

Abstract
The protozoan parasite Trypanosoma brucei is the causative agent of the cattle disease Nagana and human African sleeping sickness. Glycoproteins play key roles in the parasite’s survival and infectivity, and the de novo biosyntheses of the sugar nucleotides UDP-galactose (UDP-Gal), UDP-N-acetylglucosamine, and GDP-fucose have been shown to be essential for their growth. The only route to UDP-Gal in T. brucei is through the epimerization of UDP-glucose (UDP-Glc) by UDP-Glc 4′-epimerase. UDP-Glc is also the glucosyl donor for the unfolded glycoprotein glucosyltransferase (UGGT) involved in glycoprotein quality control in the endoplasmic reticulum and is the presumed donor for the synthesis of base J (β-d-glucosylhydroxymethyluracil), a rare deoxynucleotide found in telomere-proximal DNA in the bloodstream form of T. brucei. Considering that UDP-Glc plays such a central role in carbohydrate metabolism, we decided to characterize UDP-Glc biosynthesis in T. brucei. We identified and characterized the parasite UDP-glucose pyrophosphorylase (TbUGP), responsible for the formation of UDP-Glc from glucose-1-phosphate and UTP, and localized the enzyme to the peroxisome-like glycosome organelles of the parasite. Recombinant TbUGP was shown to be enzymatically active and specific for glucose-1-phosphate. The high-resolution crystal structure was also solved, providing a framework for the design of potential inhibitors against the parasite enzyme.
Keywords: kinetoplastids, sugar nucleotide metabolism, Trypanosoma brucei, UDP-glucose, UDP-glucose pyrophosphorylase
Introduction
Human African trypanosomiasis (HAT), commonly known as “sleeping sickness”, is a vector borne-parasitic disease caused by the kinetoplastid Trypanosoma brucei. The disease progresses through two stages following an asymptomatic period of several weeks or months. The early stage is usually characterized by malaria-like symptoms, including fatigue, headache, recurrent fever, and swollen lymph nodes. In advanced stages, the disease affects the central nervous system, causing severe neurological and mental disorders and making the individual dependent on others. Infected individuals are weakened, often for many years, causing economic loss, poverty, and social misery. HAT is completely fatal if untreated, and it constitutes a major public health problem in sub-Saharan Africa (Favre et al. 2008). Given the resurgence of both human and animal trypanosomiasis, its epidemic potential, high fatality rate, and significant impact on socioeconomic development, there is a clear need for new therapeutics to control the disease.
The trypanosome life cycle alternates between a mammalian host and the tsetse fly Glossina spp., and the different life cycle stages are adapted to survive in each host. In the host-dwelling bloodstream form of the parasite, a protective coat of 5 × 106 glycosylphosphatidylinositol (GPI)-anchored variant surface glycoprotein (VSG) homodimers is expressed on the plasma membrane. The parasite survives the immune attack of the host because it undergoes antigenic variation, a process that involves replacement of the VSG coat by another composed of antigenically different VSG molecules (Cross 1996). In addition to VSG, the bloodstream form parasite also expresses less abundant but equally essential glycoproteins, such as the transferrin receptor and the lysosomal p67 glycoprotein (Kelley et al. 1999; Alexander et al. 2002). This has lead to the investigation of potential therapeutic targets against parasite glycoprotein biosynthesis, such as enzymes of GPI and sugar nucleotide biosynthesis (Turnock et al. 2007; Turnock and Ferguson 2007).
Sugar nucleotides are the ultimate source of sugar for the majority of glycosylation reactions. They are formed in two main ways: by salvage pathways, involving activation of the sugar using a kinase and subsequent condensation with a nucleotide via a pyrophosphorylase, or by de novo pathways, involving the bioconversion of an existing sugar/sugar nucleotide. In most cases, sugar nucleotides are synthesized in the cytoplasm and used there and/or transported through specific transporters into the lumen of the Golgi apparatus and/or endoplasmic reticulum (ER) where they are used by glycosyltransferases as donor substrates in glycosylation reactions (Freeze and Elbein 2008). In T. brucei, our knowledge of sugar nucleotide biosynthesis has expanded in the last few years and it has been shown that several steps in the de novo biosynthesis of GDP-fucose (Turnock et al. 2007), UDP-galactose (UDP-Gal) (Roper et al. 2002, 2005; Urbaniak et al. 2006), and UDP-N-acetylglucosamine (UDP-GlcNAc) (Stokes et al. 2008) occur in the glycosome and are essential for parasite growth. However, several issues remain unsolved, including the pathway to and location of UDP-glucose (UDP-Glc) biosynthesis.
UDP-Glc is the sugar nucleotide acting as a donor of glucose in diverse biochemical pathways and is a central metabolite both in prokaryota and eukaryota. In eukaryotic cells, UDP-Glc is essential in the synthesis of diverse glucose-containing glycolipids, glycoproteins, and a variety of secondary metabolites (Flores-Diaz et al. 1997). Furthermore, it plays a crucial role for the “quality control” of newly synthesized glycoproteins taking place in the ER (Hammond and Helenius 1995). Thus, UDP-Glc is the glucosyl donor for the unfolded glycoprotein glucosyltransferase (UGGT) involved in the calnexin- and/or calreticulin-mediated glycoprotein quality control refolding cycles in the ER. In T. brucei, UGGT has been shown to be essential for parasite growth and survival at 40°C, and a UGGT null mutant was hypersensitive to the effects of the N-glycosylation inhibitor tunicamycin (Izquierdo et al. 2009). UDP-Glc is also the presumed donor for the synthesis of base J (β-d-glucosylhydroxymethyluracil), a rare deoxynucleotide of unknown function found in telomere-proximal DNA in the bloodstream form of T. brucei (van Leeuwen et al. 1998).
The de novo UDP-Glc biosynthetic pathway involves three steps starting from glucose: (a) generation of Glc-6-P, via a hexokinase activity; (b) transfer of the phosphate group from position 6 to position 1, through the action of a phosphoglucomutase; and (c) coupling of Glc-1P to UTP. This last, reversible, step is catalyzed by UDP-Glc pyrophosphorylase (UGP; EC 2.7.7.9).
In this paper, we confirm that the putative T. brucei UDP-Glc pyrophosphorylase (TbUGP) gene (GeneDB ID: Tb10.389.0330) encodes a functional and highly specific UGP enzyme, localize native TbUGP to the glycosomes and describe its high-resolution crystal structure in complex with UDP-Glc.
Results
Identification and expression of TbUGP
A BLASTp search of the T. brucei-predicted protein database with Homo sapiens, Saccharomyces cerevisiae, and Leishmania major UDP-Glc pyrophosphorylase (UGP) amino acid sequences (NCBI accession nos. NP_001001521, NP_011851, and XP_001682505.1, respectively) revealed a single putative TbUGP gene (GeneDB ID: Tb10.389.0330). This single putative TbUGP locus was also confirmed by Southern blotting of restriction endonuclease digested genomic DNA (data not shown). The predicted amino acid sequence of Tb10.389.0330 contains the highly conserved nucleotide binding (NB) loop (KLNGGLGTXMGX4K) and 16 of the 17 residues shown to be involved in substrate binding (SB) in the UGP crystal structures from other organisms (Figure 1) (Lamerz et al. 2006). The only residue involved in catalytic activity that is different in the T. brucei sequence is Gly221, which is an Ile in the H. sapiens and Bos taurus orthologues. TbUGP was found to have 37% amino acid identity with the human enzyme.
Fig. 1.

Alignment of UDP-Glc pyrophosphorylase predicted amino acid sequences. The sequences of T. brucei, T. cruzi, L. major, Rattus norvegicus, H. sapiens and Candida albicans were aligned using Clustal W (http://www.ebi.ac.uk/) and Jalview (http://www.jalview.org/). Residues participating in contacts with the nucleoside (black circles), phosphate (triangles), and glucose residue (square) are highlighted. The highly conserved nucleotide-binding (pyrophosphorylase motif) loop (dashed line) and substrate-binding loop (full line) are boxed
The TbUGP open reading frame was amplified from T. brucei strain 427 genomic DNA by polymerase chain reaction (PCR) using a high-fidelity polymerase, and the consensus sequence (EMBL nucleotide sequence database accession no. FN662556) predicts a 485-amino acid protein of 54,507 Da. The same sequence was also obtained when the gene was amplified from cDNA (EMBL nucleotide sequence database accession no. FN662557) using the same TbUGP 3′-primer and a 5′-spliced leader primer described in Materials and methods. T. brucei genes are generally free of introns and are transcribed in polycistronic units that are processed by trans-splicing with a common 35-nucleotide “miniexon” or “spliced leader” sequence (LeBowitz et al. 1993). Comparison of the cDNA and genomic sequences allowed us to determine the precise site of trans-splicing for this gene, which, as expected, occurs in a polypyrimidine tract, 113 bases upstream of the ATG codon. The predicted amino acid sequence for TbUGP from T. brucei strain 427 was almost identical to that in the T. brucei strain 927 genome database, except for substitution of Ser for Phe at position 54 and Ser for Asn at position 473. Neither residue is believed to be involved in the active site.
Semi quantitative RT (reverse transcription)–PCR, using RNA from T. brucei bloodstream and procyclic form, showed that the TbUGP mRNA is present in both life cycle stages (Figure 2A).
Fig. 2.
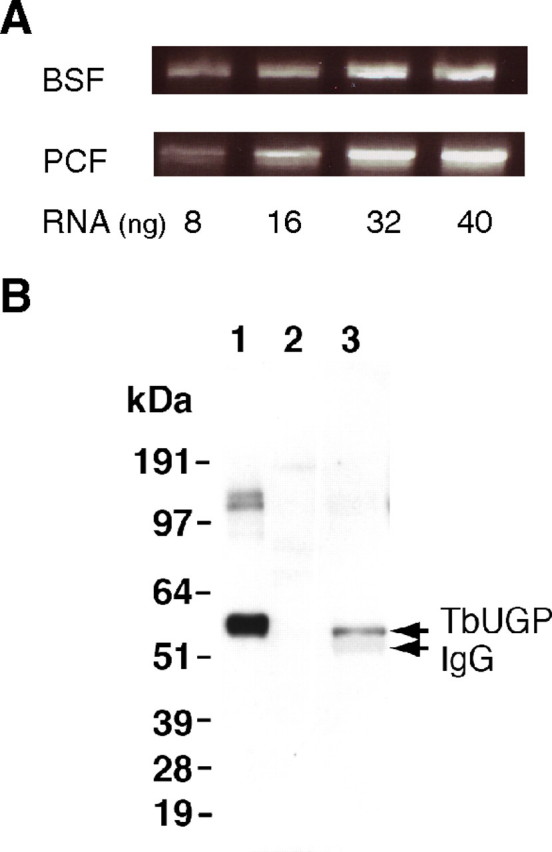
Detection of TbUGP mRNA and TbUGP protein in T. brucei. (A) RT–PCR reaction using 8, 16, 32 and 40 ng of RNA from T. brucei bloodstream (BSF) or procyclic (PCF) form parasites. (B) TbUGP western blot. Lane 1, E. coli recombinant TbUGP (25 ng); lane 2, BSF total lysate (5 × 106 cell equivalent) and lane 3, TbUGP immunoprecipitate (2 × 108 cell equivalent). The top arrow on the right indicates the native TbUGP, which has a slightly lower molecular weight than the recombinant protein due to the His6 tag, and the faint band underneath is the heavy chain of IgG used in the immunoprecipitation. The molecular weight standards (kDa) are shown on the left
Polyclonal antibodies against the recombinant enzyme (see Materials and methods) were raised in mice and affinity purified against immobilized recombinant protein. These antibodies failed to detect any relevant cross-reactivity with other proteins present in the cell (Figure 2B, lane 2) and due to the low abundance of TbUGP in the cell, it could only be detected in immunoprecipitates of 2 × 108 bloodstream form cells (Figure 2B, lane 3). These results show the monospecificity of the antibodies used in the cellular localization experiment (Figure 5) and also suggest that TbUGP protein expression level is similar to other enzymes involved in sugar nucleotide biosynthesis (Roper et al. 2002).
Fig. 5.
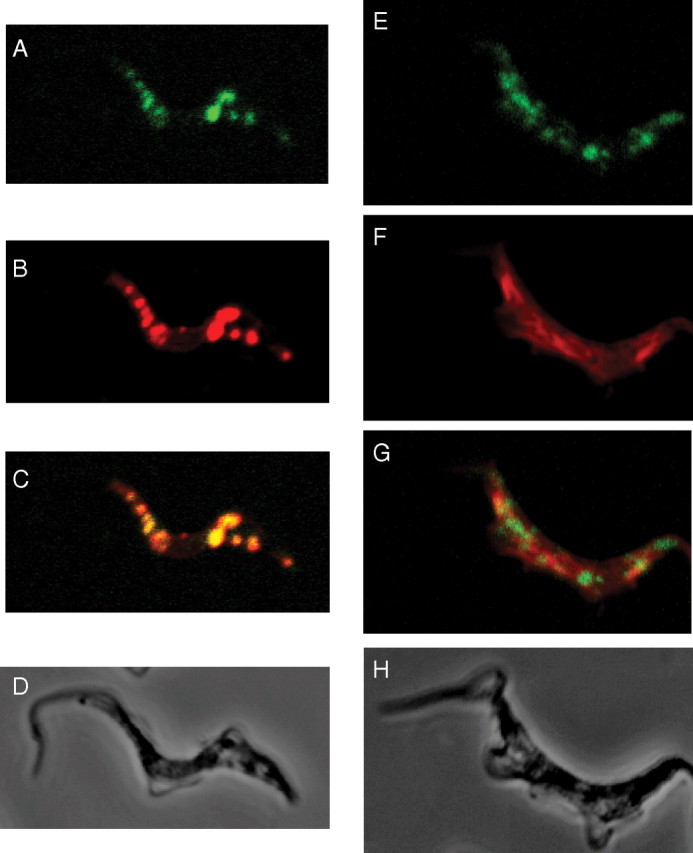
Subcellular localization of TbUGP. Wild-type bloodstream form T. brucei cells were stained with affinity-purified mouse anti-TbUGP and Alexa 488-conjugated anti-mouse antibody (green channel, panels A, E) and with rabbit anti-GAPDH and Alexa 594-conjugated anti-rabbit antibody (red channel, B) to mark the glycosomes, or rabbit anti-enolase and Alexa 594-conjugated anti-rabbit antibody (red channel, F) to mark the cytosol. Merged images are shown in panels (C, G) and corresponding phase contrast images are shown in panels (D, H)
Expression of monomeric TbUGP in Escherichia coli
The TbUGP ORF was cloned into a modified pET15b vector so that it contains a PreScission™ protease site between the protein and the His6 tag. This plasmid was expressed in E. coli and the protein purified as described in Materials and methods (Figure 3A). After purification by nickel affinity chromatography, the recombinant TbUGP eluted as a single peak by gel filtration with a retention time consistent with a monomeric 55 kDa protein. Protein with the His6 tag removed by PreScission™ protease also eluted as a monomer by gel filtration, indicating that the N-terminal His6 tag does not influence quaternary organization. Since oligomerization has a key role in the regulation of activity for other UGPs (Martz et al. 2002), UDP-Glc synthetic activity was assayed in each fraction from gel filtration and this demonstrated that the monomeric state is active. Furthermore, analysis of the pooled active fractions of recombinant His6-TbUGP protein by analytical ultracentrifugation (at 0.75 mL mg/mL) indicated that the majority of TbUGP is monomeric at this concentration (Figure 3B), although some dimer and possibly higher oligomers were detected. The ultracentrifugation data were unaffected by the presence or absence of UTP and Glc-1-P (data not shown). This result is in agreement with the data previously published for L. major and barley UGP, which were also found to be active as monomers (Martz et al. 2002; Lamerz et al. 2006; Steiner et al. 2007) but is in contrast with human UGP which is octameric (Chang et al. 1996).
Fig. 3.
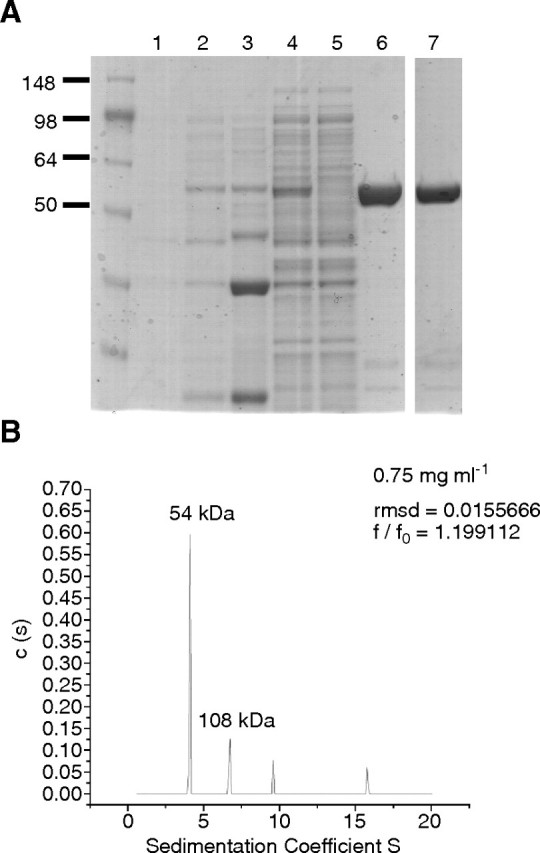
Purification and characterization of recombinant His6-TbUGP. (A) Coomassie blue-stained SDS–PAGE gel of proteins in uninduced E. coli cells (lane 1) and in IPTG-induced E. coli cells (lanes 2–7); total protein (lane 2), pellet after lysis and centrifugation (lane 3), supernatant after lysis and centrifugation (lane 4), flow through after Ni-affinity chromatography (lane 5), His6-TbUGP purified by Ni-affinity chromatography and elution with imidazole (lane 6). Lane 7, His6-TbUGP further purified by gel filtration. (B) Analytical ultracentrifugation profile of recombinant TbUGP at a final concentration of 0.75 mg/mL
TbUGP enzyme kinetics and specificity
We measured the TbUGP forward reaction (i.e., the production of UDP-Glc and pyrophosphate [PPi] from Glc-1-P and UTP) using recombinant His6-TbUGP. The reactions were started by adding the enzyme and stopped by boiling. After adding an excess of 2 mM NaOH, aliquots of the resulting solutions were analyzed by anion exchange high-performance liquid chromatography (HPLC), as described in Materials and methods, allowing the resolution and detection by ultraviolet (UV) absorbance of UDP-Glc and UTP. Using this method, we determined the optimal pH for the TbUGP forward reaction to be between pH 6.0 and 7.5 (Figure 4A) and showed that TbUGP was, as expected, divalent cation-dependent (with an optimal assay condition of 10 mM Mg2 +). The enzyme could also be activated by Mn2 + and Co2 +, but not by Zn2 + or Cu2 + (data not shown).
Fig. 4.
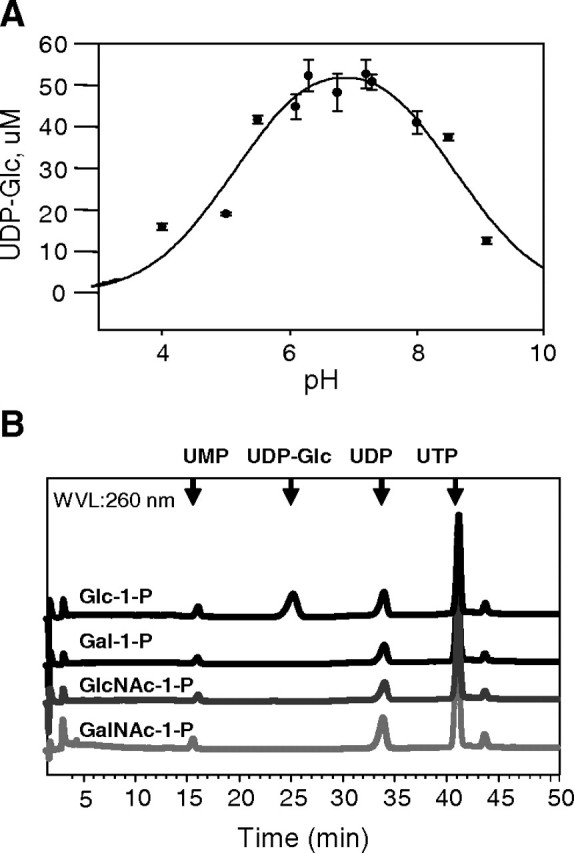
pH optimum and substrate specificity of recombinant TbUGP. (A) The pH optimum of TbUGP was determined by comparing the yields of UDP-Glc using the HPLC assay. The error bars indicate the SD values (n = 3). UDP-Glc was quantified by comparison with a UDP-Glc calibration curve. (B) The specificity of TbUGP was analyzed using the HPLC assay and different sugar-1-phosphate substrates, as indicated. Only Glc-1-P produced a UV-absorbing peak in the sugar nucleotide region of the chromatogram
The substrate specificity of TbUGP was also defined with the HPLC assay using a variety of sugar-1-phosphate substrates (Figure 4B). Using Glc-1-P as a substrate, a single peak was observed with a retention time of 25.5 min, coeluting with an authentic UDP-Glc standard. No sugar nucleotide peaks were observed in the absence of TbUGP or when GalNAc-1-P, GlcNAc-1-P or Gal-1-P was used as a substrate. These data showed that Glc-1-P was the preferred substrate of TbUGP under these conditions.
The purified recombinant TbUGP protein was then kinetically characterized for the forward reaction using a discontinuous colorimetric coupled assay (Stokes et al. 2008). In this case, the PPi product is converted to inorganic phosphate (Pi) by pyrophosphatase, and the Pi is subsequently reacted with malachite green. Using this assay, the specific activity and apparent Km values for UTP and Glc-1-P were determined. The linearities of the Lineweaver–Burke plots showed that the T. brucei enzyme obeyed simple Michaelis–Menten kinetics for both substrates. The specific activity of the recombinant TbUGP was 5.2 × 104 μmol PPi released/min mg protein, and the apparent Km values for UTP and Glc-1-P were 53 ± 8 μM and 122 ± 17 μM, respectively (data not shown). These values of are well within the normal ranges reported for UGPs from other eukaryotic organisms, including L. major UGP (Lamerz et al. 2006).
Subcellular localization of TbUGP in bloodstream form T. brucei
Localization studies were conducted using affinity-purified polyclonal mouse anti-TbUGP antibody together with either rabbit anti-GAPDH antibody (as a glycosomal marker) or rabbit anti-enolase (as a cytosolic marker). The secondary antibodies were anti-mouse Alexa 488 (green) and anti-rabbit Alexa 594 (red). The data show substantial co-localization of the anti-TbUGP with anti-GAPDH signals (Figure 5A, B, C) and an absence of co-localization of the anti-TbUGP with anti-enolase signals (Figure 5E, F, G). These data indicate that TbUGP localizes to the glycosome microbodies of bloodstream form T. brucei. Interestingly, although all of the GAPDH-staining glycosomes appear to also contain TbUGP (compare Figure 5A and B), the ratio of TbUGP to GAPDH staining may be variable. Although the majority of TbUGP staining is glycosomal, we cannot rule out that some TbUGP may be also present in the cytosol.
Crystal structure of recombinant TbUGP
The high-resolution crystal structure of a TbUGP construct lacking only the N-terminal Met-Pro sequence and complexed with UDP-Glc was solved (PDB accession no. 3GUE) by molecular replacement using the L. major UGP/UDP-Glc complex closed conformation structure (Steiner et al. 2007). The crystallographic statistics related to the structure determination of TbUGP and its complex with UDP-Glc are listed in Table I. The asymmetric unit contains two protein chains, labeled A and B. The crystal contacts between A and B are not substantial. Several crystal contact interfaces are mediated by moieties from the crystallization buffer (i.e., glycerol, PEG, and sulfate), and thus protein subunit oligomerization is not implied by the crystallographic data (Figure 6A). Several surface loops and residues were not modeled due to insufficient electron density. These include the first eight N-terminal residues in both chains and residues between Asp264 and Gln272 in monomer A. In the case of monomer B, the areas not modeled are (corresponding to PDB numbering) B61–B70, B123–124, B266–B271, and the carboxy-terminal residue B483. Because monomer A is more complete, we use herein this model for our analysis. Although the resolution is fairly high, no peaks corresponding to magnesium could be found in the electron density.
Table I.
Crystallographic data of TbUGP (PDB accession no. 3GUE)
| Data collection | ||
|---|---|---|
| Space group | C2 | |
| Cell dimensions | a (Å) | 167.7 |
| b (Å) | 77.49 | |
| c (Å) | 112.21 | |
| alpha | 90.00 | |
| beta | 117.88 | |
| gamma | 90.00 | |
| Wavelength | 1.5418 | |
| Resolution | 25–1.92 (1.99–1.92) | |
| Measured reflections | 1,716,578 | |
| Unique reflections | 96,124 | |
| Rsym | 0.120 (0.706) | |
| I/sigI | 10.38 (1.33) | |
| Completeness (%) | 98.7 (90.6) | |
| Redundancy | 3.1 (3.0) | |
| PDB code | 3GUE | |
| Refinement | ||
| Resolution | 25.0–1.92 | |
| Number of reflections | 91,276 | |
| Test set | 4791 | |
| Rwork/Rfree | 0.243/0.294 | |
| Number of atoms | 8155 | |
| Mean Bfactor | 27.931 | |
| Ramachandran favored (%) | 97.81 | |
| Ramachandran disallowed (%) | 0 | |
| Root mean square deviations | Bond lengths (Å) | 0.016 |
| Bond angles (°) | 1.519 |
Fig. 6.

Crystal structure of TbUGP in complex with UDP-Glc. (A) Representation of the dimer observed as an artifact of crystallization. (B) Illustration of the three classical UGP domains present in TbUGP: the N-terminal domain (green), the catalytic central domain (red), and the C-terminal domain (yellow). (C) Overall structure comparison of TbUGP (green) with the unbound (yellow) and bound (cyan) forms of LmUGP. Arrow 1 shows the additional 12 degree closure of TbUGP onto substrate compared to both open and closed conformations of LmUGP. The actual active site is aligned almost perfectly with the closed LmUGP structure. Arrow 2 shows the substrate-binding loop (SB loop) aligning well with the closed conformation. Arrow 3 shows the nucleotide-binding loop (NB loop), similar to the one in LmUGP. (D) Electrostatic potential of TbUGP showing the extensive basic patch covering the substrate-binding pocket. Images were generated by Pymol
The TbUGP structure is similar to those of other previously described eukaryotic UGP structures (Roeben et al. 2006; McCoy et al. 2007; Steiner et al. 2007). It contains an N-terminal domain (amino acids Pro2–Thr45, Asn164–Val188, Pro328–Ala352, green) and a C-terminal domain (Asp388–Glu485, yellow) flanking the central catalytic domain (red, Figure 6B). The central catalytic domain forms a Rossman fold consisting of an eight-stranded mixed beta-sheet, flanked by an outer layer of helices that then branches off into the C-terminal left-handed beta helix (residues 408–485), containing a strongly hydrophobic core. Density is missing in the TbUGP data for residues Asp264–Gln272, which are highly solvent exposed due to crystal packing. This missing area corresponds to a flexible, protruding loop (Arg261–Leu280) also found in the L. major structure (Figure 6C).
The structure was solved using the “closed” conformation of LmUGP, and we see that TbUGP is also in the closed conformation and appears to be even more firmly closed onto the substrate. Structure-based comparison with LmUGP (PDB accession no. 2OEG) reveals that TbUGP C-terminal domain (R331–L349) moves an additional 12 degrees towards the catalytic domain pocket compared to the LmUGP in closed configuration (Figure 6C). The active site is located in the central catalytic domain, in a pocket with distinctive positive electrostatic potential (Figure 6D).
The SB loop (Thr251–Gly258) aligns well with LmUGP which is expected since both structures were obtained in the presence of UDP-Glc. The NB loop also moves closer to the substrate pocket, however, less dramatically compared to the 12 degree motion of the C-terminus lobe. The LBH as well is compressed in towards the pocket. The overall effect of all these motions is to occlude the SB pocket, which can be seen in Figure 6C and D.
Active site of the UDP-Glc complex
The structure of TbUGP in complex with the product of the forward reaction, UDP-Glc, confirms the location of the active site of the enzyme (Figure 6B, C, D). TbUGP contains all of the crucial amino acids that were earlier implicated in catalysis or SB of UGPs from other eukaryotic species (Chang et al. 1996; Duggleby et al. 1996; McCoy et al. 2007). For instance, Lys367 of Solanum tuberosum UGP, located in the C-terminal domain and crucial for catalysis, corresponds to Lys377 in the T. brucei enzyme.
The UDP-Glc is bound in a deep cleft located at the center of the catalytic domain that consists of highly conserved residues. The SB loop (Thr251–Gly258) and the NB loop (Lys82–Lys97), both located in the central catalytic domain, are well conserved (Figure 1).
The uridine ring stays parallel to Gly85, Gly86, and Gly191, which probably fixes the ring by hydrophobic interactions. Gln163 side chain oxygen and nitrogen also contribute by interaction with the heteroatoms in the ring.
In the case of the ribose ring, O-2 seems to be interacting with the backbone nitrogen of Gly86 residue, and O-3 is interacting with the backbone nitrogen of Gly221. The rest of the interactions are involving water molecules so, as proposed for the L. major structure, analogues of UDP-Glc modified in the ribose residue could be inhibitory (Steiner et al. 2007).
The phosphates are fixed by a mixture of hydrogen bonds, with Lys377 stabilizing both the α-phosphate and the oxygen in the phosphate diester link. The α-phosphate also interacts closely with a water molecule and a glycerol that appears inside the active site (which is probably replaced by water in solution). The β-phosphate interactions include several water molecules, Lys 256 and His192.
The glucose residue is fixed via several hydrogen bonds, as observed in other UGPs. In this case, the OH-4 of glucose is involved in three different possible interactions, with Gly258 (with both the CO and N from the amide bond) and with Asn303; the OH-2 interacts with Glu281 and His192.
Accession numbers
Nucleotide sequences have been deposited in the EMBL nucleotide sequence database. Accession no. FN662556 corresponds to TbUGP ORF. Accession no. FN662557 corresponds to the sequence obtained by gene amplification from cDNA, as described in Materials and methods.
Coordinate and structure factors for TbUGP in complex with UDP-Glc have been deposited in the RCSB Protein Data Bank (www.pdb.org) with accession number 3GUE.
Discussion
UGPs have been purified and characterized from a wide range of prokaryotic and eukaryotic organisms, including Mycobacterium tuberculosis (Lai et al. 2008), Arabidopsis thaliana (McCoy et al. 2007), and B. taurus (Stevens and Phelps 1976). The eukaryotic enzymes have no significant sequence similarity to their prokaryotic counterparts and, whereas other enzymes of sugar nucleotide metabolism in T. brucei, like UDP-Glc 4′-epimerase, appear to have a bacterial origin (Roper et al. 2002), TbUGP clearly belongs of the eukaryotic family of UGPs and no putative bacterial UGP homologues could be found in the T. brucei genome. The kinetoplastid UGPs belong to their own phylogenic clade (Lamerz et al. 2006).
There is nothing particularly remarkable about the enzymatic properties of TbUGP, apart for its strict specificity for Glc-1-P which differentiates it from the UGPs of the parasite’s human and bovine hosts that can also utilize Gal-1-P for the synthesis of UDP-Gal (Turnquist et al. 1974). The molecular geometries observed in the TbUGP crystal structure around the OH-2 and OH-4 of the glucose residue of bound UDP-Glc are probably responsible for its strict substrate specificity. Thus, the tight network of interactions with the OH-4 of the glucose residue explains why UDP-Gal (an epimer of UDP-Glc at the OH-4 position) is not a substrate, and the interactions of the OH-2 of the glucose residue with residues Glu281 and His192 provide the steric constraints that prevent the binding of 2-deoxy-2-N-acetamido sugars (as in UDP-GlcNAc and UDP-GalNAc). The strict sugar-specific substrate specificity of TbUGP is reminiscent of other T. brucei sugar nucleotide biosynthetic enzymes, for example, TbGalE (UDP-Glc 4′-epimerase) and TbUAP (UDP-GlcNAc pyrophosphorylase) interconvert and utilize exclusively UDP-Glc/UDP-Gal (Roper et al. 2005) and GlcNAc-1-P (Stokes et al. 2008), respectively, whereas their mammalian counterparts can also interconvert and utilize UDP-GlcNAc/UDP-GalNAc and GalNAc-1-P, respectively.
Another difference between the parasite and human UGPs relates to quaternary structure: TbUGP, like L. major UGP, is predominantly monomeric and fully active in its monomeric state whereas the human enzyme, like that of other mammals and of fungi, is active as an octamer (Turnquist et al. 1974; Chang et al. 1996; Roeben et al. 2006). Interestingly, both AGX1, the extensively characterized dimeric human UDP-GlcNAc pyrophosphorylase (Mio et al. 1998), and Arabidopsis UGP (McCoy et al. 2007) have a C-terminal domain loop responsible for dimerization, and in S. cerevisiae UGP, this feature has been shown to be essential for stabilization of the octameric active form. The absence of a comparable C-terminal domain loop in TbUGP may explain its natural monomeric state.
Sequestration by oligomerization has been suggested to be important in the regulation of UGPs, particularly in plants (Chen et al. 2007). Other possible methods of regulation include O-GlcNAc-ylation, as found for human UGP (Wells et al. 2003), differences in N-terminal acetylation status, as demonstrated for UGPase isoforms in rice (Chen et al. 2007), and phosphorylation, as shown by the down-regulation of yeast UGP by phosphorylation by a serine/threonine kinase (Roeben et al. 2006). Since cytosolic O-GlcNAc-ylation does not occur in T. brucei, this may be ruled out as a regulatory mechanism for TbUGP. Furthermore, a recent analysis of the bloodstream form T. brucei phosphoproteome (Nett et al. 2009) did not identify TbUGP as a phosphoprotein (although its low abundance and glycosomal location might have prevented its detection). It may be that TbUGP and metabolic flux into the UDP-hexose pathways, like the non-allosteric regulation of glycolysis (Haanstra et al. 2008), is principally regulated by glycosomal compartmentation in T. brucei.
Glycosomes of trypanosomatids belong to the microbody family of organelles, containing the glyoxysomes of plants and peroxisomes of all other eukaryotes. The organelles resemble each other with respect to morphology and metabolic functions, but a unique feature of trypanosomatid glycosomes is their essential role in carbohydrate metabolism, i.e., glycolysis, gluconeogenesis, glycerol metabolism, the pentose-phosphate pathway (Michels et al. 2006), and much of sugar nucleotide biosynthesis (Roper et al. 2002; Turnock et al. 2007; Stokes et al. 2008), including TbUGP (this paper). Glycosomal proteins are targeted to these organelles via a peroxisomal targeting sequence (PTS), which can be located in the C-terminus (PTS-1) or N-terminus (PTS-2) of the protein. Internal PTS sequences have also been proposed as another targeting mechanism, with T. cruzi phosphoglucomutase as an example (Penha et al. 2009). TbUGP does not contain traditional trypanosome PTS sequences (Opperdoes and Szikora 2006). However, several internal PTS-1-like sequences can be found, such as -NRV- (N164–V166), -GKL- (G203–L205), -SNG- (S219–G221), or -SDL- (S380–L382). Of these four sequences, SNG is in the active site, and only GKL seems to be exposed to the solvent in the crystal structure. Nevertheless, considering that the apo-structure will be different to the protein in complex with UDP-Glc (Steiner et al. 2007), none of these sequences can be discounted until appropriate mutagenesis experiments are performed. In cases where no targeting sequence exists, it has been proposed that proteins can traffic to the glycosome by “piggybacking” on other PTS-targeted proteins (Purdue and Lazarow 2001; Titorenko et al. 2002). One such candidate for TbUGP is UDP-Glc 4′-epimerase, TbGalE, which has a TKL C-terminal PTS-1 sequence and which works in partnership with TbUGP in the biosynthesis of UDP-Gal (Roper et al. 2002). In any case, it would now appear that all enzymes involved in the UDP-hexose biosynthetic pathway exist in the glycosome, since hexokinase (Glc → Glc-6-P), TbUGP (Glc-1-P → UDP-Glc), and TbGalE (UDP-Glc ↔ UDP-Gal) have all been experimentally localized there. The only missing link is the identity and location of the T. brucei phosphoglucose mutase (Glc-6-P → Glc-1-P) activity, for which there is no obvious candidate gene in T. brucei, although the T. cruzi enzyme has been identified and localized to glycosomes (Penha et al. 2009). The fact that UDP-Gal, UDP-Glc, and UDP-GlcNAc are all synthesized in the glycosome and used in the ER and/or nucleus reinforces the hypothesis that glycosomal sugar nucleotide transporters and/or antiporters may exist (Stokes et al. 2008).
The related parasite L. major has a salvage pathway whereby it can take up galactose and convert it, via galactokinase and Isselbacher pathway UDP-sugar pyrophosphorylase, to UDP-Gal (Damerow et al. 2010). In contrast, this salvage pathway is completely absent in T. brucei, and the organism is entirely dependent on the de novo synthesis of UDP-Gal for glycoprotein biosynthesis (Roper et al. 2002; Urbaniak et al. 2006). We may therefore assume that TbUGP, which provides UDP-Glc for epimerization to UDP-Gal, is also essential and a potential drug target for human African sleeping sickness. Indeed, TbUGP may be a superior target to the TbGalE epimerase because UDP-Glc is also required as the glucosyl donor for the UGGT and for the synthesis of base J (β-d-glucosylhydroxymethyluracil) (van Leeuwen et al. 1998; Izquierdo et al. 2009). The crystal structure of TbUGP described here will help in the design of TbUGP inhibitors and in assessing their modes of binding. The therapeutic potential of TbUGP as a drug target will obviously depend on our ability to find drug-like parasite-specific TbUGP inhibitors.
Materials and methods
DNA isolation and manipulation
Plasmid DNA was purified from E. coli (DH5α) using the Qiagen Miniprep or Maxiprep kit as appropriate. Gel extraction was performed using QIAquick kits. Custom oligonucleotides were obtained from the University of Dundee oligonucleotide facility. T. brucei genomic DNA was isolated from 1 × 108 bloodstream form cells using DNAzol (Helena Biosciences, Gateshead, UK). All T. brucei cell cultures are mycoplasma free.
Cloning and sequencing of TbUGP
The TbUGP open reading frame identified in the T. brucei genome database (Tb10.389.0330) was amplified by PCR from genomic DNA with Platinum Taq DNA Polymerase High Fidelity (Invitrogen, Carlsbad, CA) using a forward primer containing an NdeI site (underlined), 5′-ggaattccatatgccgctaaaccctcc-3′, and a reverse primer containing a XhoI site (underlined), 5′-cgcctcgagctactcgactaccacaacct-3′. The products of three separate PCR reactions were cloned into a modified pET15B (Invitrogen) vector with a PreScission™ protease site replacing the thrombin cleavage site (a kind gift of Jon Urch and Daan van Aalten, College of Life Sciences, University of Dundee), and a representative clone from each PCR was sequenced in both directions. All three clones were identical.
The TbUGP sequence was also obtained when the gene was amplified from cDNA using the same TbUGP 3′-primer and a 5′-spliced leader primer 5′-gcccgctattattagaacagtttctgta-3′.
All plasmids were verified by sequencing (DNA Sequencing Service, College of Life Sciences, University of Dundee; www.dnaseq.co.uk).
RT–PCR
Total RNA from bloodstream form (BSF) and procyclic form (PCF) T. brucei was extracted using the RNAeasy extraction kits with on-column DNAse digestion (RNAse-free DNAse; Qiagen, Valencia, CA). RNA samples were treated with Omniscript reverse transcriptase (Qiagen) to generate cDNA. The cDNAs were then amplified by PCR using Taq polymerase and TbUGP ORF primers (described in Cloning and sequencing of TbUGP) and Dol-P-Man synthetase primers (forward 5′-aatggatgcggaccttcagcacccac-3′, reverse 5′-tagaaccgtgagcgcggtgccatac-3′) as a constitutively expressed positive control.
Protein expression and purification
The aforementioned modified pET15B plasmid containing the TbUGP ORF was used to transform E. coli BL21 (DE3) cells. Selected clones were grown aerobically at 37°C in Lysogeny broth (LB) medium containing ampicillin (100 μg/mL) to a culture density of OD 0.6. The expression of His6-TbUGP was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. Cells were grown overnight at 16°C and harvested by centrifugation. The E. coli cells were resuspended in 50 mM Tris–HCl (pH 7.7), 0.15 M NaCl (buffer A), containing 50 mM imidazole, 1 mg/mL lysozyme, and Complete Protease Inhibitor Cocktail Tablets (Roche Diagnosis, GmbH, Mannheim, Germany). After lysis in a French press, the lysate was cleared by centrifugation (20,000 × g, 30 min, 4°C) and the supernatant passed through a 0.45-μm filter. Purification of His6-TbUGP was performed using a 5-mL His-Trap column (GE Healthcare, Uppsala, Sweden) previously equilibrated with 30 mL of buffer A containing 50 mM imidazole. After sample loading, the column was washed and His6-TbUGP eluted by application of a 50–350-mM imidazole gradient (AKTA purifier, GE Healthcare, Uppsala, Sweden). The fractions obtained were checked by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE), and those containing His6-TbUGP were pooled and concentrated using a Vivaspin concentrator (Sartorius Stedi, UK Ltd., Epson, UK 10 kDa molecular weight cut off) at 4°C. As a second step, recombinant His6-TbUGP was loaded onto a Superdex 200 HR30 HPLC column (GE Healthcare, Uppsala, Sweden). Proteins were eluted at 0.5 mL/min in buffer A and monitored by absorbance at 280 nm. Fractions (2 mL) were collected and the presence of His6-TbUGP in peak fractions verified by SDS–PAGE. The final yield was 10 mg/L culture. After gel filtration, His6-TbUGP can be stored at − 80% in buffer A, 10% glycerol for several months. To cleave the His6 tag, the fusion protein was digested with GST-PreScission protease (a kind gift of Daan Van Aalten, University of Dundee) in 50 mM Tris pH 7.7, 0.150 mM NaCl, and 1 mM dithiothreitol (DTT) for 16 h at 4°C. Recombinant proteins were identified in the Proteomics and Mass Spectrometry Facility, College of Life Sciences, University of Dundee.
For crystallization, TbUGP was cloned into a modified pET15b vector containing a TEV protease site and transformed into E. coli BL21(DE3)-V2R-pRARE2 cells. Selected cells were inoculated into 10 mL of LB with ampicillin/chloramphenicol (50 μg/mL and 25 μg/mL, respectively) and incubated with shaking at 250 rpm overnight at 37°C. The culture was transferred into 50 mL of TB with 50 μg/mL ampicillin in a 250-mL shaking flask and incubated at 37°C for 3 h. Then the culture was transferred into 1.8 L of TB with 50 μg/mL kanamycin and 0.3 mL of antifoam (Sigma, Dorset, UK) and cultured using the LEX system (Harbinger Biotechnology and Engineering, Ontario Canada) to an OD600 of ~ 5, induced with 0.5 mM IPTG overnight at 15°C and harvested by centrifugation. Pellets were resuspended in binding buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES] pH 7.5, 500 mM NaCl, 5 mM imidazole, and 5% glycerol) with the addition of protease inhibitors (1 mM benzamidine and 1 mM phenylmethyl sulfonyl fluoride). Prior to mechanical lysis, each pellet from 1 L of culture was pretreated with 0.5% 3-([3-cholamidopropyl]-dimethylammonio)-1-propanesulfonate and 500 units of benzonase for 30 min at room temperature. Cells were mechanically lysed with a microfluidizer (Microfluidizer Processor, M-110EH), and the cell lysate was centrifuged using a Beckman JA-25.50 rotor at ~ 75,000 × g (24,000 rpm) for 20 min at 10°C. The cleared lysate was loaded onto a column pre-packed with 10 g DE52 (Whatman) anion exchange resin (previously activated with 2.5 M NaCl and equilibrated with binding buffer), and subsequently onto a 3-mL Ni-NTA (Qiagen) column pre-equilibrated with binding buffer. After the lysate was loaded, the DE52 was further washed with 20 mL of binding buffer. The Ni-NTA column was then washed with 200 mL of wash buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 30 mM imidazole, and 5% glycerol) at 2–2.5 mL/min. After washing, the protein was eluted with 15 mL of an elution buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 250 mM imidazole, and 5% glycerol). To remove the His6 tag, the protein was treated with TEV protease and dialysed in 10 mM HEPES, 500 mM NaCl, 5 mM imidazole, and 5 mM DTT overnight. The sample was passed through the Ni-NTA column one more time before being loaded onto a Sephadex S200 26/60 gel filtration column (GE Healthcare) pre-equilibrated with 10 mM HEPES, pH 7.5 and 500 mM NaCl, 5 mM DTT. The collected fractions corresponding to the correct eluted protein peak were concentrated using a 15-mL Amicon Ultra centrifugal filter device (Millipore, Billerica, MA).
Analytical ultracentrifugation
Recombinant TbUGP (0.75 mg/mL in buffer A) was analyzed by sedimentation velocity using a Beckman Optima XL-1 Analytical Ultracentrifuge with an AN50-Ti rotor at 32,000 rpm at 20°C. Absorbance data (72 scans at 280 nm) were collected and analyzed using the SEDFIT program (Schuck 2004). TbUGP was assumed to be globular, and its density was predicted from its amino acid composition.
UDP-Glc pyrophosphorylase activity assays
Two methods were used to assay TbUGP, an HPLC assay and a discontinuous coupled colorimetric assay, as previously described for T. brucei UDP-GlcNAc pyrophosphorylase (Stokes et al. 2008).
Briefly, the HPLC assay used 0.05 μg His6-TbUGP incubated in 100 μL of the HPLC assay buffer (50 mM Tris–HCl pH 7.2, 250 μM UTP, 10 mM MgCl2, 1 mM DTT, 250 μM Glc-1-P) for 8 min, terminated by boiling for 5 min. The samples were diluted in 2 mM NaOH and analyzed using a Dionex high-pH anion exchange chromatography HPLC system. To determine substrate specificity, Glc-1-P was substituted by Gal-1-P, GlcNAc-1-P, or GalNAc-1-P (all at 250 μM). For metal ion dependence, MgCl2 was replaced with CuCl2, ZnCl2, CoCl2, or MnCl2. For pH dependence, the Tris–HCl buffer was replaced with a 50-mM sodium acetate for pH 4.0–6.0 and 50-mM N-cyclohexyl-3-aminopropanesulfonic acid buffer for pH higher than 9.0.
The TbUGP colorimetric assay was performed with 0.0125 μg of His6-TbUGP in a 96-well plate format (Nunc™) in 12.5 mM Tris–HCl pH 7.2, NaCl 150 mM, 500 μM UTP, 500 μM Glc-1-P, 10 mM MgCl2, 1 mM DTT, bovine serum albumin (BSA) 0.1 mg/mL, and 1 U/mL pyrophosphatase (Sigma) to a final volume of 50 μL. The reaction was left for 8 min at room temperature and terminated by the addition of 100 μL of the color reagent (0.2% ammonium molybdate, 0.5% Triton X-100, 0.7 N HCl, 0.03% malachite green). Absorbance at 620 nm was measured after 20 min using a SpectraMax 340 PC (Molecular Devices, Sunnyvale, CA).
TbUGP localization
Two Balb/c adult mice were used to raise polyclonal antibodies against His6-tagged TbUGP protein (about 0.1 mg per mouse) with Freund’s complete adjuvant. Each mouse received two further immunizations with Freund’s incomplete adjuvant over 2 months. Antibodies were then affinity purified on CNBr-Sepharose-immobilized TbUGP that had had its His6 tag removed with PreScission™ protease. Similar protocol was used to immunize two New Zealand rabbits with 0.5 mg of purified TbUGP protein per animal.
Wild-type bloodstream form T. brucei cells were grown in HMI-9 medium to a density of 1 × 106 cells/mL, harvested by centrifugation and resuspended in trypanosome dilution buffer (0.1 M Na2HPO4, 0.01 M NaH2PO4, 0.025 M KCl, 0.4 M NaCl, 5 mM MgSO4, 0.1 M glucose adjusted to pH 7.45 with HCl) to a density of 4 × 107 cells/mL. Aliquots (15 μL) were added to 13 mm coverslips, left at 4°C for 15 min, and fixed in 1 mL 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min followed by three 5-min washes in 2 mL PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. Samples were then blocked in 5% fish skin gelatin (FSG) in PBS containing 10% normal goat serum. The coverslips were incubated with immunopurified mouse anti-TbUGP (0.5 μg/mL) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antiserum at 1:2000 dilution or rabbit anti-enolase antiserum at 1:4000 dilution in 1% FSG in PBS, 0.05% TX-100. Both anti-GAPDH and anti-enolase were kind gifts of Paul Michels (Catholic University of Louvain). Samples were then washed as above in PBS and incubated with 50 μL of Alexa 594-conjugated anti-mouse IgG and Alexa 488-conjugated anti-rabbit IgG for 1 h. Coverslips were mounted on glass slides, sealed with Hydromount containing 2.5% DABCO and left to dry in the dark for 30 min. Microscopy was performed on a Zeiss LSM 510 META confocal microscope.
Immunoprecipitation and western blotting of TbUGP
T. brucei bloodstream form cells (2 × 108 cells) purified from rat infected blood were washed with trypanosome dilution buffer and hypotonically lysed in 200 μL of water containing 0.1 mM 1-chloro-3-tosylamido-7-amino-2-heptanone and 1 μg/mL leupeptin. The lysate was adjusted to final concentrations of 0.03% SDS and 1% Triton X-100, and an EDTA-free protease inhibitor tablet (Roche) was added, incubated at 4°C for 15 min and centrifuged (20,000 × g for 30 min). The pellet was discarded and the supernatant was incubated (2 h, 4°C) with 12 μg of affinity-purified rabbit anti-TbUGP previously coupled to 50 μL of 30 mg/mL protein G Dynabeads (Invitrogen). The beads were recovered by placing on the magnet for 2 min and washed three times with cold 20 mM Tris–HCl pH 7.2, 0.15 M NaCl, containing 0.03% SDS and 1% Triton X-100. The bound antigen was eluted with 50 mM sodium citrate pH 2.8 and immediately neutralized with 1.2 M Tris–HCl pH 8.8. The elution was boiled in SDS-sample buffer containing 0.1 M DTT, loaded onto 4–12% gradient NuPage gel (Invitrogen) for SDS–PAGE, and western blotted onto nitrocellulose. Briefly, the membrane was blocked with 5% (w/v) BSA, 0.05% (w/v) Tween-20 (Sigma), 0.15 M NaCl in 50 mM Tris–HCl pH 7.4, and subsequently probed with 5 μg/mL affinity-purified mouse anti-TbUGP as the primary antibody, washed and then incubated with 1:10,000 horse radish peroxidase-conjugated anti-mouse IgG (Jackson Immunoresearch, Stratech, Newmarket Suffolk, UK). After washing, the membrane was developed by chemiluminescent detection (ECL-plus, GE Healthcare).
Crystallization and data collection
Crystals of TbUGP were obtained from a hanging drop vaporization method consisting of 17 mg/mL protein mixed one to one with a crystallization buffer of 22% PEG 3350, 0.1 M ammonium sulfate, and 0.1 M Bis–Tris pH 5.5. Crystals were flash-cooled in crystallization buffer supplemented with 18% glycerol.
X-ray diffraction data were collected on a home source Rigaku FRE with wavelength 1.5418 Å. An initial native dataset was collected using the Rigaku software then indexed and scaled using the HKL2000 package (Otwinowski et al. 2003). A molecular replacement solution was found using the program BALBES (Long et al. 2008), which found a solution using a model from the UPG-bound form of UGP from L. major (PDB accession no. 2OEG-A). This initial dataset was partially twinned and subsequent refinement statistics were poor. Additional crystals were screened and a second dataset was collected on a less pathogenic crystal. The BALBES model was used again and refinement statistics with this dataset were much improved. The final model was built by iterative rounds of manual model building in Coot (Emsley and Cowtan 2004) followed by refinement in the ccp4 program refmac5.2 (Collaborative Computational Project Number 4, 1994). The final model has overall good geometry, with 98% of all residues favored Ramachandran regions and no outliers. Coordinates and structure factors have been deposited to the RCSB protein databank (PDB accession no. 3GUE). Final statistics are shown in Table I. Ligand interactions, close contacts, and oligomerization state within the crystal were analyzed using the program PISA (Krissinel and Henrick 2007) and crystal figures created with the Pymol package (DeLano 2005).
Acknowledgments
The authors are grateful to the University of Dundee Proteomics Facility and to Mark Agacan, supported by Wellcome Strategic Award 083481, for performing the proteomics and AUC experiments, and to Alan R. Prescott (Division of Cell Biology and Immunology, University of Dundee) for valuable help with the microscopy. We would also like to thank Paul Michels for providing antibodies anti-GAPDH and anti-enolase, Daan van Aalten for providing vectors and reagents, and Waldemar Striker for his critical reading of this manuscript.
Conflict of interest statement
No conflict of interest has been declared by the authors.
Abbreviations
- BSA
bovine serum albumin
- DTT
dithiothreitol
- HAT
Human African trypanosomiasis
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LB
Lysogeny broth
- LmUGP
Leishmania major UDP-Glucose pyrophosphorylase
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- RT
reverse transcription
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TbUGP
Trypanosoma brucei UDP-Glucose pyrophosphorylase
- UDP-Gal
UDP-galactose
- UDP-Glc
UDP-glucose
- UDP-GlcNAc
UDP-N-acetylglucosamine
- UGGT
unfolded glycoprotein glucosyltransferase
- UV
ultraviolet
- VSG
variant surface glycoprotein
Funding
This work was supported by a Wellcome Trust programme grant (grant number 085622). The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation and Genome Canada, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck, the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust.
References
- Alexander DL, Schwartz KJ, Balber AE, Bangs JD. Developmentally regulated trafficking of the lysosomal membrane protein p67 in Trypanosoma brucei. J Cell Sci. 2002;115:3253–3263. doi: 10.1242/jcs.115.16.3253. [DOI] [PubMed] [Google Scholar]
- Chang H-Y, Peng H-L, Chao YC, Duggleby RG. The importance of conserved residues in human liver UDP-glucose pyrophosphorylase. Eur J Biochem. 1996;236:723–728. doi: 10.1111/j.1432-1033.1996.t01-1-00723.x. [DOI] [PubMed] [Google Scholar]
- Chen R, Zhao X, Shao Z, Zhu L, He G. Multiple isoforms of UDP-glucose pyrophosphorylase in rice. Physiol Plant. 2007;129:725–736. [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cross GA. Antigenic variation in trypanosomes: Secrets surface slowly. Bioessays. 1996;18:283–291. doi: 10.1002/bies.950180406. [DOI] [PubMed] [Google Scholar]
- Damerow S, Lamerz A-C, Haselhorst T, Fuhring J, Zarnovican P, von Itzstein M, Routier FH. Leishmania UDP-sugar pyrophosphorylase: The missing link in galactose salvage? J Biol Chem. 2010;285:878–887. doi: 10.1074/jbc.M109.067223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The case for open-source software in drug discovery. Drug Discov Today. 2005;10:213–217. doi: 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
- Duggleby RG, Chao YC, Huang JG, Peng H-L, Chang H-Y. Sequence differences between human muscle and liver cDNAs for UDP-glucose pyrophosphorylase and kinetic properties of the recombinant enzymes expressed in Escherichia coli. Eur J Biochem. 1996;235:173–179. doi: 10.1111/j.1432-1033.1996.00173.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fevre EM, Wissmann BV, Welburn SC, Lutumba P. The burden of human African trypanosomiasis. PLoS NeglTrop Dis. 2008;2:e333. doi: 10.1371/journal.pntd.0000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Diaz M, Alape-Giron A, Persson B, Pollesello P, Moos M, von Eichel-Streiber C, Thelestam M, Florin I. Cellular UDP-glucose deficiency caused by a single point mutation in the UDP-glucose pyrophosphorylase gene. J Biol Chem. 1997;272:23784–23791. doi: 10.1074/jbc.272.38.23784. [DOI] [PubMed] [Google Scholar]
- Freeze HH, Elbein AD. Glycosylation precursors. In: Varki AC, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2nd ed. New York: Cold Spring Harbor Laboratory Press; 2008. [PubMed] [Google Scholar]
- Haanstra JR, van Tuijl A, Kessler P, Reijnders W, Michels PA, Westerhoff HV, Parsons M, Bakker BM. Compartmentation prevents a lethal turbo-explosion of glycolysis in trypanosomes. Proc Natl Acad Sci USA. 2008;105:17718–17723. doi: 10.1073/pnas.0806664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Helenius A. Quality control in the secretory pathway. Curr Opin Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Izquierdo L, Atrih A, Rodrigues JA, Jones DC, Ferguson MAJ. Trypanosoma brucei UDP-Glucose:glycoprotein glucosyltransferase has unusual substrate specificity and protects the parasite from stress. Eukaryot Cell. 2009;8:230–240. doi: 10.1128/EC.00361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RJ, Alexander DL, Cowan C, Balber AE, Bangs JD. Molecular cloning of p67, a lysosomal membrane glycoprotein from Trypanosoma brucei. Mol Biochem Parasitol. 1999;98:17–28. doi: 10.1016/s0166-6851(98)00155-8. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Lai X, Wu J, Chen S, Zhang X, Wang H. Expression, purification, and characterization of a functionally active Mycobacterium tuberculosis UDP-glucose pyrophosphorylase. Protein Expr Purif. 2008;61:50–56. doi: 10.1016/j.pep.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Lamerz A-C, Haselhorst T, Bergfeld AK, von Itzstein M, Gerardy-Schahn R. Molecular cloning of the Leishmania major UDP-glucose pyrophosphorylase, functional characterization, and ligand binding analyses using NMR Spectroscopy. J Biol Chem. 2006;281:16314–16322. doi: 10.1074/jbc.M600076200. [DOI] [PubMed] [Google Scholar]
- LeBowitz JH, Smith HQ, Rusche L, Beverley SM. Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev. 1993;7:996–1007. doi: 10.1101/gad.7.6.996. [DOI] [PubMed] [Google Scholar]
- Long F, Vagin AA, Young P, Murshudov GN. BALBES: A molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martz F, Wilczynska M, Kleczkowski LA. Oligomerization status, with the monomer as active species, defines catalytic efficiency of UDP-Glucose pyrophosphorylase. Biochem J. 2002;367:295–300. doi: 10.1042/BJ20020772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy JG, Bitto E, Bingman CA, Wesenberg GE, Bannen RM, Kondrashov DA, Phillips GN., Jr Structure and dynamics of UDP-Glucose pyrophosphorylase from Arabidopsis thaliana with bound UDP-glucose and UTP. J Mol Biol. 2007;366:830–841. doi: 10.1016/j.jmb.2006.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels PA, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta. 2006;1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Mio T, Yabe T, Arisawa M, Yamada-Okabe H. The eukaryotic UDP-N-acetylglucosamine pyrophosphorylases. Gene cloning, protein expression, and catalytic mechanism. J Biol Chem. 1998;273:14392–14397. doi: 10.1074/jbc.273.23.14392. [DOI] [PubMed] [Google Scholar]
- Nett IRE, Martin DMA, Miranda-Saavedra D, Lamont D, Barber JD, Mehlert A, Ferguson MAJ. The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol Cell Proteomics. 2009;8:1527–1538. doi: 10.1074/mcp.M800556-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opperdoes FR, Szikora J-P. In silico prediction of the glycosomal enzymes of Leishmania major and trypanosomes. Mol Biochem Parasitol. 2006;147:193–206. doi: 10.1016/j.molbiopara.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Borek D, Majewski W, Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- Penha LL, Sant’Anna CB, Mendonça-Previato L, Cunha-e-Silva NL, Previato JO, Lima APCA. Sorting of phosphoglucomutase to glycosomes in Trypanosoma cruzi is mediated by an internal domain. Glycobiology. 2009;19:1462–1472. doi: 10.1093/glycob/cwp121. [DOI] [PubMed] [Google Scholar]
- Purdue PE, Lazarow PB. Peroxisome biogenesis. Annu Rev Cell Dev Biol. 2001;17:701–752. doi: 10.1146/annurev.cellbio.17.1.701. [DOI] [PubMed] [Google Scholar]
- Roeben A, Plitzko JM, Körner R, Böttcher UM, Siegers K, Hayer-Hartl M, Bracher A. Structural basis for subunit assembly in UDP-glucose pyrophosphorylase from Saccharomyces cerevisiae. J Mol Biol. 2006;364:551–560. doi: 10.1016/j.jmb.2006.08.079. [DOI] [PubMed] [Google Scholar]
- Roper JR, Güther ML, Macrae JI, Prescott AR, Hallyburton I, Acosta-Serrano A, Ferguson MAJ. The suppression of galactose metabolism in procylic form Trypanosoma brucei causes cessation of cell growth and alters procyclin glycoprotein structure and copy number. J Biol Chem. 2005;280:19728–19736. doi: 10.1074/jbc.M502370200. [DOI] [PubMed] [Google Scholar]
- Roper JR, Güther ML, Milne KG, Ferguson MAJ. Galactose metabolism is essential for the African sleeping sickness parasite Trypanosoma brucei. Proc Natl Acad Sci USA. 2002;99:5884–5889. doi: 10.1073/pnas.092669999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P. A model for sedimentation in inhomogeneous media. I. Dynamic density gradients from sedimenting co-solutes. Biophys Chem. 2004;108:187–200. doi: 10.1016/j.bpc.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Steiner T, Lamerz A-C, Hess P, Breithaupt C, Krapp S, Bourenkov G, Huber R, Gerardy-Schahn R, Jacob U. Open and closed structures of the UDP-glucose pyrophosphorylase from Leishmania major. J Biol Chem. 2007;282:13003–13010. doi: 10.1074/jbc.M609984200. [DOI] [PubMed] [Google Scholar]
- Stevens RA, Phelps CF. Studies on ligand binding to bovine liver uridine diphosphate glucose pyrophosphorylase. Biochem J. 1976;159:65–70. doi: 10.1042/bj1590065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes MJ, Güther MLS, Turnock DC, Prescott AR, Martin KL, Alphey MS, Ferguson MAJ. The synthesis of UDP-N-acetylglucosamine is essential for bloodstream form Trypanosoma brucei in vitro and in vivo and UDP-N-acetylglucosamine starvation reveals a hierarchy in parasite protein glycosylation. J Biol Chem. 2008;283:16147–16161. doi: 10.1074/jbc.M709581200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko VI, Nicaud J-M, Wang H, Chan H, Rachubinski RA. Acyl-CoA oxidase is imported as a heteropentameric, cofactor-containing complex into peroxisomes of Yarrowia lipolytica. J Cell Biol. 2002;156:481–494. doi: 10.1083/jcb.200111075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnock DC, Ferguson MAJ. Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot Cell. 2007;6:1450–1463. doi: 10.1128/EC.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnock DC, Izquierdo L, Ferguson MAJ. The de novo synthesis of GDP-fucose is essential for flagellar adhesion and cell growth in Trypanosoma brucei. J Biol Chem. 2007;282:28853–28863. doi: 10.1074/jbc.M704742200. [DOI] [PubMed] [Google Scholar]
- Turnquist RL, Gillett TA, Hansen RG. Uridine diphosphate glucose pyrophosphorylase. Crystallization and properties of the enzyme from rabbit liver and species comparisons. J Biol Chem. 1974;249:7695–7700. [PubMed] [Google Scholar]
- Urbaniak MD, Turnock DC, Ferguson MAJ. Galactose starvation in a bloodstream form Trypanosoma brucei UDP-glucose 4′-epimerase conditional null mutant. Eukaryot Cell. 2006;5:1906–1913. doi: 10.1128/EC.00156-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Kieft R, Cross M, Borst P. Biosynthesis and function of the modified DNA base beta-D-glucosyl-hydroxymethyluracil in Trypanosoma brucei. Mol Cell Biol. 1998;18:5643–5651. doi: 10.1128/mcb.18.10.5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L, Whelan SA, Hart GW. O-GlcNAc: A regulatory post-translational modification. Biochem Biophys Res Commun. 2003;302:435–441. doi: 10.1016/s0006-291x(03)00175-x. [DOI] [PubMed] [Google Scholar]