

Abstract
The mineralocorticoid aldosterone is indispensable for the control of blood pressure and fluid volume in mammals. It acts in large part to increase the abundance and activity of the epithelial Na+ channel (ENaC), which mediates apical Na+ entry in the distal parts of the kidney tubules. Aldosterone acts through the mineralocorticoid receptor to alter the transcription of specific genes, including SGK1 and GILZ1. Recent evidence suggests that these key aldosterone-regulated factors function within a unique multi-protein ENaC-regulatory-complex that governs the net cell surface expression and activity of the channel. Another aldosterone-induced protein, CNK3 (connector enhancer of kinase suppressor of Ras 3), also stimulates ENaC and has all of the features of a scaffolding protein. With these observations in mind, we discuss the possibility that CNK3 coordinates the dynamic assembly of the ENaC-regulatory-complex, and promotes context-appropriate aldosterone signal transduction in the regulation of epithelial Na+ transport.
Keywords: Aldosterone signaling, ENaC-regulatory complex
1. Introduction
Aldosterone and its principal receptor, the mineralocorticoid receptor (MR), play essential roles in mammalian physiology, and pathophysiology (Bhargava & Pearce, 2004). Although multiple tissues express MR, and are impacted by aldosterone, effects on epithelial ion transport in the kidney and colon are paramount. The regulation of transepithelial sodium (Na+) transport in the kidney is central to the maintenance of extracellular fluid volume and blood pressure. The epithelial Na+ channel, ENaC, mediates apical entry of Na+ in the aldosterone-sensitive distal nephron, and constitutes the rate-limiting step for Na+ reabsorption by the kidney (Thomas & Itani, 2004). MR targets include transporter or channel subunits themselves such as αENaC and the α-subunit of the basolateral Na,K-ATPase, as well as regulatory proteins, such as the PI3K/mTOR-dependent kinase, SGK1 (serum and glucocorticoid-induced kinase 1) (McCormick et al, 2005), the small chaperone, GILZ (glucocorticoid-induced leucine zipper protein) (Muller et al, 2003), and possibly K-WNK1 (the kidney isoform of ‘with-no-lysine’ kinase 1) (Naray-Fejes-Toth et al, 2004) and K-Ras (Stockand et al, 1999). Most of these regulatory targets profoundly influence the fundamental signaling mechanisms that control Na+ transport in the kidney tubules. ENaC itself is regulated by a wide variety of factors through changes in plasma membrane abundance or inherent activity. These multiple modes of regulation of expression, protein-protein interactions and processing of ENaC are integrated to result in net membrane incorporation and activity of the channel. In this review, we recall the role of two important MR targets, SGK1 and GILZ1, in mediating the aldosterone response, and discuss how they are organized into a single protein complex, which we have termed the ENaC-regulatory complex (ERC). We also address the role of the recently identified aldosterone-induced protein, CNK3 (connector enhancer of kinase suppressor of Ras 3; also an MR target) in ENaC regulation, and discuss its potential function as a scaffold for the formation of the ERC.
2. Aldosterone-induced early genes cooperate in mediating the response to hormone
2.1. SGK1 integrates multiple cellular signals to facilitate aldosterone effects on Na+ transport
Over the last decade, the aldosterone-regulated serine-threonine kinase SGK1 (Fig. 1A), has emerged as a key modulator of ENaC in kidney cortical collecting duct (CCD) cells. SGK1 was originally cloned from breast cancer cells, as a glucocorticoid-induced gene (Webster et al, 1993b), but its first physiologically relevant function was identified when it was independently cloned as an aldosterone-induced gene, and shown to stimulate ENaC-mediated Na+ transport (Chen et al, 1999; Naray-Fejes-Toth et al, 1999; Shigaev et al, 2000). Both its expression level and activity are regulated by hormonal and non-hormonal factors which modulate its gene transcription (Bhargava et al, 2004), protein stability (Arteaga et al, 2006; Soundararajan et al, 2010b), phosphorylation state (Garcia-Martinez & Alessi, 2008; Lu et al, 2010; Park et al, 1999; Wang et al, 2001), and access to substrates (Pao et al, 2007; Soundararajan et al, 2009). By phosphorylating a variety of ENaC-regulatory proteins, SGK1 alters ENaC expression, trafficking and activity, and stimulates Na+ transport in the CCD.
Figure 1. Annotated mouse SGK1 (A), GILZ1 (B) and CNK3 (C) protein schematics showing key motifs and interaction domains.
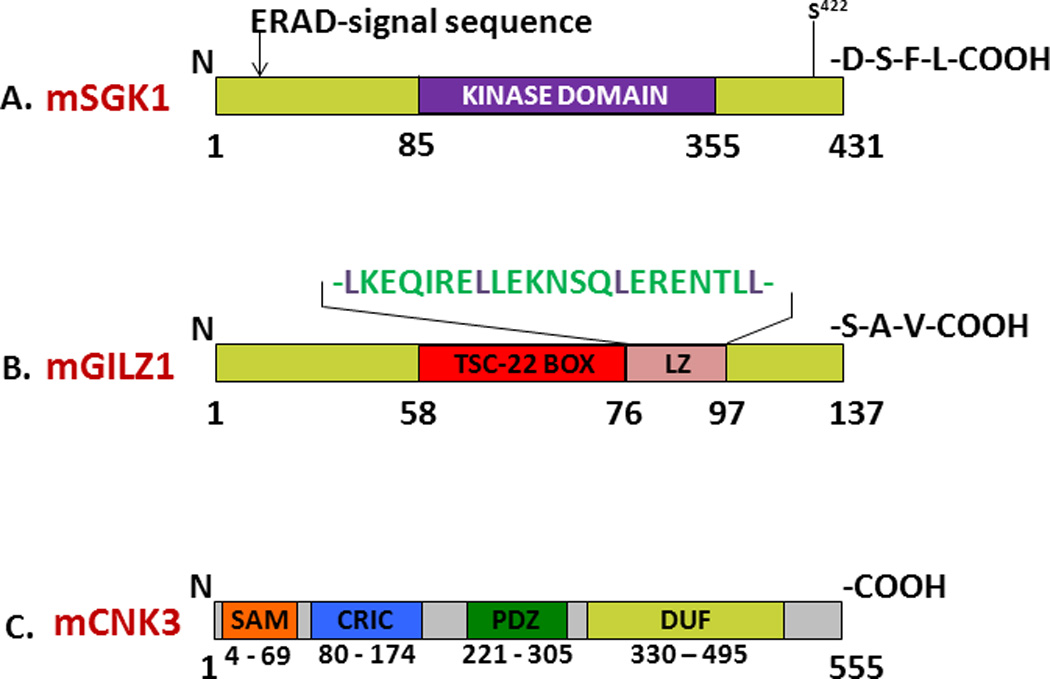
A. SGK1 is an important aldosterone-regulated protein kinase that stimulates renal ENaC function. The ER-associated degradation signal (or ‘degron’) lies within the N-terminus of SGK1, upstream of its kinase domain. The Ser residue within the hydrophobic motif that is critical for mTORC2-dependent kinase activation is depicted. SGK1 also possesses a standard Class I PDZ domain-interaction motif at its C-terminus. B. GILZ1 is a small aldosterone-induced chaperone that plays a central role in protein trafficking and signaling. The TSC-22 signature box defines region of homology with the TGFβ-stimulated clone-22 protein and other family members. The leucine zipper (LZ) motif mediates GILZ dimerization. Also shown is the sequence of the Class I PDZ domain-interaction motif at the C-terminus. C. CNK3 is a recently identified MR-target shown to be critical for ENaC activity. As with other members of the CNK family, CNK3 contains a sterile-α-motif (SAM), a conserved-region-in-CNK (CRIC) domain, a classic PDZ domain, and a downstream region commonly referred to as the domain-of-unknown-function or DUF.
SGK1 stimulation of ENaC involves negative regulation of the E3 ubiquitin ligase, Nedd4-2 (neural precursor cell expressed, developmentally down-regulated 4-2). Nedd4-2 interacts with the C-terminal tails of ENaC subunits, decreases surface expression of the channel via channel ubiquitinylation, and hence inhibits Na+ currents (Staub et al, 1996). SGK1 physically interacts with Nedd4-2, phosphorylates and inhibits it (Debonneville et al, 2001; Flores et al, 2005; Snyder et al, 2002) within an ENaC-regulatory complex (Soundararajan et al, 2009), and hence indirectly enhances cell surface expression of ENaC (Alvarez de la Rosa & Canessa, 2003; Alvarez de la Rosa et al, 1999). Nedd4-2-independent mechanisms of SGK1 stimulation of ENaC have also been demonstrated. For example, SGK1 was shown to directly phosphorylate a serine residue in the intracellular C-terminal tail of αENaC, which directly activates channels at the cell surface (Diakov & Korbmacher, 2004). Also, SGK1 has been implicated in the stimulation of ENaC via phosphorylation of WNK4, a kinase mutated in Familial Hyperkalemic Hypertension (Ring et al, 2007). A third mechanism of ENaC stimulation by SGK1 involves up-regulation of components of the Na+ transport machinery per se, primarily αENaC (Kempe et al, 2010). SGK1 plays an important role in aldosterone-sensitive αENaC transcription in vivo via inhibition of a transcriptional repression element, the disruptor of telomeric silencing alternative splice variant a (Dot1a)-ALL1-fused gene from chromosome 9 (Af9) complex (Pearce & Kleyman, 2007; Zhang et al, 2007). SGK1 also regulates other transporters in the distal nephron, including the sodium chloride co-transporter (NCC) and possibly K channels (Fejes-Toth et al, 2008; Vallon & Lang, 2005; Vallon et al, 2009). The ability of SGK1 to regulate in vivo renal Na+ reabsorption is well illustrated by the impaired Na+ retention of gene-targeted mice lacking functional SGK1 (Fejes-Toth et al, 2008; Wulff et al, 2002). The downstream actions of SGK1 on specific targets such as Nedd4-2, WNK4, Raf1, and others have been well characterized, particularly as they pertain to ENaC expression, stability, trafficking and open probability (Loffing & Korbmacher, 2009; McCormick et al, 2008; Pearce & Kleyman, 2007; Soundararajan et al, 2010b; Vallon et al, 2009).
Interestingly, SGK1 is a short-lived protein. Many tissues and cell types express abundant SGK1 mRNA, however the protein is barely detectable because its half-life is short (<30 min) (Arteaga et al, 2006; Webster et al, 1993a). Following synthesis, SGK1 is rapidly targeted to the endoplasmic reticulum (ER), where ER-associated ubiquitin ligases CHIP and HRD1 aid in its ubiquitinylation and subsequent proteasome-mediated degradation (Arteaga et al, 2006; Bogusz et al, 2006). How is SGK1 then redirected from the ER and targeted appropriately to ENaC? A clue to this was obtained from a recent report that suggested a novel role for yet another aldosterone-induced ENaC-regulator, GILZ, which protects SGK1 from rapid ER-associated degradation (ERAD), by controlling its protein-protein interactions and availability in specific subcellular compartments (Soundararajan et al, 2010b) (more below).
2.2. GILZ1 and transepithelial Na+ transport
GILZ is a ubiquitously expressed protein that is well known for its ability to interact with a myriad of cellular factors and influence key processes such as transformation, differentiation, proliferation, ion transport and apoptosis, to just name a few. There are four distinct isoforms of GILZ, termed GILZ1-4 in order of their discovery (Soundararajan et al, 2007). The transcripts arise as splice variants from a single gene, Tsc22d3, located on the mouse X chromosome. A large intron spanning almost 55 kb contains multiple response elements (six GREs and three FHREs), most of which have been reported to be functional (Asselin-Labat et al, 2005; Asselin-Labat et al, 2004; Wang et al, 2004). The different splice variants are involved in controlling distinct aspects of cellular physiology, and modulate distinct signaling pathways (Soundararajan et al, 2007). GILZ1 is the most potent isoform relevant to stimulation of Na+ transport (Soundararajan et al, 2007). This original 137 amino acid-long variant was initially reported by Riccardi and colleagues in 1997 (D'Adamio et al, 1997). GILZ1 shares considerable homology with other members of the TGFβ-stimulated clone 22 (TSC-22) family of leucine zipper proteins, of which there are at least six others (Ayroldi & Riccardi, 2009). It consists of a central leucine zipper, an N-terminal domain which includes the TSC-22 signature box, and a C-terminal proline- and glutamic acid-rich region (Fig. 1B). Raf-1 was one of the earliest binding partners discovered for GILZ1 (Ayroldi et al, 2002). The Raf-1 interaction domain within GILZ1 has been broadly localized to the Ras-binding domain within the N-terminus (the precise sub-domains/residues important for interaction are unknown), while the central leucine zipper appears essential for homo-dimerization (Ayroldi et al, 2007; Riccardi et al, 1999). Interestingly, GILZ1 also contains a PSD-95 - DLG-1 - ZO-1 (PDZ) domain-binding motif (or PBM) at the C-terminal end (Fig. 1B), a characteristic feature of many proteins involved in forming complexes with larger integral membrane proteins. The interesting link between GILZ1 and transepithelial Na+ transport was explored following the extensive series of reports that demonstrated that extracellular signal-regulated kinase 1/2 (ERK1/2) abrogates ENaC function on one hand, and that GILZ1 inhibits the inhibitory Raf-1/MEK1/2/ERK1/2 signaling cascade, on the other (Ayroldi et al, 2002; Cao et al, 2005; Hendron et al, 2002; Michlig et al, 2005; Schild et al, 1996; Shi et al, 2002).
As its name implies, GILZ1 is rapidly induced by glucocorticoids in T lymphocytes, in which it inhibits anti-CD3-induced IL-2 production, IL2-receptor expression, Fas and Fas-ligand up-regulation, and cell death consequent to CD3-induced activation (Ayroldi et al, 2001; D'Adamio et al, 1997; Riccardi et al, 1999; Riccardi et al, 2000). In kidney cortical collecting duct (CCD), as well as in mpkCCDc14 cells, a highly differentiated mouse CCD cell line, GILZ1 is robustly induced by aldosterone (Robert-Nicoud et al, 2001). Studies in multiple model systems have shown that GILZ1 stimulates ENaC cell surface expression and activity, at least in part by inhibiting ERK1/2 (Soundararajan et al, 2009; Soundararajan et al, 2007; Soundararajan et al, 2010b; Soundararajan et al, 2005). Importantly, the ENaC-stimulatory effects of GILZ1 are more readily appreciable only in the context of activated ERK1/2.
Transepithelial Na+ transport requires the coordinate functioning of multiple different hormone-regulated components. Recent evidence supports the idea that many of these aldosterone-regulated factors function within a unique multi-protein assembly (called the ENaC-regulatory complex or ERC) that governs the net cell surface expression and activity of the channel (Soundararajan et al, 2009; Soundararajan et al, 2010a; Soundararajan et al, 2010b).
2.3. The ENaC-Regulatory Complex or ERC
In the absence of aldosterone, ENaC activity is limited by Raf-1 and Nedd4-2, the two major negative regulatory components governing ENaC. Raf-1 is the master regulator of the inhibitory ERK1/2 signaling cascade, and Nedd4-2 is the E3 ubiquitin ligase that targets ENaC for proteasome-mediated degradation following ubiquitinylation. Aldosterone-induced GILZ1 plays a unique role in relieving this “doublein-hibition”. GILZ1 appears to physically interact with ENaC, Raf-1 as well as with Nedd4-2, thereby facilitating the assembly of the ERC (Soundararajan et al, 2009; Soundararajan et al, 2010b). In addition to direct inhibition of Raf-1 and Nedd4-2, GILZ1 also physically interacts with SGK1, alters its subcellular localization and selectively recruits it into the ERC (Soundararajan et al, 2009). GILZ1 recruitment of SGK1 to the ERC is a key factor in protecting SGK1 from rapid ER-associated proteasome-mediated degradation. Within the ERC, GILZ1 and SGK1 synergize to markedly stimulate ENaC surface expression and activity (Soundararajan et al, 2009). By modulating its subcellular localization and enabling SGK1 to preferentially interact with its target substrates, Nedd4-2 and Raf-1 within the ERC on one hand, and by simultaneously inhibiting the interaction of SGK1 with the E3 ubiquitin ligases CHIP and HRD1 on the other, GILZ1 stabilizes SGK1 levels, and allows SGK1 to perform its ENaC-stimulatory activities in a synergistic manner (Soundararajan et al, 2010b).
GILZ1 is thus an adaptor protein, which interacts with—and modulates the activity of—multiple components of the ENaC regulatory machinery. In the absence of aldosterone, when GILZ1 and SGK1 levels are low, Nedd4-2 and Raf-1 are associated with, and inhibit ENaC. In the presence of aldosterone, GILZ1 and SGK1 levels rise, and they are recruited into the complex, where together they inhibit Raf-1 and Nedd4-2 activity, and therefore stimulate ENaC. Thus by promoting the association of PI3K- and Raf-1 pathway components into a highly-specific ion transport regulatory “complex”, GILZ1 is able to selectively govern ENaC surface expression and activity.
2.3.1. Functional significance of the ERC
Hormonal control of transepithelial Na+ transport utilizes pleiotropic signaling pathways, such as the PI3K and the Raf/MEK/ERK1/2 cascades, which simultaneously impact on numerous cell functions including cell proliferation, differentiation and apoptosis. The ERC provides for a novel mechanism for organizing these specific signals, and transmitting them appropriately. Through this mechanism, ion transport can be selectively stimulated while having little or no effect on context-inappropriate targets. Harnessing and channeling of signals through such multi-protein complexes permits the cell to perform coordinate regulation of several simultaneous, possibly divergent, signals at multiple levels, with a remarkable degree of specificity.
This is of considerable physiological significance. Within the mammalian kidney, ERK1/2 expression can be detected in all nephron segments, and it plays an important role in many physiological (O'Brien et al, 2004) and pathological processes (Nagao et al, 2003). Consistent with tonic inhibition of Na+ transport by the ERK pathway, in contrast to other nephron segments, ERK1/2 appears to have basal activity in distal nephron cells (Fujita et al, 2004; Masaki et al, 2004). Components of the MAPK cascade (Raf-1, MEK1/2 and ERK1/2, in this case) exist in a multi-protein complex (Kyriakis, 2007). Recent evidence suggests the involvement of Nedd4-2 in this complex as well (Soundararajan et al, 2009). Together, these observations are consistent with the hypothesis that tonic ENaC inhibition by ERK1/2 and Nedd4-2 is implicated in setting baseline Na+ transport, and that aldosterone stimulates Na+ transport in part by increasing GILZ1 expression, which inhibits ERK1/2 signaling on one hand, and on the other, acts in concert with aldosterone-induced SGK1 to enhance ENaC-mediated Na+ current. As discussed in the next section, recent evidence also suggests that CNK3 functions through a similar mechanism, and indeed might be part of the ERC.
2.4 The ENaC-regulator CNK3
GILZ1 interacts with several ENaC-regulatory proteins, as well as with ENaC itself, and plays a key role in recruiting activating proteins into the complex. It is therefore of considerable interest to identify the spectrum of GILZ1-interacting partners or GILZIPs.
One such interesting candidate is the recently identified MR target gene CNKSR3 (connector enhancer of kinase suppressor of Ras 3) commonly referred to as CNK3. CNK3 is the third isoform of the mammalian CNK protein family, whose members possess a modular structure and contain common protein-protein interaction domains: an N-terminal sterile α motif (SAM), followed by a conserved region in CNK (CRIC) and a PDZ domain (Fig. 1C). In contrast to its homologs, CNK3 is smaller and lacks the part corresponding to their C-terminal moieties that harbor a pleckstrin homology (PH) domain (Douziech et al, 2003; Therrien et al, 1998). Interestingly, IPCEF1 (also known as PIP3-E or CNK3B) corresponds to the C-terminus and is separated by less than 100 kb from the CNK3 locus on the same chromosome (Douziech et al, 2003). This led to the hypotheses that they might originate from the same gene and code for a hypothetical full length CNK3 protein. However, no overlapping transcripts have been identified and gene annotation programs predicted them as independent loci (Claperon & Therrien, 2007). In keeping with this prediction, unlike CNK3, IPCEF1 is not regulated by aldosterone in HEK-293 MR-expressing cells (unpublished data), further supporting the view that both loci are regulated in an independent manner.
CNK3, like SGK1 and GILZ1, is rapidly induced by physiological concentrations of aldosterone, and its promoter harbors two functional MR binding loci in close vicinity to the transcription start site, suggesting a direct mode of regulation (Ziera et al, 2009). Evidence that CNK3 might be involved in the mechanism of Na+ reabsorption came from qPCR expression analyses along different nephron segments microdissected from mice kidneys that revealed that CNK3 is highly expressed in the connecting tubule (CNT) and the cortical collecting duct (CCD), the prime target segments of aldosterone-regulated Na+ retention in the kidney (Loffing et al, 2001). In fact, CNK3 expression correlates with, and is required for, ENaC-mediated Na+ transport in renal epithelial cells (Ziera et al, 2009). The first study addressing the mechanistic aspects of CNK3 function revealed that CNK3 expression significantly interferes with the activation of the Raf-1/MEK1/2/ERK1/2 signaling cascade (Ziera et al, 2009), a mechanism that is reminiscent of how GILZ1 indirectly stimulates ENaC activity (Soundararajan et al, 2005).
Recently, Lim et al. isolated protein complexes of all three human CNK isoforms from HEK 293T cells and determined binding partners using mass spectrometry (Lim et al, 2010). Although these CNK complexes were seen to possess several common binding partners, key differences in their composition suggested functional diversity. Interestingly, no CNK3-selective binding target was identified under the conditions studied. Peptides from all three subunits of the heterotrimeric serine/threonine phosphatase PP2A were detected as the most abundant binding partners of CNK3 (Lim et al, 2010). It is conceivable that PP2A regulates the phosphorylation of CNK3 itself or that of one of its binding targets and thereby controls the composition and stoichiometry of an extended scaffolding complex. For instance, the phosphorylation of Ser-162 within the second PDZ domain of the scaffold protein Na+/H+ exchange regulatory factor (NHERF-1) negatively regulates its interaction with, and consequently the activity of, the CFTR chloride ion channel (Raghuram et al, 2003). The modulation of CNK3 through phosphorylation in addition to its transient (hormone-dependent) expression pattern in the collecting duct would confer CNK3 with ideal regulatory properties to dynamically respond to the ever-changing external milieu that the kidney is exposed to. CNK3 possesses, among other protein-protein interaction motifs, a PDZ domain, an attribute that has been shown to be critical for the formation of higher-order signaling networks at the plasma membrane (reviewed in (Brone & Eggermont, 2005; Nourry et al, 2003)). Interestingly, both SGK1 and GILZ1 harbor consensus Class I PBMs at their C-termini (Figs. 1A, B). It is possible that they interact with CNK3 via its classic PDZ domain. At this juncture, it is important to note that SGK1 has been previously reported to interact with PDZ domains of other large scaffold proteins such as NHERF2 (Yun et al, 2002). It is interesting to speculate whether aldosterone-induced CNK3 acts to assemble various ENaC-regulatory components in close vicinity of the channel and thereby exerts its stimulatory effects on channel function. A similar mechanism has been suggested for CNK1 as a positive regulator of insulin signaling. CNK1 helps to localize cytohesins at the cell surface to generate a PIP2-rich microenvironment, which is critical for PI3K/AKT signal transduction (Lim et al, 2010).
2.5 A coordinate working model for aldosterone-dependent ENaC regulation
Aldosterone-induced transepithelial Na+ transport via ENaC involves the coordinate functioning of stimulatory signaling proteins such as SGK1, GILZ1 and CNK3, with inhibitory proteins, such as Nedd4-2 and ERK1/2. Our current hypothesis involves CNK3 as a central hormone-induced scaffolding platform, that possibly aids in the assembly of the ENaC-regulatory complex, thereby promoting appropriate signal transduction (Fig. 2). It is not clear at this point which of these interactions are direct, or what is the full scope of this dynamic multi-protein complex. The precise composition of the ERC, and the molecular mechanisms governing the role of CNK3 and its scaffolding functions in the context of aldosterone signaling are also important issues that remain to be elucidated.
Figure 2. Schematic depiction of hypothetical mechanism for ERC regulation of ENaC.
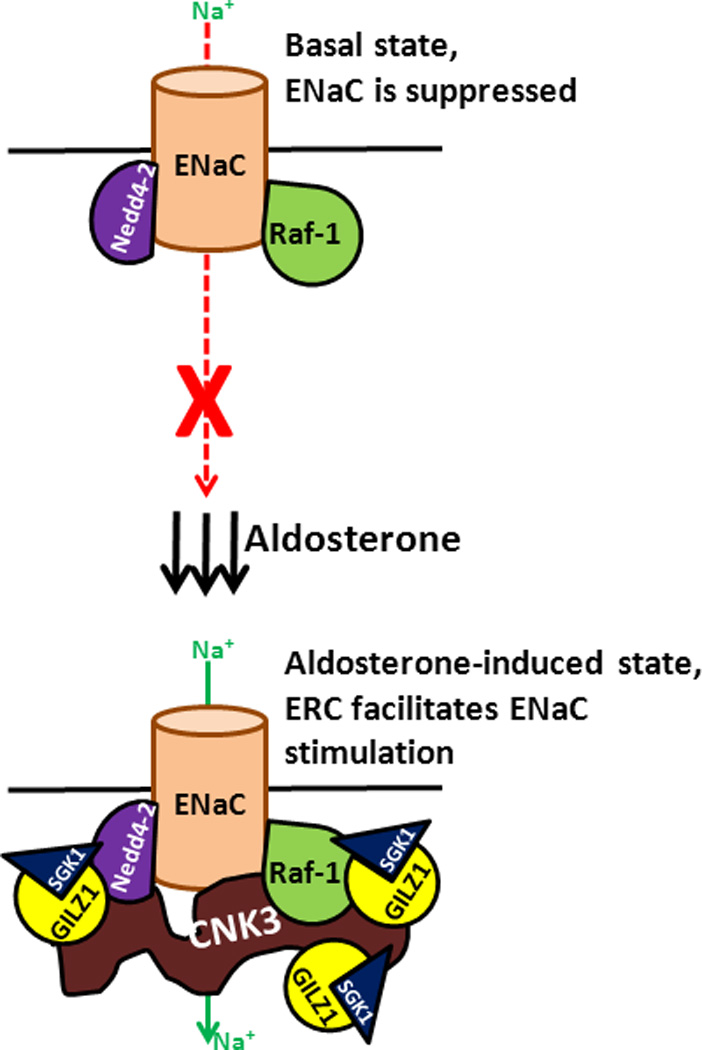
In the absence of aldosterone, ENaC activity is limited by Raf-1 and Nedd4-2, the two major negative regulatory components governing ENaC, within the ERC. In the absence of GILZ1 and SGK1, this regulatory complex is thus inhibitory. Aldosterone-induced SGK1 and GILZ1 act together to stimulate ENaC by interfering with the inhibitory components of this complex. Aldosterone simultaneously also markedly upregulates the expression of CNK3, a PDZ domain-containing scaffold protein that assists in the assembly and functioning of an ENaC-stimulatory multi-protein complex, that includes GILZ1 and SGK1. The net result is increased cell surface expression and activity of ENaC. Red and green arrows represent Na+ movement through the channel, while black arrows represent aldosterone action. Dashed red arrow shows ENaC in the “inhibited” state (red ‘X’, top panel), while solid green arrow in the bottom panel shows the activated or “disinhibited” state of ENaC.
Highlights.
>The ENaC-regulatory complex (ERC). >Its composition and function in regulation of ENaC. >Role of GILZ1 as a key aldosterone-induced modifier of complex composition. >CNK3 as a molecular scaffold assisting in assembly and functioning of the ERC. >Hypothetical model for aldosterone-induced ENaC activity.
Acknowledgements
This work is supported by National Institutes of Health grants K01-DK078679 (to RS), R01-DK56695 (to DP), and R01-DK85101 (to DP).
Abbreviations
- GILZ1
glucocorticoid-induced leucine zipper protein-1
- CCD
cortical collecting duct
- ER
endoplasmic reticulum
- ENaC
epithelial sodium channel
- SGK1
serum- and glucocorticoid-induced kinase-1
- CHIP
C-terminus of Hsc (heat-shock cognate protein) 70-interacting protein
- HRD1
HMG-CoA reductase degradation protein-1
- ERK
extracellular signal-regulated kinase
- MEK
mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- ERAD
ER-associated degradation
- PI3K
phosphatidylinositide 3’-kinase
- Nedd4-2
neural precursor cell expressed, developmentally downregulated protein
- CNK3
connector enhancer of kinase suppressor of Ras 3
- PDZ
PSD-95 - DLG-1 - ZO-1 domain
- PBM
PDZ domain-binding motif
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez de la Rosa D, Canessa CM. Role of SGK in hormonal regulation of epithelial sodium channel in A6 cells. Am J Physiol Cell Physiol. 2003;284:C404–C414. doi: 10.1152/ajpcell.00398.2002. [DOI] [PubMed] [Google Scholar]
- Alvarez de la Rosa D, Zhang P, Naray-Fejes-Toth A, Fejes-Toth G, Canessa CM. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999;274:37834–37839. doi: 10.1074/jbc.274.53.37834. [DOI] [PubMed] [Google Scholar]
- Arteaga MF, Wang L, Ravid T, Hochstrasser M, Canessa CM. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitin-conjugation machinery. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11178–11183. doi: 10.1073/pnas.0604816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselin-Labat ML, Biola-Vidamment A, Kerbrat S, Lombes M, Bertoglio J, Pallardy M. FoxO3 mediates antagonistic effects of glucocorticoids and interleukin-2 on glucocorticoid-induced leucine zipper expression. Mol Endocrinol. 2005;19:1752–1764. doi: 10.1210/me.2004-0206. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, Pallardy M. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. 2004;104:215–223. doi: 10.1182/blood-2003-12-4295. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, D'Adamio F, Riccardi C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. Faseb J. 2009;23:3649–3658. doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. The Journal of clinical investigation. 2007;117:1605–1615. doi: 10.1172/JCI30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002;22:7929–7941. doi: 10.1128/MCB.22.22.7929-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava A, Pearce D. Mechanisms of mineralocorticoid action: determinants of receptor specificity and actions of regulated gene products. Trends Endocrinol Metab. 2004;15:147–153. doi: 10.1016/j.tem.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Bhargava A, Wang J, Pearce D. Regulation of epithelial ion transport by aldosterone through changes in gene expression. Molecular and cellular endocrinology. 2004;217:189–196. doi: 10.1016/j.mce.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Bogusz AM, Brickley DR, Pew T, Conzen SD. A novel N-terminal hydrophobic motif mediates constitutive degradation of serum- and glucocorticoid-induced kinase-1 by the ubiquitin-proteasome pathway. Febs J. 2006;273:2913–2928. doi: 10.1111/j.1742-4658.2006.05304.x. [DOI] [PubMed] [Google Scholar]
- Brone B, Eggermont J. PDZ proteins retain and regulate membrane transporters in polarized epithelial cell membranes. Am J Physiol Cell Physiol. 2005;288:C20–C29. doi: 10.1152/ajpcell.00368.2004. [DOI] [PubMed] [Google Scholar]
- Cao L, Owsianik G, Becq F, Nilius B. Chronic exposure to EGF affects trafficking and function of ENaC channel in cystic fibrosis cells. Biochem Biophys Res Commun. 2005;331:503–511. doi: 10.1016/j.bbrc.2005.03.201. [DOI] [PubMed] [Google Scholar]
- Chen SY, Bhargava A, Mastroberardino L, Meijer OC, Wang J, Buse P, Firestone GL, Verrey F, Pearce D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2514–2519. doi: 10.1073/pnas.96.5.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claperon A, Therrien M. KSR and CNK: two scaffolds regulating RAS-mediated RAF activation. Oncogene. 2007;26:3143–3158. doi: 10.1038/sj.onc.1210408. [DOI] [PubMed] [Google Scholar]
- D'Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. Embo J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel's alpha-subunit. J Biol Chem. 2004;279:38134–38142. doi: 10.1074/jbc.M403260200. [DOI] [PubMed] [Google Scholar]
- Douziech M, Roy F, Laberge G, Lefrancois M, Armengod AV, Therrien M. Bimodal regulation of RAF by CNK in Drosophila. EMBO J. 2003;22:5068–5078. doi: 10.1093/emboj/cdg506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes-Toth G, Frindt G, Naray-Fejes-Toth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. American journal of physiology. 2008;294:F1298–F1305. doi: 10.1152/ajprenal.00579.2007. [DOI] [PubMed] [Google Scholar]
- Flores SY, Loffing-Cueni D, Kamynina E, Daidie D, Gerbex C, Chabanel S, Dudler J, Loffing J, Staub O. Aldosterone-induced serum and glucocorticoid-induced kinase 1 expression is accompanied by Nedd4-2 phosphorylation and increased Na+ transport in cortical collecting duct cells. J Am Soc Nephrol. 2005;16:2279–2287. doi: 10.1681/ASN.2004100828. [DOI] [PubMed] [Google Scholar]
- Fujita H, Omori S, Ishikura K, Hida M, Awazu M. ERK and p38 mediate high-glucose-induced hypertrophy and TGF-beta expression in renal tubular cells. American journal of physiology. 2004;286:F120–F126. doi: 10.1152/ajprenal.00351.2002. [DOI] [PubMed] [Google Scholar]
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Hendron E, Patel P, Hausenfluke M, Gamper N, Shapiro MS, Booth RE, Stockand JD. Identification of cytoplasmic domains within the epithelial Na+ channel reactive at the plasma membrane. J Biol Chem. 2002;277:34480–34488. doi: 10.1074/jbc.M204615200. [DOI] [PubMed] [Google Scholar]
- Kempe DS, Siraskar G, Frohlich H, Umbach AT, Stubs M, Weiss F, Ackermann TF, Volkl H, Birnbaum MJ, Pearce D, Foller M, Lang F. Regulation of renal tubular glucose reabsorption by Akt2/PKBss. American journal of physiology. 2010 doi: 10.1152/ajprenal.00592.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM. The integration of signaling by multiprotein complexes containing Raf kinases. Biochim Biophys Acta. 2007;1773:1238–1247. doi: 10.1016/j.bbamcr.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Lim J, Zhou M, Veenstra TD, Morrison DK. The CNK1 scaffold binds cytohesins and promotes insulin pathway signaling. Genes Dev. 2010;24:1496–1506. doi: 10.1101/gad.1904610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- Loffing J, Zecevic M, Feraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. American journal of physiology. 2001;280:F675–F682. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, Shokat KM, Ashrafi K, Pearce D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol. 2010;21:811–818. doi: 10.1681/ASN.2009111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Nikolic-Paterson DJ. Activation of the extracellular-signal regulated protein kinase pathway in human glomerulopathies. J Am Soc Nephrol. 2004;15:1835–1843. doi: 10.1097/01.asn.0000130623.66271.67. [DOI] [PubMed] [Google Scholar]
- McCormick JA, Bhalla V, Pao AC, Pearce D. SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology (Bethesda) 2005;20:134–139. doi: 10.1152/physiol.00053.2004. [DOI] [PubMed] [Google Scholar]
- McCormick JA, Yang CL, Ellison DH. WNK kinases and renal sodium transport in health and disease: an integrated view. Hypertension. 2008;51:588–596. doi: 10.1161/HYPERTENSIONAHA.107.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michlig S, Harris M, Loffing J, Rossier BC, Firsov D. Progesterone down-regulates the open probability of the amiloride-sensitive epithelial sodium channel via a Nedd4-2-dependent mechanism. J Biol Chem. 2005;280:38264–38270. doi: 10.1074/jbc.M506308200. [DOI] [PubMed] [Google Scholar]
- Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between alphaENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+ J Am Soc Nephrol. 2003;14:1107–1115. doi: 10.1097/01.asn.0000061777.67332.77. [DOI] [PubMed] [Google Scholar]
- Nagao S, Yamaguchi T, Kusaka M, Maser RL, Takahashi H, Cowley BD, Grantham JJ. Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int. 2003;63:427–437. doi: 10.1046/j.1523-1755.2003.00755.x. [DOI] [PubMed] [Google Scholar]
- Naray-Fejes-Toth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Toth G. sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial na+ channels. J Biol Chem. 1999;274:16973–16978. doi: 10.1074/jbc.274.24.16973. [DOI] [PubMed] [Google Scholar]
- Naray-Fejes-Toth A, Snyder PM, Fejes-Toth G. The kidney-specific WNK1 isoform is induced by aldosterone and stimulates epithelial sodium channel-mediated Na+ transport. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17434–17439. doi: 10.1073/pnas.0408146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourry C, Grant SG, Borg JP. PDZ domain proteins: plug and play! Sci STKE. 2003;2003:RE7. doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- O'Brien LE, Tang K, Kats ES, Schutz-Geschwender A, Lipschutz JH, Mostov KE. ERK and MMPs sequentially regulate distinct stages of epithelial tubule development. Dev Cell. 2004;7:21–32. doi: 10.1016/j.devcel.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Pao AC, McCormick JA, Li H, Siu J, Govaerts C, Bhalla V, Soundararajan R, Pearce D. NH2 terminus of serum and glucocorticoid-regulated kinase 1 binds to phosphoinositides and is essential for isoform-specific physiological functions. American journal of physiology. 2007;292:F1741–F1750. doi: 10.1152/ajprenal.00027.2007. [DOI] [PubMed] [Google Scholar]
- Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3- kinase-stimulated signaling pathway. Embo J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce D, Kleyman TR. Salt, sodium channels, and SGK1. The Journal of clinical investigation. 2007;117:592–595. doi: 10.1172/JCI31538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuram V, Hormuth H, Foskett JK. A kinase-regulated mechanism controls CFTR channel gating by disrupting bivalent PDZ domain interactions. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9620–9625. doi: 10.1073/pnas.1633250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi C, Cifone MG, Migliorati G. Glucocorticoid hormone-induced modulation of gene expression and regulation of T-cell death: role of GITR and GILZ, two dexamethasone-induced genes. Cell Death Differ. 1999;6:1182–1189. doi: 10.1038/sj.cdd.4400609. [DOI] [PubMed] [Google Scholar]
- Riccardi C, Zollo O, Nocentini G, Bruscoli S, Bartoli A, D'Adamio F, Cannarile L, Delfino D, Ayroldi E, Migliorati G. Glucocorticoid hormones in the regulation of cell death. Therapie. 2000;55:165–169. [PubMed] [Google Scholar]
- Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4025–4029. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Nicoud M, Flahaut M, Elalouf JM, Nicod M, Salinas M, Bens M, Doucet A, Wincker P, Artiguenave F, Horisberger JD, Vandewalle A, Rossier BC, Firsov D. Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2712–2716. doi: 10.1073/pnas.051603198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. Embo J. 1996;15:2381–2387. [PMC free article] [PubMed] [Google Scholar]
- Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, Garty H. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem. 2002;277:13539–13547. doi: 10.1074/jbc.M111717200. [DOI] [PubMed] [Google Scholar]
- Shigaev A, Asher C, Latter H, Garty H, Reuveny E. Regulation of sgk by aldosterone and its effects on the epithelial Na(+) channel. American journal of physiology. 2000;278:F613–F619. doi: 10.1152/ajprenal.2000.278.4.F613. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8. doi: 10.1074/jbc.C100623200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Melters D, Shih IC, Wang J, Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7804–7809. doi: 10.1073/pnas.0809892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan R, Pearce D, Hughey RP, Kleyman TR. Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem. 2010a;285:30363–30369. doi: 10.1074/jbc.R110.155341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan R, Wang J, Melters D, Pearce D. Differential Activities of Glucocorticoid-induced Leucine Zipper Protein Isoforms. J Biol Chem. 2007;282:36303–36313. doi: 10.1074/jbc.M707287200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Wang J, Melters D, Pearce D. Glucocorticoid-induced Leucine Zipper 1 Stimulates the Epithelial Sodium Channel by Regulating Serum- and Glucocorticoid-induced Kinase 1 Stability and Subcellular Localization. J Biol Chem. 2010b;285:39905–39913. doi: 10.1074/jbc.M110.161133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundararajan R, Zhang TT, Wang J, Vandewalle A, Pearce D. A novel role for glucocorticoid-induced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem. 2005;280:39970–39981. doi: 10.1074/jbc.M508658200. [DOI] [PubMed] [Google Scholar]
- Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. Embo J. 1996;15:2371–2380. [PMC free article] [PubMed] [Google Scholar]
- Stockand JD, Spier BJ, Worrell RT, Yue G, Al-Baldawi N, Eaton DC. Regulation of Na(+) reabsorption by the aldosterone-induced small G protein K-Ras2A. J Biol Chem. 1999;274:35449–35454. doi: 10.1074/jbc.274.50.35449. [DOI] [PubMed] [Google Scholar]
- Therrien M, Wong AM, Rubin GM. CNK, a RAF-binding multidomain protein required for RAS signaling. Cell. 1998;95:343–353. doi: 10.1016/s0092-8674(00)81766-3. [DOI] [PubMed] [Google Scholar]
- Thomas CP, Itani OA. New insights into epithelial sodium channel function in the kidney: site of action, regulation by ubiquitin ligases, serum- and glucocorticoid-inducible kinase and proteolysis. Current opinion in nephrology and hypertension. 2004;13:541–548. doi: 10.1097/00041552-200409000-00010. [DOI] [PubMed] [Google Scholar]
- Vallon V, Lang F. New insights into the role of serum- and glucocorticoid-inducible kinase SGK1 in the regulation of renal function and blood pressure. Curr Opin Nephrol Hypertens. 2005;14:59–66. doi: 10.1097/00041552-200501000-00010. [DOI] [PubMed] [Google Scholar]
- Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl-cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. American journal of physiology. 2009;297:F704–F712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Barbry P, Maiyar AC, Rozansky DJ, Bhargava A, Leong M, Firestone GL, Pearce D. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. American journal of physiology. 2001;280:F303–F313. doi: 10.1152/ajprenal.2001.280.2.F303. [DOI] [PubMed] [Google Scholar]
- Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15603–15608. doi: 10.1073/pnas.0407008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MK, Goya L, Firestone GL. Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J Biol Chem. 1993a;268:11482–11485. [PubMed] [Google Scholar]
- Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993b;13:2031–2040. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, Jansen M, Schlunz M, Klingel K, Loffing J, Kauselmann G, Bosl MR, Lang F, Kuhl D. Impaired renal Na(+) retention in the sgk1-knockout mouse. The Journal of clinical investigation. 2002;110:1263–1268. doi: 10.1172/JCI15696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CC, Chen Y, Lang F. Glucocorticoid activation of Na(+)/H(+) exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J Biol Chem. 2002;277:7676–7683. doi: 10.1074/jbc.M107768200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. The Journal of clinical investigation. 2007;117:773–783. doi: 10.1172/JCI29850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziera T, Irlbacher H, Fromm A, Latouche C, Krug SM, Fromm M, Jaisser F, Borden SA. Cnksr3 is a direct mineralocorticoid receptor target gene and plays a key role in the regulation of the epithelial sodium channel. Faseb J. 2009;23:3936–3946. doi: 10.1096/fj.09-134759. [DOI] [PubMed] [Google Scholar]