

Abstract
The molecular mechanisms that contribute to the initiation and progression of head and neck squamous cell carcinoma (HNSCC) have not been completely delineated. Our observations indicate that defects in the TGF-β and PI3K/Akt signaling pathways are common in human HNSCCs. Conditional activation of the PI3K/Akt pathway due to Pten deletion in the mouse head and neck epithelia gives rise to hyperproliferation, but only a few lesions progress to HNSCC. However, Pten-deficient mice developed full-penetrance HNSCC in combination with type I TGF-β receptor (Tgfbr1) deletion. Molecular analysis revealed enhanced cell proliferation, decreased apoptosis, and increased expression of CCND1 in the basal layer of the head and neck epithelia, as well as in the tumors of Tgfbr1/Pten double conditional knockout (2cKO) mice. Furthermore, neoplastic transformation involves senescence evasion and is associated with an increased number of putative cancer stem cells (CSCs). In addition, the NF-κB pathway activation, myeloid derived suppressor cell (MDSC) infiltration, angiogenesis, and immune suppression in the tumor microenvironment, all of which are characteristic of human HNSCCs, contribute significantly to head and neck carcinogenesis in 2cKO mice. These tumors display pathology and multiple molecular alterations resembling human HNSCCs. This suggests that the Tgfbr1/Pten 2cKO mouse model is suitable for preclinical intervention, and that it has significant implications in the development of diagnostic cancer biomarkers and effective strategies for prevention and treatment of HNSCCs.
Keywords: TGF-β, PI3K/Akt, Head and Neck Squamous Cell Carcinoma (HNSCC), Conditional Knockout, Cancer Mouse Model
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most common types of human cancer (Jemal et al., 2010). The underlying cellular and molecular mechanisms that contribute to the initiation and progression of HNSCC have not been completely delineated (Mao et al., 2004). Tobacco, alcohol consumption, and human papillomavirus (HPV) are the major risk factors associated with the development of HNSCC (Licitra et al., 2006; Curado et al., 2009). These risk factors, together with genetic susceptibility, result in the accumulation of multiple genetic and epigenetic alterations in a multistep process of cancer development (Deshpande et al., 2008). In addition, the tumor microenvironment also contributes significantly to head and neck carcinogenesis (Yang et al., 2010). Recent advances in the understanding of the oncogenesis of HNSCC have revealed multiple deregulated signaling pathways. Transforming Growth Factor-β (TGF-β) and PTEN/PI3K/Akt/mTOR pathways are among the most frequently altered signaling routes. Both pathways play central roles in numerous cellular processes including metabolism, cell growth, apoptosis (programmed cell death), survival, and differentiation, which ultimately contribute to HNSCC progression (Molinolo et al., 2009).
TGF-β belongs to a superfamily of multi-functional cytokines that regulate cell proliferation, differentiation, migration, adhesion, and apoptosis, thereby influencing important physiological processes such as embryonic development, immune function, and carcinogenesis (Derynck et al., 2003; Massagué, 2008). The three mammalian TGF-β isoforms, TGF-β1, -β2, and -β3, exert their functions through a cell surface receptor complex composed of type I (TGFBR1) and type II (TGFBR2) serine/threonine kinase receptors. Upon ligand binding, TGFBR2 recruits and phosphorylates TGFBR1, which in turn phosphorylates Smad2 or Smad3. Phosphorylated Smad2 or Smad3 binds to Smad4, and then these complexes translocate from the cytoplasm into the nucleus. This results in the transcriptional activation of TGF-β-responsive genes that mediate the effects of TGF-β at the cellular level. In addition to SMAD-mediated signaling, receptor activation also induces other downstream targets, including Ras, RhoA, TAK1, MEKK1, PI3K, and PP2A, to produce the full spectrum of TGF-β responses (Moustakas et al., 2009; Zhang et al., 2009).
The effects of TGF-β signaling on carcinogenesis largely depend on the tissue of origin and the tumor type. In most types of human cancer, TGF-β plays a paradoxical role in cancer development by acting as a tumor suppressor during the early stages (Engle et al., 1999), and as a tumor promoter during the later stages (Piek et al., 2001; Tang et al., 2003). Several reports have noted that mutations and polymorphisms of TGFBR1 and Smads are associated with HNSCC (Chen et al., 2001; Xie et al., 2003; Pasche et al., 2005), suggesting that TGF-β functions as a potent tumor suppressor. However, it is not clear whether alterations in TGF-β signaling act alone or in concert with alterations in other pathways to promote a pro-oncogenic phenotype in advanced late-stage HNSCC.
The PI3K/Akt pathway is important for suppressing apoptosis and promoting cell growth and proliferation. In HNSCC, hyperactivation of PI3K can be induced by mutations or by enhanced activity of its upstream activators, including the activation of Ras oncoproteins or inactivation of PTEN (phosphatase and tensin homolog deleted on chromosome 10) (Molinolo et al., 2009). PTEN is a potent tumor suppressor gene and a negative regulator of the PI3K/Akt pathway. While PTEN mutations were identified in 0-16% of HNSCCs, loss of PTEN expression was observed in 29% of tongue cancers, and loss of heterozygosity (LOH) of the PTEN locus was identified in 40% of HNSCCs (Henderson et al., 1998; Shao et al., 1998; Lee et al., 2001). Additionally, 47% of HNSCC cases showed at least one molecular alteration in the PI3K/Akt pathway, including PI3KCA and AKT2 amplification, p110α overexpression, and PTEN protein downregulation. This suggests the critical role of the PTEN/PI3K/Akt signaling pathways in the carcinogenesis of HNSCC (Pedrero et al., 2005). The studies from our previous mouse model indicate that there may be negative crosstalk between the TGF-β tumor suppressor and the PI3K/Akt pathways (Bian et al., 2009).
To further characterize the interactions between TGF-β signaling and the PI3K/Akt pathway during the development of HNSCC, we conditionally deleted Tgfbr1 and Pten to enhance PI3K/Akt pathway activation in mouse head and neck epithelia using the Cre-LoxP approach. Here we show that loss of Tgfbr1, in combination with Pten deletion, results in cellular senescence evasion, cancer-related inflammation, and expansion of the cancer stem cells in the basilar epithelial layer. This also causes full-penetrance HNSCCs that resemble human HNSCCs in both their pathological and molecular characteristics.
Results
Loss of TGFBR1 and PTEN is a common event in human HNSCCs
To determine whether deletion in the TGF-β and PTEN/PI3K/Akt signaling pathways often occur together in a subset of human HNSCCs, we examined TGFBR1 mRNA expression in ten human HNSCC cell lines. Human oral keratinocytes (HOK) were used as the normal control. The qRT-PCR results revealed that the mRNA expression levels of TGFBR1 were reduced in all ten HNSCC cell lines compared to HOK cells, and in 6/10 (60%) of the cell lines the reduction was more than 50% (Figure 1a). Increased phosphorylation of Akt (p-AktSer473) was found in 7/10 (70%) of the same HNSCC cell lines by Western blot (Figure 1b). Using immunostaining, we performed tissue array analysis on 20 human HNSCC samples and 6 normal controls. The TGFBR1 protein level was found to be undetectable or decreased in 10/20 (50%) of the HNSCC samples, while the PTEN protein level was found to be undetectable or decreased in 16/20 (80%) of the HNSCC samples, as compared to normal controls. A similar decrease was also observed in phosphorylated Smad2, an activated mediator of TGF-β signaling (9/20, 45%), and there was an increase in p-Akt, a downstream target inhibited by PTEN (12/20, 60%) (Figure 1c). In total, 8 out of 20 HNSCC samples (40%) exhibited concurrent TGFBR1 and PTEN loss. Together, the results from human HNSCC cell lines and tumors suggest that loss of TGFBR1 and PTEN is a common event in human HNSCCs.
Figure 1.
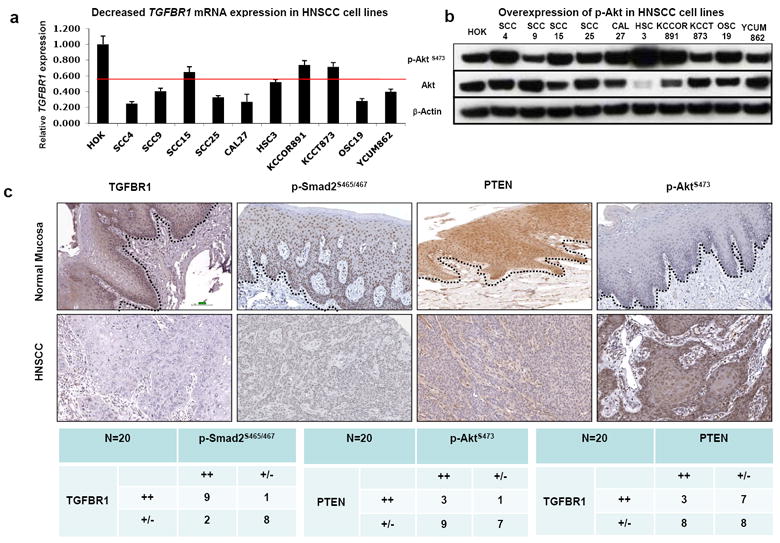
Loss of TGFBR1 and activation of PTEN/PI3K/Akt pathway in HNSCC samples. (a) TGFBR1 mRNA significantly reduced in HNSCC cell lines by qRT-PCR. Human oral keratinocytes (HOK) were used as normal control. (b) Western blot analysis demonstrates that p-Akt was overexpressed in most of the HNSCC cell lines. (c) Tissue array analysis revealed that TGFBR1, p-Smad2, and PTEN expression were reduced, while p-Akt was overexpressed in most human HNSCCs by IHC. The dotted lines delineate the adjacent epithelial compartment (magnification, 200X). Bar=100um.
Loss of Tgfbr1 in the head and neck epithelia, together with activation of the PTEN/PI3K/Akt pathway by Pten deletion, results in SCCs in mice with complete penetrance
The inducible Tgfbr1 or Pten conditional knockout mice were generated by crossing Tgfbr1 flox or Pten flox mice with K14-CreERtam mice, respectively. An inducible head- and neck-specific double knockout mouse model was generated by crossing K14-CreERtam;Tgfbr1flox/flox (Tgfbr1 cKO) mice with Ptenflox/flox mice. Untreated Tgfbr1 cKO mice or Pten cKO mice and Tamoxifen (Tam) treated Tgfbr1 cKO mice appeared normal. However, upon inducing Cre activity with Tam, all Pten cKO mice displayed a thickened skin and hyperkeratosis on the ears and muzzle area (n =24). After one year, 10% (3/31) of the Tgfbr1 cKO mice and 21% (5/24) of the Pten cKO mice developed SCCs. Thus, our results indicate that Pten deletion can give rise to hyperproliferation in the head and neck epithelia, but that loss of Tgfbr1 or Pten alone is not sufficient for promoting early or frequent tumor formation in the head and neck epithelia of these mice.
In contrast with individual Tgfbr1 or Pten cKO mice, Tgfbr1/Pten 2cKO mice began developing benign papillomas as early as 4 weeks after receiving Tam treatment. The tissue-specific deletion of Pten, together with Tgfbr1 in mice head and neck epithelia, causes multiple hyperproliferative and/or tumor lesions in the ears, vibrissae, skin, and paw (Supplementary Figure S1a and b). A histological comparison with the normal squamous epithelium revealed that the skin in the Tgfbr1/Pten 2cKO mouse showed hyperplastic and pseudopapillary epithelium. A thickening of the epithelium in the mouth was also observed. Additionally, the tongue was covered with hyperplastic mucosa (Supplementary Figure S1c and d). By 10 weeks, all 42 (100%) of the Tgfbr1/Pten 2cKO mice developed spontaneous tumors. Only 2 out of 38 (5.3%) Tgfbr1 and Pten floxed homozygous (Tgfbr1f/f;Ptenf/f) control littermates developed tumors. No tumors developed in the heterozygous mice (K14-CreERtam;Tgfbr1f/+;Ptenf/+, n = 31) during the same time period (Supplementary Figure S1e).
The Tgfbr1/Pten 2cKO mice and controls (Tgfbr1f/f;Ptenf/f) were dissected 10 weeks after Tam treatment. Genomic DNA was extracted from all major organs and tissues as well as from the tumors that developed in these mice. Cre-mediated recombination of the Tgfbr1flox/flox and Ptenflox/flox alleles was assessed using a PCR-based assay. Deletions of Tgfbr1 and Pten were detected in all of the tumors as well as in the buccal mucosa (BM), tongue (Tg), toes (To), and ear (Er), but not in the esophagus (Es), forestomach (FS), back skin (SK), or any other nonstratified epithelial organs of the Tgfbr1/Pten 2cKO mice (Supplementary Figure S2). No recombination was detected prior to Tam administration (data not shown). Tgfbr1 and Pten mRNA expression was examined by quantitative RT-PCR (qRT-PCR). The expression levels of Tgfbr1 mRNA in Tgfbr1f/f;Ptenf/f mice were normalized as 1.00 ± 0.34 in the buccal mucosa and 1.00 ± 0.25 in the tongue. The mRNA expression levels were significantly reduced to a mean of 0.38 ± 0.04 in the buccal mucosa (p<0.01) and 0.48 ± 0.04 in the tongue of Tgfbr1/Pten 2cKO mice (p<0.01) (Supplementary Figure S3a). Pten mRNA in Tgfbr1f/f;Ptenf/f mice was normalized as 1.00 ± 0.36 in the buccal mucosa and 1.00 ± 0.09 in the tongue. Pten mRNA expression levels were significantly reduced to a mean of 0.27 ± 0.09 in the buccal mucosa (p<0.01) and 0.62 ± 0.13 in the tongue of Tgfbr1/Pten 2cKO mice (p<0.01) (Supplementary Figure S3a). Using immunostaining, the Tgfbr1 and Pten protein levels were found to be significantly decreased in the tongues of Tgfbr1/Pten 2cKO mice, as compared to those of Tgfbr1f/f;Ptenf/f mice. p-Akt, a downstream target inhibited by Pten, was overexpressed (Supplementary Figure S3b), suggesting that, upon oral administration of Tam, inactivation of TGF-β signaling by Tgfbr1 deletion and activation of PI3k/Akt signaling by Pten loss were localized in the oral epithelia. Because these mice lick Tam applied by oral application, it is spread to the front limbs and to the head and neck epithelium, resulting in deletion of the pathways in these areas as well. The results were further confirmed by Western blot (Supplementary Figure S3c). The TGF-β signaling pathway is mediated by both Smad-dependent and Smad-independent signals. Using Western blot, we observed that even though Tgfbr1 is deleted in the tumors, the total levels of the TGF-β activated kinase-1 (TAK1) are elevated. In addition, increased activation of TAK1 occurs in tissues where TGF-β signaling is abrogated and so Smad-independent signaling still occurs, thus bypassing the loss of Tgfbr1. The elevated TAK1 in the tumors is probably a result of inflammatory signaling, as attenuated TGF-β signaling causes activation of the pro-inflammatory NF-κB pathway. No significant changes could be seen in the levels of Smad7, an inhibitory Smad that is part of a negative feedback loop during TGF-β signaling. Our results demonstrated that loss of Tgfbr1 does not result in downregulation of Smad-independent signaling (Supplementary Figure S4).
Tgfbr1/Pten 2cKO mice developed tumors in the oral cavity, bilateral ears, periorbital region, muzzle area, and the skin around the head and neck area (Figure 2a and b). SCCs were also observed in the perianal region of the Tgfbr1/Pten 2cKO mice. Histopathological analysis of the tumors revealed that they were composed of atypical cells that grow in solid sheets or occasionally differentiated keratin pearls (Figure 2c and d). These pathological changes are indistinguishable from human HNSCCs. Western blot analysis showed that these tumors exhibited multiple molecular alterations commonly found in human HNSCCs, including overexpression of EGFR and p-Akt, and activation of nuclear factor-κB (NF-κB) and the signal transducer and activator of transcription-3 (Stat3) pathways (Molinolo et al., 2009) (Figure 2e). Our results suggest that tumors which developed in Tgfbr1/Pten 2cKO mice are similar to human HNSCCs at both the pathological and molecular levels. Although three months after Tam treatment was an insufficient timeframe in which to detect metastases in any of the mice that were analyzed, the mice had to be euthanized due to the fast growth of the primary tumors.
Figure 2.
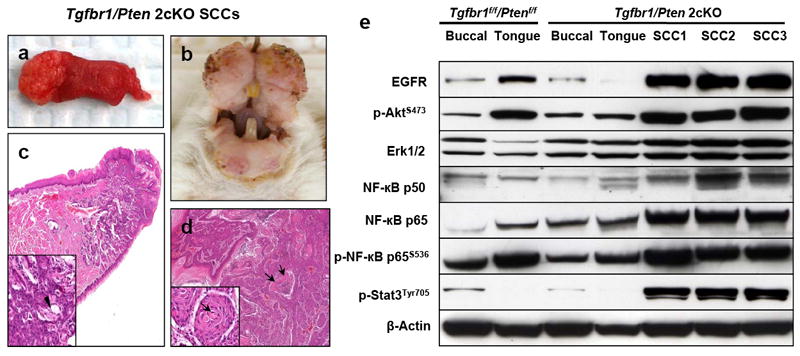
Tumors that developed in Tgfbr1/Pten 2cKO mice displayed pathology and multiple molecular alterations that resembled human HNSCCs. (a) Tumors developed in the tongue and oral cavity (b) of Tgfbr1/Pten 2cKO mice. (c) Pathological sections of tongue squamous cell carcinomas. The neoplasia affects the tip of the tongue and infiltrates deeply into the skeletal muscle layer. At higher magnification (inset), the tumor is composed of atypical cells that grow in solid sheets and occasionally differentiating keratin pearls (arrow head). (d) SCC growing into the oral cavity adopts a pseudopapillary structure and solid sheets, frequently showing central necrosis (arrows). At a higher magnification (inset), the tumor is composed of malignant cells that grow into solid sheets and differentiating keratin pearls (arrow). (Magnifications, 20X and 200X for main figure and inset, respectively). (e) Western blot analysis showed tumors exhibiting multiple molecular alterations commonly found in human HNSCCs, including overexpression of EGFR and p-Akt, as well as activation of NF-κB and the signal transducer and activator of transcription-3 (Stat3) pathways.
Absence of Tgfbr1 results in enhanced cell proliferation and inhibition of apoptosis
The TGF-β pathway modulates both cell growth and apoptosis. Using BrdUrd assays, we found that four weeks after Tam treatment there was a significantly increased number of proliferative cells in Tgfbr1/Pten 2cKO mice head and neck epithelia and SCCs, when compared to those of Tgfbr1f/f;Ptenf/f mice (p<0.001) (Figure 3a and b). Decreased apoptosis was also observed (p<0.001), indicating that an imbalance between cell proliferation and apoptosis occurs early in the head and neck epithelia of Tgfbr1/Pten 2cKO mice (Figure 3c and d). Immunostaining also revealed that CCND1 (Cyclin D1), an oncogene and a promoter of G0/G1 cell cycle progression inducible by NF-κB (Guttridge et al., 1999; Loercher et al., 2004), was increased in the tongue and SCCs of Tgfbr1/Pten 2cKO mice compared to those in Tgfbr1f/f;Ptenf/f mice (p<0.001) (Figure 3e and f). Our results indicate that loss of Tgfrb1 leads to enhanced cell proliferation, as well as inhibition of apoptosis in SCCs that developed in Tgfbr1/Pten 2cKO mice and normal Tgfbr1/Pten 2cKO mice head and neck epithelia.
Figure 3.
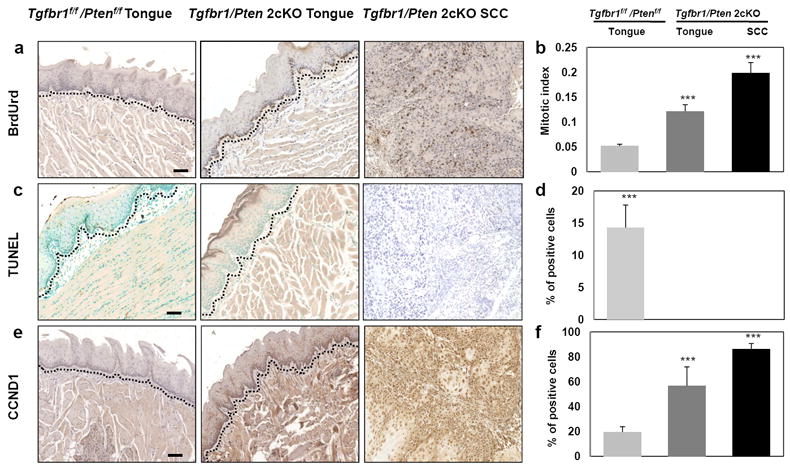
Enhanced cell proliferation and inhibition of apoptosis in Tgfbr1/Pten 2cKO mice. (a) A significantly increased number of proliferative cells in the tongue and SCCs of Tgfbr1/Pten 2cKO mice by BrdU assays. (c) Decreased apoptosis was also observed in the tongues and HNSCCs of Tgfbr1/Pten 2cKO mice compared to those of Tgfbr1f/f/Ptenf/f mice by TUNEL assays. (e) CCND1 was overexpressed in the tongues of Tgfbr1/Pten 2cKO mice and its expression was even more remarkable in the HNSCCs. Percentage of positive cells in the tongue or HNSCCs of Tgfbr1/Pten 2cKO mice compared to those of Tgfbr1f/f/Ptenf/f mice. (b, d, and f). Columns, average of three to five immunostained sections; the dotted lines delineate the adjacent epithelial compartment. (magnification, 200X). Bar=100um. **, P<0.01; ***, P<0.001.
TGF-β defects block Akt-induced senescence and result in overproduction of epithelial stem cells
Loss of the tumor suppressor protein PTEN, which functions by inhibiting the PI3K/Akt pathway, has been found to induce senescence in the prostate (Chen et al., 2005). TGF-β1 also plays an important role in growth suppression and senescence of the mouse keratinocytes (Vijayachandra et al., 2009). We hypothesized that the reason for the prevention of tumor development in Pten cKO mice is due to Akt-induced senescence. To determine whether loss of TGF-β signaling can block premature senescence induced by Akt activation, we examined the expression of SA-βgal staining in tumors from Tgfbr1/Pten 2cKO mice (n=3). Compared to the expression of this senescence marker in tumors from Pten cKO mice, SA-βgal expression in tumors from double knockout mice was significantly decreased in malignant as well as hyperplastic/dysplastic lesions (p<0.001) (Figure 4a and b). Akt-induced senescence has been associated with the activation of TP53-dependent mechanisms (Chen et al., 2005; Kim et al., 2007). The results from a previous study suggested that TP53-mediated senescence was responsible for the reduced malignant conversion of oral lesions in mice displaying constitutive Akt activity (myrAkt) (Moral et al., 2009). While the Western blot results revealed that TP53 was undetectable in association with increased p-Akt, we did detect an increase in the expression of the CDKN2D (p19Arf) gene. This is important for both the upstream activation of TP53, and the downstream cell cycle inhibitor CDKN1A (p21Cip1) in tumors from Pten cKO and Tgfbr1/Pten 2cKO mice that may be induced by the TP53 family member TP73 in the absence of TP53 (Rocco et al., 2006) (Figure 4c). Our results suggest that Tgfbr1/Pten 2cKO tumors escape senescence by inactivation of TP53 or other mechanisms, despite CDKN2D (p19Arf) and CDKN1A (p21Cip1) expression. Interestingly, increased expression of Id1, a transcriptional repressor previously reported to inactivate TP53 and PTEN and thereby enhance PI3K-Akt signaling (Lee et al., 2009), was detected in the tumors of Tgfbr1/Pten 2cKO mice but not in the tumors of Pten cKO mice (Figure 4c). Id1 is a negatively regulated downstream target of TGF-β signaling, and thus inactivation of TGFBR1 can cause overexpression of Id1. Id1 was found to render cells refractory to CDKN1A-dependent cell cycle arrest (Swarbrick et al., 2008). Therefore, senescence in carcinomas may be subverted by inactivation of any member of the p19-p53-p21 pathway, including TP53 and CDKN1A (p21Cip1), or by overexpression of Id1. Activated Akt has been found to interact directly with Smad3 (Conery et al., 2004; Remy et al., 2004). In SCCs of Tgfbr1/Pten 2cKO mice with the loss of TGF-β signaling, the same phenomenon was observed by immunoprecipitation indicating that Smad3 may also mediate the cross-talk between PI3K/Akt and TGF-β signaling pathways (Supplementary Figure S5). Altogether our results suggest that the neoplastic transformation of SCCs in Tgfbr1/Pten 2cKO mice was due to evasion of senescence, apoptosis, and increased proliferation.
Figure 4.
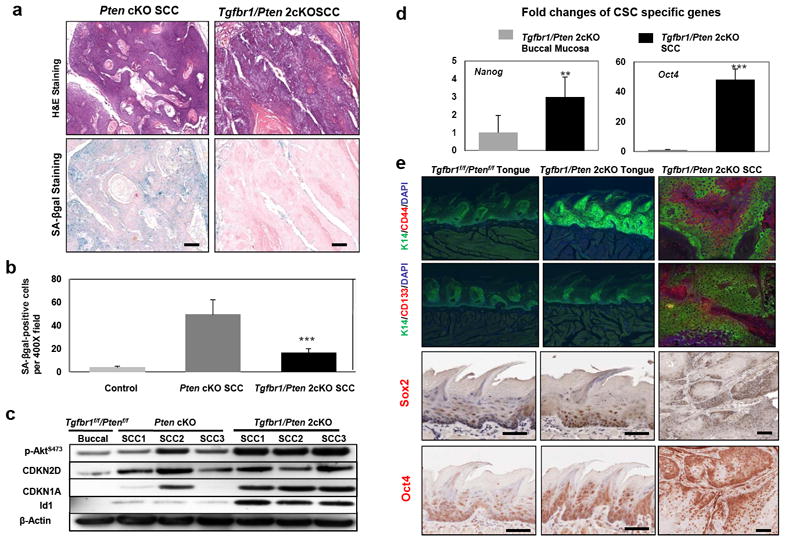
TGF-β defects block Akt-induced senescence, which results in overproduction of epithelial stem cells. (a) H&E and SA-βgal staining show the presence of many senescent cells in the HNSCC of Pten cKO mice, but an absence in the HNSCC of Tgfbr1/Pten 2cKO mice. (b) Quantitative analysis of SA-βgal positive HNSCCs in Pten cKO mice and Tgfbr1/Pten 2cKO mice. ***, P<0.001. (c) Western blot analysis revealed that the mechanism which leads to 2cKO SCCs bypassing cellular senescence is not through inactivation of the TP53-dependent pathway, but rather through overexpression of Id1. (d) Significantly increased mRNA expression of cancer stem cell specific genes Nanog and Oct4 in the HNSCC of Tgfbr1/Pten 2cKO mice by qRT-PCR. **, P<0.01; ***, P<0.001. E, CD44, CD133, Sox2, and Oct4 are overexpressed in Tgfbr1/Pten 2cKO HNSCCs by immunofluorescent and immunohistochemical staining (magnification, 200X). Bar=100um.
The initiation, growth, recurrence, and metastasis of HNSCC have all been related to a small subpopulation of tumor-initiating cells or cancer stem cells (CSC). A recent study has shown that TGF-β can suppress tumorigenesis through its effects on the putative cancer stem or on the early progenitor cells in a breast cancer xenograft model (Tang et al., 2007). Here, we found that loss of Tgfbr1 and Pten significantly increased mRNA expression of stem cell transcription factors Nanog (p<0.01) and Oct4 (p<0.001) in the epithelia of Tgfbr1/Pten 2cKO HNSCCs (Figure 4d). Using immunostaining, we observed that CD44 and CD133, the two most commonly demonstrated markers for the CSC subpopulation in HNSCC (Chen, 2009; Prince et al., 2007), as well as Sox2 and Oct4, are overexpressed within Tgfbr1/Pten 2cKO HNSCCs (Figure 4e). The increased expression of these CSC transcription factors and markers, together with an increase in the pool of proliferating cells in the basilar compartment, suggest that there is an expansion of the cancer stem cell population in these tumors.
Loss of TGF-β signaling in head and neck epithelia enhances chemokine production and recruitment of tumor promoting myeloid derived suppressor cells (MDSCs)
NF-κB is the master regulator of inflammation. One of the most important mechanisms underlying TGF-β regulation of cancer-related inflammation in the tumor microenvironment operates through NF-κB pathway. Either Tgfb1 overexpression or Tgfbr2 deletion in the mouse oral mucosa causes NF-κB activation in the surrounding tumor stroma (Lu et al., 2004; Cohen et al., 2009). Recent work has further associated the infiltration of bone marrow–derived inflammatory cells with tumor invasion (Yang et al., 2010). Deletion of Tgfbr2 in the mammary epithelium showed increased recruitment of F4/80+ cells, increased expression of pro-inflammatory cytokines, and altered composition of the fibrovascular stroma, thus promoting further tumor progression (Bierie et al., 2008). Because both the TGF-β and the PTEN/PI3K/Akt pathways are known to be involved in inflammation and have overlapping pro-inflammatory cytokine targets, we analyzed expression of the cytokine genes in 2cKO mouse tumors. To identify a set of overlapping targets of cancer-related inflammatory genes, we performed a mouse cytokine PCR array which profiles the expression of 84 key cytokine genes involved in inflammation or pre-inflammation of mouse head and neck cancer. By selecting cut-off of P<0.05 and fold changes >2, we confirmed an increase of 18 cytokines in the Tgfbr1/Pten 2cKO HNSCC compared with the wide-type tongue. Not surprisingly, a variety of genes encoding inflammatory cytokines are expressed, including Tnf, Il1a, Il1b, Il13, Il15, and Il17b with cytokine receptors Il1r1, Il1r2, Il6ra, Cxcr2, Cxcr3, IL13ra1, Tnfrsf1a. The mRNA expression of chemokines Ccl4, Ccl2, Ccl3, Cxcl1, and Cxcl5, and chemokine receptor Cxcr3, was increased in the SCC tissues of Tgfbr1/Pten 2cKO mice, which have been linked to chemoattractants myeloid derived suppressor cells (Figure 5a). To further confirm the increased cytokine by PCR array, we validated the expression of important cytokines such as Il1b, Il1a, Tnf, Cxcl1, Cxcl5 and Ptgs2 by quantitative PCR primer (Supplementary Figure S6). We also performed immunohistochemical analysis to determine the CD11b+, Gr-1+, and F4/80+ cell populations within our tumor samples. The results indicated that there was a significant increase in the amount of CD11b+ (Figure 5b), as well as in the Gr-1+ and F4/80+ cell populations (data not shown) of the HNSCCs that developed in the Tgfbr1/Pten 2cKO mice (p<0.01). These results, together with the finding that the NF-κB pathway was significantly activated in the Tgfbr1/Pten 2cKO HNSCCs (Figure 2e), suggest that when TGF-β and PTEN signaling is lost in HNSCCs, the decreased TGF-β1 signaling leads to upregulation of NF-κB and its regulated proinflammatory factors such as Cxcl1, Cxcl5, and Ptgs2 (Cohen et al., 2009). These factors then contribute in turn to the recruitment of bone marrow–derived cell populations known to promote tumor progression.
Figure 5.
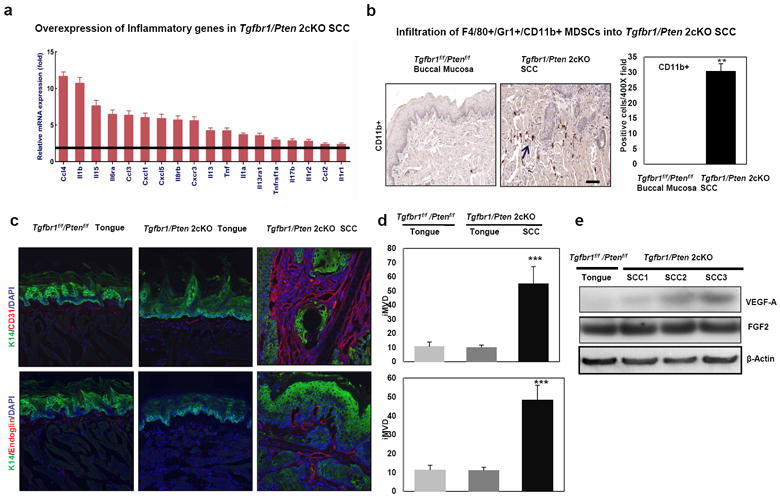
Enhanced chemokine production and recruitment of tumor promoting myeloid derived suppressor cells (MDSCs) in HNSCCs of Tgfbr1/Pten 2cKO mice. (a) Overexpression of inflammatory genes in Tgfbr1/Pten 2cKO SCC by Cytokine RT-PCR Array.(b) A significant increase in the amount of CD11b+, cell populations in the HNSCCs that developed in the Tgfbr1/Pten 2cKO mice by immunostaining. (c) Significantly increased expression of CD31 and Endoglin (CD105) in the stroma surrounding HNSCCs of Tgfbr1/Pten 2cKO mice. No expression was detected in the normal tongues of Tgfbr1f/f/Ptenf/f or Tgfbr1/Pten 2cKO mice. (d) percentage of intratumoral microvessel density (iMVD) indicated by CD31 or Endoglin (CD105)-stained microvessels per 200 X field in tumor stroma of Tgfbr1/Pten 2cKO mice. Columns, three immunostained sections. **, P<0.01; ***, P<0.001 (magnifications, 200X). (e) Western blot analysis revealed that the expression of VEGFA was increased in SCCs of Tgfbr1/Pten 2cKO mice.
MDSCs promote tumor invasion through increased angiogenesis, and we further analyzed the expression levels of vascular markers CD31 and Endoglin (CD105) in tumor stroma. Using immunofluorescent staining, we found that CD31 and Endoglin (CD105) expression, indicated by microvessel density (iMVD), was significantly increased in SCCs. This suggests an increase in angiogenesis in the tumor microenvironment (p<0.001) (Figure 5c and d). The results from Western blot further confirmed that VEGF-A was up-regulated in the SCCs of the 2cKO mice (Figure 5e).
In addition to promoting growth and invasion of the primary tumor, MDSCs have been shown to contribute to tumor associated immunosuppression. On one hand, these MDSCs can suppress antigen and nonantigen-specific CD8+ T-cells (Kusmartsev et al., 2005). On the other hand, these cells can support proliferation of regulatory T cells (Tregs) (Huang et al., 2006). We analyzed the immune status of the Tgfbr1/Pten 2cKO mice using flow cytometry analysis. Compared with their control littermates, Tgfbr1/Pten 2cKO mice showed a substantially reduced number of CD8+ effector T cells in the jugular lymph nodes. In contrast, the frequency of Tregs (CD4+CD25+Foxp3+) was increased, consistent with active immune suppression in Tgfbr1/Pten 2cKO mice (Supplementary Figure S7). A proposed model for how alterations in TGF-β and PTEN/PI3K/Akt signaling promote HNSCCs in mice is shown in Figure 6.
Figure 6.
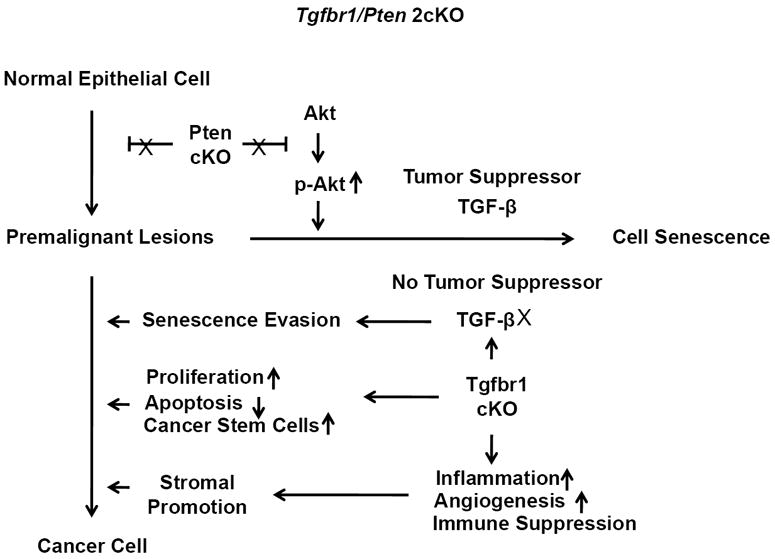
A schematic presentation of the proposed TGF-β and PTEN/PI3K/Akt signaling alteration that promotes HNSCC in mice. Activation of the PI3K/Akt pathway due to Pten deletion initiates tumor formation by increasing proliferation in the head and neck epithelia. However, Pten deletion alone is not sufficient to induce invasive HNSCC due to the induction of premature senescence by p-Akt in the presence of tumor suppressors such as TGF-β. In combination with the additional loss of Tgfbr1, which results in a loss of tumor inhibition by TGF-β signaling, premalignant cells cannot undergo cellular senescence. They will therefore progress into cancer cells through mechanisms which include increased cell proliferation, decreased apoptosis, senescence evasion and overproduction of epithelial stem cells. Decreased Tgfbr1 can also increase chemokine production in tumor stroma, which leads to enhanced recruitment of MDSCs. This results in increased inflammation, angiogenesis, and immune suppression, which contribute significantly to mouse head and neck carcinogenesis.
Discussion
TGF-β is a potent growth inhibitor for epithelial cells (Massagué et al., 2006). Inactivation by mutations or by the experimental deletion of components of the TGF-β signaling pathway has been shown to promote tumorogenesis in a variety of organ systems, including HNSCCs (Bierie et al., 2006; Levy et al., 2006). The results from our previous study indicated that inducible deletion of Tgfbr1 alone in head and neck epithelia is not sufficient for SCC formation in mice. This finding, together with the fact that DMBA treatment facilitates tumor development in these mice, suggests that rather than initiation, the loss of Tgfbr1 may play a more crucial role in tumor progression in the mouse HNSCCs (Bian et al., 2009). There is growing evidence which suggests that many of the pro-oncogenic responses to TGF-β are either Smad-independent or require cooperation between the Smad pathways and alternative pathways, with relatively low activation of the Smad pathway (Wakefield et al., 2002). Recent reports showed that decreased Tgfbr1 expression in Tgfbr1 cKO mice leads to increased cell proliferation and cell survival through activation of the PI3K/Akt pathway, suggesting that there is negative cross-talk between the TGF-β tumor suppressor and the PI3k/Akt pathways (Wang et al., 2008; Bian et al., 2009). The PTEN/PI3K/Akt signaling pathways play critical roles in the carcinogenesis of HNSCC (Pedrero et al., 2005). Hyperactivation of PI3K can be induced by inactivation of PTEN (Molinolo et al., 2009). In mice, excision of Pten results in hyperproliferative and neoplastic lesions that recapitulate features of Cowden’s disease (Squarize et al., 2008). Herein, we have shown that defects in the TGF-β and PI3K/Akt signaling pathways are common in human HNSCCs. Activation of the PI3K/Akt pathway due to Pten deletion initiates tumor formation by increasing proliferation in the head and neck epithelia. However, Pten deletion alone is not sufficient to induce invasive HNSCC due to the induction of premature senescence by p-Akt in the presence of the tumor suppressor TGF-β. In combination with the additional loss of Tgfbr1 which blocks tumor inhibition by TGF-β signaling, premalignant cells cannot undergo cellular senescence and will progress into cancer cells.
Senescence in carcinoma can be subverted by the inactivation of any member of the p19-p53-p21 pathway or by overexpression of Id1 (Chen et al., 2005; Swarbrick et al., 2008). The overexpression of CDKN2D (p19Arf) and CDKN1A (p21Cip1) in tumors of the Tgfbr1/Pten 2cKO mice, together with the increased expression of Id1 in these tumors, suggested that overexpression of Id1 due to Tgfbr1 deletion likely plays a role in bypassing cellular senescence in 2cKO SCCs. How Id1 acts to prevent cellular senescence in these tumors warrants further investigation.
Molecular analysis of Tgfbr1/Pten 2cKO mice revealed enhanced cell proliferation, loss of apoptosis, and increased expression of CCND1 (Cyclin D1) in the basal layer of the head and neck epithelia, as well as in the tumors of 2cKO mice. This suggests that the inhibition of head and neck cancer development by Tgfbr1 and Pten is mainly due to collaborative regulation of the cell cycle. The early onset and full penetrance of HNSCC in 2cKO mice, together with the increased number of putative cancer stem cells in tumors of 2cKO mice, suggests that Tgfbr1 and Pten function cooperatively to regulate tissue homeostasis in mouse head and neck epithelia. However, the molecular mechanism by which Tgfbr1 and Pten regulate cancer stem cell self-renewal and differentiation needs to be further investigated.
It is widely believed that TGF-β can affect cancer progression through both autocrine and paracrine effects. Autocrine effects of TGF-β in premalignant epithelial cells are tumor suppressive, while more advanced cancer cells with a functional TGF-β receptor complex may exhibit tumor-promoting autocrine effects due to a convergence of TGF-β signaling with other signaling pathways. Paracrine effects of TGF-β, which are generally tumor promoting, include stimulation of inflammation and angiogenesis, escape from immunosurveillance and recruitment of myofibroblasts (De Wever et al., 2003). In the current study we found that, upon deletion of Tgfbr1/Pten in mouse head and neck epithelia, the NF-κB pathway is activated and genes that are known to promote inflammation in HNSCCs, such as Cxcl1, Cxcl5, and Ptgs2, are overexpressed. There is also enhanced recruitment of MDSCs, which results in increased angiogenesis through upregulation of VEGF-A as well as immune suppression in the tumor stroma. Similar results have been observed in other mouse models when TGF-β signaling was disrupted (Lu et al., 2006; Bierie et al., 2008). These findings suggest that the tumor microenvironment also contributes significantly to mouse head and neck carcinogenesis. Therefore, on one hand the deletion of Tgfbr1 in mouse head and neck epithelia prevents TGF-β1 from exerting its tumor suppressive effects on tumor initiating cells and prevents senescence caused by Pten deletion. On the other hand, inactivation of TGF-β signaling by Tgfbr1 loss would certainly enhance cancer-related inflammation and the recruitment of MDSCs in the tumor microenvironment for tumor progression.
Given the complex nature of HNSCC in vivo, the most powerful tools that can be used to study head and neck carcinogenesis are transgenic mouse models. Here we present a novel mouse model for HNSCC research which targets deletion of Tgfbr1 and Pten in mouse head and neck epithelia. 100% of the 2cKO mice developed spontaneous tumors within 10 weeks after induction of Cre activity with Tamoxifen. The emerging tumors were not only pathologically indistinguishable from their human counterparts, but they also showed several major molecular alterations taking place in human HNSCCs, including those affecting EGFR, Erk, NF-κB, and Stat3 (Molinolo et al., 2009). Previous studies have linked inactivation of TGF-β and the activation of these signaling pathways and transcription factors with the expression of key proliferative and pro-inflammatory factors such as CCND1, Cxcl1, Cxcl5 and Ptgs2, as well as the related pathologic alterations detected here (Cohen et al., 2009). These findings indicated that the HNSCCs of 2cKO mice displayed pathology and multiple molecular alterations that mimicked human HNSCCs, suggesting that this mouse model is suitable for developing strategies for preclinical intervention. Furthermore, this suggests that the mouse model has significant implications for the development of diagnostic cancer biomarkers as well as effective therapeutic approaches targeting both the TGF-β and the PI3K/Akt pathways for the treatment of HNSCCs. Alternative models targeting TGF-β signaling have also been generated (White et al., 2010). TGFBR2 deletion, when combined with a tumor-initiating Ras mutation, causes HNSCC in mice (Lu et al., 2006). Downstream conditional deletion of Smad4 in mouse oral keratinocytes resulted in the development of HNSCC in mice starting at 29 weeks of age. These tumors histologically resembled human HNSCCs and metastasized to regional lymph nodes (Bornstein et al., 2009). However, tumors that developed in those mouse models were either not full-penetrance or took a long time to appear.
In summary, we have demonstrated that activation of the PI3K/Akt pathway due to Pten deletion initiates tumor formation by increasing proliferation in the head and neck epithelia. However, Pten deletion alone is not sufficient for inducing invasive HNSCC due to the induction of premature senescence by p-Akt. TGF-β is a major tumor suppressor and Pten deletion, in combination with the additional loss of Tgfbr1, results in premalignant cells progressing into cancer cells through senescence evasion and the expansion of epithelial stem cells. Decreased Tgfbr1 can also activate the NF-κB pathway, causing increased chemokine production in tumor stroma which leads to enhanced recruitment of MDSCs. This results in increased inflammation, angiogenesis, and immune suppression, which contribute significantly to mouse head and neck carcinogenesis. Our results indicate that the loss of TGF-β signaling and PTEN in head and neck epithelia promotes HNSCCs through regulation of both premalignant cells and interactions with the tumor microenvironment in these mice. The findings of our study have significant implications regarding the development of diagnostic cancer biomarkers, as well as effective preventive and therapeutic strategies targeting both the TGF-β and the PI3K/Akt pathways for the treatment of HNSCCs.
Material and Methods
Tissue samples and cancer cell lines
Formalin-fixed and paraffin-embedded HNSCC tissue arrays were obtained from Cybrdi (Rockville, MD). Each array contained HNSCC tumor tissues from 20 individuals spotted in triplicate, plus normal mucosa tissues from 6 normal subjects, spotted in duplicate. Human tongue SCC lines Cal27, SCC4, SCC9, SCC15, and SCC25 were purchased from ATCC. KCCOR891, HSC-3, KCCT-873, OSC19, and YCUM862 were kindly provided by Drs. Koji Kawakami of Kyoto University, Japan and Raj Puri from US FDA. Human oral keratinocytes (HOK) (ScienCell Research Laboratories, Carlsbad, CA) were used as normal control.
Generation of Tgfbr1/Pten 2cKO mice
The Tgfbr1/Pten 2cKO mice (K14-CreERtam;Tgfbr1f/f;Ptenf/f) were generated from crosses between Tgfbr1 cKO mice (K14-CreERtam;Tgfbr1flax/flax) and Ptenfoxp/foxp mice. The Tgfbr1/Pten 2cKO mice and their controls (Tgfbr1f/f;Ptenf/f and K14-CreERtam;Tgfbr1f/+;Ptenf/+ mice) were from the same litter and therefore had the same mixed genetic background. The treatment procedures of Tamoxifen and additional details are provided in the Supplementary Data.
Histology, immunostaining and BrdU labeling
Immunohistochemical staining (IHC), quantifications of IHC slides, intratumoral microvessel density (iMVD), and BrdUrd labeling are described in the Supplementary Data.
Western blot analysis
Normal buccal mucosa and tongue from 5 pairs of Tgfbr1f/f/Ptenf/f and Tgfbr1/Pten 2cKO mice, together with tumors that developed in Tgfbr1/Pten 2cKO mice, were carefully dissected. A total of 40 μg of protein from each sample were denatured and then loaded in each lane of NuPAGE 4-12% Bis-Tris precast gel. Additional details are provided in the Supplementary Data.
Additional Methods
Information on TUNEL assay, Senescence assay, Cre-mediated recombination assessment, quantitative real-time PCR, mouse cytokine RT-PCR array, immunoprecipitation and flow cytometry analysis is detailed in the Supplementary Data.
Statistical analysis
Statistical differences in the levels of mRNA expression between controls and experimental samples, and the difference between the immunohistochemical quantifications of each group, were determined using the Student’s t-test.
Supplementary Material
Acknowledgments
We would like to thank Dr Stefan Karlsson for providing us with the Tgfbr1 flox mice, Drs Raj Puri and Lalage Wakefield for critical reading of the manuscript, Drs Juan Du, Ping Zhang, Zhong Chen, and Anita Terse for technical help, and Shelagh Johnson for expert editorial assistance. This work was supported by the Divisions of Intramural Research, National Institute of Dental and Craniofacial Research (Z01-DE-000698), and National Institute on Deafness and Communication Disorders (ZIA-DC-000073), NIH.
Footnotes
Conflict of interest: The authors declare no conflict of interest
References
- Bian Y, Terse A, Du J, Hall B, Molinolo A, Zhang P, et al. Progressive tumor formation in mice with conditional deletion of TGF-beta signaling in head and neck epithelia is associated with activation of the PI3K/Akt pathway. Cancer Res. 2009;69:5918–26. doi: 10.1158/0008-5472.CAN-08-4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–20. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Bierie B, Stover DG, Abel TW, Chytil A, Gorska AE, Aakre M, et al. Transforming growth factor-beta regulates mammary carcinoma cell survival and interaction with the adjacent microenvironment. Cancer Res. 2008;68:1809–19. doi: 10.1158/0008-5472.CAN-07-5597. [DOI] [PubMed] [Google Scholar]
- Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119:3408–19. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Yan W, Wells RG, Rimm DL, McNiff J, Leffell D, et al. Novel inactivating mutations of transforming growth factor-beta type I receptor gene in head-and-neck cancer metastases. Int J Cancer. 2001;93:653–61. doi: 10.1002/ijc.1381. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZG. The cancer stem cell concept in progression of head and neck cancer. J Oncol. 2009;894064 doi: 10.1155/2009/894064. Epub 2009 Dec 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J, Chen Z, Lu SL, Yang XP, Arun P, Ehsanian R, et al. Attenuated transforming growth factor beta signaling promotes nuclear factor-kappaB activation in head and neck cancer. Cancer Res. 2009;69:3415–24. doi: 10.1158/0008-5472.CAN-08-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery AR, Cao Y, Thompson EA, Townsend CM, Jr, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–72. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- Curado MP, Hashibe M. Recent changes in the epidemiology of head and neck cancer. Curr Opin Oncol. 2009;21:194–200. doi: 10.1097/CCO.0b013e32832a68ca. [DOI] [PubMed] [Google Scholar]
- De Wever O, Mareel M. Role of tissue stroma in cancer cell invasion. J Pathol. 2003;200:429–47. doi: 10.1002/path.1398. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Deshpande AM, Wong DT. Molecular mechanisms of head and neck cancer. Expert Rev Anticancer Ther. 2008;8:799–809. doi: 10.1586/14737140.8.5.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor β1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–86. [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson YC, Wang E, Clayman GL. Genotypic analysis of tumor suppressor genes PTEN/MMAC1 and p53 in head and neck squamous cell carcinomas. Laryngoscope. 1998;108:1553–6. doi: 10.1097/00005537-199810000-00024. [DOI] [PubMed] [Google Scholar]
- Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–31. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–77. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumorassociated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175:4583–92. doi: 10.4049/jimmunol.175.7.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JI, Soria JC, Hassan KA, El-Naggar AK, Tang X, Liu DD, et al. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch Otolaryngol Head Neck Surg. 2001;127:1441–5. doi: 10.1001/archotol.127.12.1441. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kang MB, Jang SH, Qian T, Kim HJ, Kim CH, et al. Id-1 activates Akt-mediated Wnt signaling and p27(Kip1) phosphorylation through PTEN inhibition. Oncogene. 2009;28:824–31. doi: 10.1038/onc.2008.451. [DOI] [PubMed] [Google Scholar]
- Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Licitra L, Perrone F, Bossi P, Suardi S, Mariani L, Artusi R, et al. High-risk human papillomavirus affects prognosis in patients with surgically treated oropharyngeal squamous cell carcinoma. J Clin Oncol. 2006;24:5630–6. doi: 10.1200/JCO.2005.04.6136. [DOI] [PubMed] [Google Scholar]
- Loercher A, Lee TL, Ricker JL, Howard A, Geoghegen J, Chen Z, et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64:6511–23. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- Lu SL, Reh D, Li AG, Woods J, Corless CL, Kulesz-Martin M, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405–10. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5:311–6. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- Massagué J, Gomis RR. The logic of TGF-β signaling. FEBS Lett. 2006;580:2811–20. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-β in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–34. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moral M, Segrelles C, Lara MF, Martínez-Cruz AB, Lorz C, Santos M, et al. Akt activation synergizes with Trp53 loss in oral epithelium to produce a novel mouse model for head and neck squamous cell carcinoma. Cancer Res. 2009;69:1099–108. doi: 10.1158/0008-5472.CAN-08-3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. The regulation of TGF-β signal transduction. Development. 2009;136:3699–714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- Pasche B, Knobloch TJ, Bian Y, Liu J, Phukan S, Rosman D, et al. Somatic acquisition and signaling of TGFBR1*6A in cancer. JAMA. 2005;294:1634–46. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, et al. Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005;114:242–8. doi: 10.1002/ijc.20711. [DOI] [PubMed] [Google Scholar]
- Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–65. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- Piek E, Roberts AB. Suppressor and oncogenic roles of transforming growth factor-beta and its signaling pathways in tumorigenesis. Adv Cancer Res. 2001;83:1–54. doi: 10.1016/s0065-230x(01)83001-3. [DOI] [PubMed] [Google Scholar]
- Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–8. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006;9:45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Shao X, Tandon R, Samara G, Kanki H, Yano H, Close LG, et al. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. Int J Cancer. 1998;77:684–8. doi: 10.1002/(sici)1097-0215(19980831)77:5<684::aid-ijc4>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden’s disease by mTOR inhibition with rapamycin. Cancer Res. 2008;68:7066–72. doi: 10.1158/0008-5472.CAN-08-0922. [DOI] [PubMed] [Google Scholar]
- Swarbrick A, Roy E, Allen T, Bishop JM. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc Natl Acad Sci U S A. 2008;105:5402–7. doi: 10.1073/pnas.0801505105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR, et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest. 2003;112:1116–24. doi: 10.1172/JCI18899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Yoo N, Vu M, Mamura M, Nam JS, Ooshima A, et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. 2007;67:8643–52. doi: 10.1158/0008-5472.CAN-07-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayachandra K, Higgins W, Lee J, Glick A. Induction of p16ink4a and p19ARF by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol Carcinog. 2009;48:181–6. doi: 10.1002/mc.20472. [DOI] [PubMed] [Google Scholar]
- Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–9. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- Wang J, Yang L, Yang J, Kuropatwinski K, Wang W, Liu XQ, et al. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008;68:3152–60. doi: 10.1158/0008-5472.CAN-07-5348. [DOI] [PubMed] [Google Scholar]
- White RA, Malkoski SP, Wang XJ. TGF-β signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29:5437–46. doi: 10.1038/onc.2010.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Bharathy S, Kim D, Haffty BG, Rimm DL, Reiss M. Frequent alterations of Smad signaling in human head and neck squamous cell carcinomas: a tissue microarray analysis. Oncol Res. 2003;14:61–73. doi: 10.3727/000000003108748612. [DOI] [PubMed] [Google Scholar]
- Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–7. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. Non-Smad pathways in TGF- β signaling. Cell Res. 2009;19:128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.