [11].Castella M., Buckberg G.D., Tan Z. Neurologic preservation by Na+-H+ exchange inhibition prior to 90 minutes of hypothermic circulatory arrest. Ann Thorac Surg. 2005;79:646–654. doi: 10.1016/j.athoracsur.2004.07.007. [DOI] [PubMed] [Google Scholar]
- [12].Ayoub I.M., Kolarova J., Yi Z., Trevedi A., Deshmukh H., Lubell D.L., Franz M.R., Maldonado F.A., Gazmuri R.J. Sodium-hydrogen exchange inhibition during ventricular fibrillation: beneficial effects on ischemic contracture, action potential duration, reperfusion arrhythmias, myocardial function, and resuscitability. Circulation. 2003;107:1804–1809. doi: 10.1161/01.CIR.0000058704.45646.0D. [DOI] [PubMed] [Google Scholar]
- [13].Kuribayashi Y., Itoh N., Horikawa N., Ohashi N. SM-20220, a potent Na+/H+ exchange inhibitor, improves consciousness recovery and neurological outcome following transient cerebral ischaemia in gerbils. J Pharm Pharmacol. 2000;52:441–444. doi: 10.1211/0022357001774057. [DOI] [PubMed] [Google Scholar]
- [14].Scholz M., Wimmer-Greinecker G., Kleine P., Dzemali O., Martens S., Moritz A., Matheis G. Cariporide (HOE642) limits S-100B release during cardiac surgery. J Cardiovasc Pharmacol. 2003;41:468–473. doi: 10.1097/00005344-200303000-00016. [DOI] [PubMed] [Google Scholar]
- [15].Mentzer R.M., Jr., Bartels C., Bolli R., Boyce S., Buckberg G.D., Chaitman B., Haverich A., Knight J., Menasché P., Myers M.L., Nicolau J., Simoons M., Thulin L., Weisel R.D., EXPEDITION Study Investigators Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–1270. doi: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- [16].Yokoyama H., Yasutake M., Avkiran M. Alpha1-adrenergic stimulation of sarcolemmal Na+-H+ exchanger activity in rat ventricular myocytes: evidence for selective mediation by the alpha1A-adrenoceptor subtype. Circ Res. 1998;82:1078–1085. doi: 10.1161/01.res.82.10.1078. [DOI] [PubMed] [Google Scholar]
- [17].Trummer G., Foerster K., Buckberg G.D., Benk C., Heilmann C., Mader I., Feuerhake F., Liakopoulos O., Brehm K., Beyersdorf F. Successful resuscitation after prolonged periods of cardiac arrest: a new field in cardiac surgery. J Thorac Cardiovasc Surg. 2010;139:1325–1332. doi: 10.1016/j.jtcvs.2009.08.046. [DOI] [PubMed] [Google Scholar]
- [18].Buckberg G.D. Strategies and logic of cardioplegic delivery to prevent, avoid, and reverse ischemic and reperfusion damage. J Thorac Cardiovasc Surg. 1987;93:127–139. [PubMed] [Google Scholar]