

Abstract
In periodontal disease, host recognition of bacterial constituents, including lipopolysaccharide (LPS), induces p38 MAPK activation and subsequent inflammatory cytokine expression, favoring osteoclastogenesis and increased net bone resorption in the local periodontal environment. In this paper, we discuss evidence that the p38/MAPK-activated protein kinase-2 (MK2) signaling axis is needed for periodontal disease progression: an orally administered p38α inhibitor reduced the progression of experimental periodontal bone loss by reducing inflammation and cytokine expression. Subsequently, the significance of p38 signaling was confirmed with RNA interference to attenuate MK2-reduced cytokine expression and LPS-induced alveolar bone loss. MAPK phosphatase-1 (MKP-1), a negative regulator of MAPK activation, was also critical for periodontal disease progression. In MPK-1-deficient mice, p38-sustained activation increased osteoclast formation and bone loss, whereas MKP-1 overexpression dampened p38 signaling and subsequent cytokine expression. Finally, overexpression of the p38/MK2 target RNA-binding tristetraprolin (TTP) decreased mRNA stability of key inflammatory cytokines at the posttranscriptional level, thereby protecting against periodontal inflammation. Collectively, these studies highlight the importance of p38 MAPK signaling in immune cytokine production and periodontal disease progression.
1. Innate Immunity and Periodontal Disease
1.1. Host-Microbe Interaction
Within the oral cavity exists a biofilm colonized by more than 500 different microbial species, very few of which are actually associated with periodontal disease [1–3]. These periopathogenic gram-negative bacteria contain multiple virulence factors, including lipopolysaccharide (LPS), which can induce the host inflammatory response. In periodontal diseases initiation and progression, such an inflammatory response to bacterial biofilm is exaggerated, resulting in leading to overproduction of inflammatory cytokines that cause gingival inflammation, bleeding, extracellular matrix degradation, bone resorption, and tooth loss [4–6].
Over the past two decades, how host-microbe interactions contribute to both disease initiation and associated tissue destruction have been elucidated. Epidemiological data indicate different intraindividual susceptibilities to periodontal disease, despite the long-term presence of oral biofilm [7–9]. Moreover, increased susceptibility and greater severity of periodontal disease were observed in individuals with impaired immune responses [10, 11]. The most significant development in periodontitis research has been the fundamental role of innate immunity in initiating immune responses and regulating adaptive (antigen-specific) responses [5]. The innate immune response recognizes and responds to all colonizing microbes, both commensal and pathogenic. The modest cytokine response to commensal bacteria stimulation in the periodontium is necessary for priming host immunity and maintaining tissue integrity, and the amplified immune response is induced when the microbial composition of plaque, in which pathogenic bacteria are greatest, changed [12, 13].
In the current paradigm, Toll-like receptors (TLRs) link the host and microbes and are considered essential for LPS-induced signaling. LPS, one of the main pathogen-associated molecular patterns (PAMPs) of pathogenic bacteria, is recognized by the host through TLRs, resulting in activation of multiple downstream cell signaling cascades [14]. To date, the TLR family includes 13 members, which is consistent with the range of PAMPs expressed by infective microorganisms. These receptors not only recognize various PAMPs and activate innate immune response, but they can also bind to endogenous molecules derived from damaged tissue and contribute to innate inflammation as well as the adaptive immune response [15]. Within the periodontium, innate immunity is comprised of resident immune cells such as monocytes/macrophages, neutrophils, dendritic cells, and nonimmune resident cells such as periodontal fibroblasts and gingival epithelial cells. Accordingly, all of these cell types express various TLRs to identify and respond temporarily to PAMPs [16–18]. In periodontal tissues, TLR2 and TLR4 expression has been positively correlated with disease severity, suggesting that these receptors have an increased capacity to signal and influence downstream cytokine expression [19–21]. All TLRs are single-pass transmembrane proteins containing a common extracellular N-terminal leucine-rich domain and a conserved intracellular C-terminal domain. The N-terminal domain is responsible for the recognition of the ligands and the C-terminal tail is shown to be homologous with the intracellular domain of the interleukin-1 receptor type I, currently designated as the Toll/IL-1 receptor (TIR) domain [22]. The classic intracellular signaling pathways activated by TLR engagement are highly conserved. The TLR-PAMP interaction recruits specific adaptor molecules which then bind the interleukin (IL)-1 receptor associated kinase (IRAK), initiating a chain of signaling transduction. In the TLR pathway, at least four adaptor proteins, including myleloid differentiation primary-response protein 88 (MyD88), TIR domain-containing adaptor-inducing interferon β (TRIF), MyD88 adapter-like/TIR domain-containing adaptor protein (Mal/TIRAP), and TRIF-related adaptor molecule (TRAM), contain TIR domains that can be recruited by activated TLRs. Each of these adaptor molecules interact with the various TLRs, an event thought to be responsible for signal transduction branching and significant TLR signaling flexibility by allowing crosstalk with other pathways, including MAP kinase, PKR, and Notch pathways [23–27] (see Figure 1).
Figure 1.
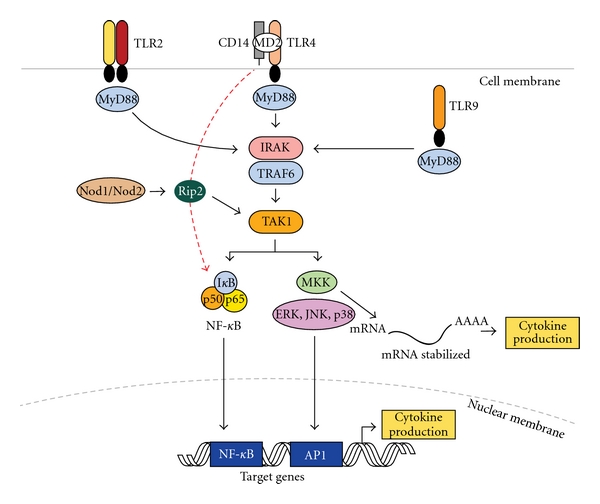
Pattern recognition receptors and innate immune signaling. TLR-2, TLR-4, and TLR-9 are depicted as examples of TLR receptors expressed in cells of the periodontal tissues. Upon ligand binding, all TLRs (except TLR3) recruit adaptor protein MyD88 and activate common upstream activator (IRAK/TRAF6 and TAK1) of NF-kB and MAP kinases. TLR-4 may also activate NF-kB independent of MyD88 and with delayed kinetics (red dotted line). Nod1/Nod2 are cytosolic PRRs that recognize peptidoglycan fragments of the bacterial wall and may amplify TLR-induced activation of signaling pathways. Activated NF-kB and MAP kinases translocate to the nucleus and bind to their motifs (NF-kB, AP-1, resp.) in the promoter of target genes (including early-response and inflammatory genes) and induce their transcription into mRNA which will ultimately lead to increased cytokine production. p38 MAP kinase also involved posttranscriptional regulation of pro-inflammatory genes (for example, IL-6, Cox-2) by modulation of mRNA stability in the cytoplasm. (TLR: toll-like receptor, CD14: cluster of differentiation 14 molecule; MD2: myeloid differentiation protein 2; MyD88: myeloid differentiation primary response gene 88; IRAK: interleukin-1 receptor-associated kinase; TRAF6: TNF receptor associated factor 6; TAK1: TGF-beta activated kinase 1; MKK: mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase: JNK: c-Jun N-terminal kinase; AP-1: activator protein-1).
Of the TLR signaling pathways, the most studied is the recognition of LPS by a macromolecular complex involving CD14, MD2, and TLR4 [6, 28, 29]. In response to LPS, complex formation triggers MyD88 and, in turn, recruits IRAK and TRAF6. The phosphorylated IRAK/TRAF6 complex then dissociates from the receptor complex to a new complex with transforming growth factor β-activated kinase (TAK1) along with TAK-1 and -2 binding proteins (TAB1 and -2) which phosphorylate TAK1. TAK1, in turn, phosphorylates both the inhibitor of nuclear factor κB-(IκB-) kinase complex (IKK complex) and mitogen-activated protein kinase (MAPK) kinases-3 and -6 (MKK3, MKK6) [30]. The IKK complex then phosphorylates IκB, which allows nuclear factor-kappa B (NF-kB) transcription factors (p50/p65) to translocate to the nucleus and bind to the promoter regions of many proinflammatory cytokine and chemokine genes and activate their transcription [31, 32]. Similarly, MKK3/6 can phosphorylate p38 and c-jun N-terminal kinase (JNK) MAPK to activate activator protein-1 (AP-1) transcription factors (TFs) and initiate gene expression. In addition, p38 can phosphorylate RNA-binding proteins, which can stabilize cytokine mRNA and thus amplify cytokine production [33], as depicted in Figure 1.
1.2. Genesis of Inflammation in Periodontal Tissues
Periodontal tissue destruction is initiated by pathogens that colonize in the periodontal pocket, a unique microenvironment in the subgingiva that fosters the growth of anaerobic bacteria and spirochetes. These microorganisms produce harmful byproducts and enzymes, such as collagenases and proteases, which degrade the extracellular matrix to produce nutrients for their growth. Resultant tissue damage creates pathways for invasion, followed by irritation and inflammation of host tissues, eliciting a host immune response. This local immune response in gingival tissues involves recruitment of inflammatory cells, generation of cytokines and prostanoids, elaboration of lytic enzymes, and osteoclast activation [34–36].
Within the inflamed periodontium exist various resident immune and nonimmune cells: periodontal ligament cells, fibroblasts, osteoblasts, osteoclasts, neutrophils, antigen-presenting cells such as dendritic cells, macrophages, T cells, and B cells. These resident cell types recognize and interact with bacterial constituents (e.g., cytoplasmic membranes, peptidoglycans, outer membrane proteins, lipopolysaccharide, capsules, and cell-surface fimbriae) to generate proinflammatory mediators, such as IL-1, tumor necrosis factor (TNF)-α, IL-6, and prostaglandin E2 (PGE2) [37–39]. These secreted molecules can recruit more nonresident cells (e.g., neutrophils, macrophages, and lymphocytes) to infiltrate tissues and initiate the innate immune response [40, 41]. They also further induce their own expressions through numerous autocrine or paracrine mechanisms and thus perpetuate and amplify chronic periodontal inflammation. Additionally, cellular components and byproducts of tissue breakdown in the periodontium can be recognized and trigger signal cytokine secretion, possibly creating a self-sustaining positive feedback circuit, leading to loss of tissue function and exacerbated clinical disease. With overexpression of proinflammatory cytokines, other inflammatory enzymes and mediators, such as matrix metalloproteinases (MMPs) and receptor activator of nuclear factor-kB ligand (RANKL), are upregulated to produce irreversible soft and hard tissue damage.
Within periodontal lesions, activated monocytes, macrophages, and fibroblasts all produce cytokines, such as TNF-α, L-1β, IL-6, and PGE2, which are significantly elevated in diseased periodontal sites, compared with healthy or inactive sites [42–48] and have been positively correlated with increased probing depth and attachment loss [49–51]. In the gingival crevicular fluid, elevated IL-6, TNF-α, and IL-1β were reported in persons afflicted with periodontitis [52, 53]. Elevated IL-6 is higher in recurrent periodontitis cases and increased GCF correlates with gram-negative fimbriae [53–55].
Periodontal disease manifests as an intimate combination of inflammation and bone resorption, leading to the eventual tooth loss. Bone is a dynamic tissue that constantly undergoes a remodeling process in which bone resorption and bone deposition are balanced.When chronic inflammation occurs in bone and other mineralized tissue, this balance is disrupted to favor net bone loss. In periodontitis, alveolar bone resorption occurs when inflammatory mediators in the overlying soft tissues reach a certain threshold at a critical distance from the bone surface and activate pathways leading to bone resorption [56].
The RANK/RANK ligand (RANKL)/osteoprotegerin (OPG) system which controls osteoclast development, differentiation, activation, and function, is a key mediator of bone loss in periodontal disease. Through interactions with its cognate receptor RANK on the cell surface of osteoclasts and osteoclast precursors, RANKL stimulates differentiation and maturation of cells from the monocyte/macrophage lineage to form functional osteoclasts and then induces osteoclastogenesis and subsequent bone resorption. OPG, a soluble member of the tumor necrosis family produced by osteoblasts, marrow stromal cells, and other cells, can bind to RANKL as a decoy receptor and inhibit osteoclast differentiation to balance this system, decreasing RANKL functional activity. Thus, the RANKL/OPG ratio eventually determines bone turnover [6, 57, 58].
Even though RANKL expression may require different signaling pathways depending on the nature of extracellular stimulation, cell type, and even cell differentiation state, RANKL expression has been shown to increase inflamed periodontal tissues from various cell types, including osteoblastic cells, bone marrow stromal cells, endothelial cells, mononuclear cells, and periodontal ligament fibroblasts [59–63]. More recently, many secreted proinflammatory cytokines and other mediators have been shown to converge and stimulate RANKL in these cell types. Meanwhile, most of these same molecules decreased OPG production. Such molecules include IL-1β, TNF-α, IL-6, IL-8, IL-11, IL-17, MMPs, and PGE2, all of which are upregulated in periodontal tissues. With alveolar bone loss, the periodontal pocket deepens and subgingival plaque biofilm further accumulates. The subgingival microflora becomes more anaerobic and the host response becomes more destructive and chronic.
2. Pathobiology of Periodontal Disease Progression
2.1. MAPK Cell Signaling
The innate immune system serves as a first defense against invading pathogenic organisms and its interaction with microbial components activates multiple signaling cascades, including MAP kinase pathways. Among the various signaling molecules that regulate inflammatory pathways, kinases are important because their aberrant or upregulated expression significantly contributes to the regulation of inflammatory disease. MAP kinases are highly conserved serine/threonine protein kinases in eukaryotes. MAP kinase BMK-1/ERK5 has been reported to respond to both growth factors and stressful stimuli, contributing to cell cycle progression and proliferation, but three other MAP kinases, p38, c-Jun N-terminal kinases JNK, and extracellular signal-regulated kinases ERK, are the most studied [64].
Different cell stimuli preferentially activate distinct MAP kinases and endow them with different functions. Many growth factor and G protein linked receptors, cell adhesion, phorbol esters, and some oncogenes are linked to activation of the ERK MAP kinases, which are involved in cellular chemotaxis, cell cycle progression and mitogenesis, oncogenic transformation and metastasis, neuronal differentiation and survival, and in processes underlying memory and learning. Inflammatory cytokines (such as IL-1 and TNF-α), trophic factor deprivation, and a number of cell stress-inducing factors (such as heat shock, osmotic shock, ultraviolet radiation, and oxygen radicals) preferentially lead to activation of JNK/SAPK and p38 MAP kinases. In all, MAPK signal transduction cascades play a pivotal regulatory role in the biosynthesis of numerous cytokines, chemokines, and other inflammatory mediators that are necessary for the immune system to combat pathogenic infections.
The MAPKs are organized in modules (MAPKKK→MAPKK→MAPK) sequentially activated by a cascade of dual phosphorylation events at tyrosine/threonine residues. Beginning with the activation of upstream MAP kinase kinase kinase (MKKK), MAP kinase kinase (MKK) is further activated by MKKK at two serine residues. MKK in turn activates MAP kinase by phosphorylating the MAPKs at the adjacent threonine and tyrosine residues localized within a conserved activation loop motif, TxY, between the kinase subdomains VII and VIII (where x represents glutamate in ERK, proline in JNK/SAPK, and glycine in p38 MAPKs) [65]. Furthermore, MKK-3 and -6 are specific for p38 whereas MKK-4 also activates JNKs. Once activated, the MAPKs can target an array of downstream substrate proteins for phosphorylation, including the “downstream” serine/threonine kinases (which themselves become activated), cytoskeletal elements, cell death regulators, and many nuclear receptors and transcription factors (including AP-1 (homodimer or heterodimer of the proteins c-fos and c-jun), NF-κB, or CAAT-enhancer-binding protein) which facilitate gene transcription. For example, NF-κB can bind to the promoter regions of many proinflammatory cytokine and chemokine genes and activate their transcription. In addition to the regulation of the expression of inflammatory mediators, MAPKs are also implicated in the regulation of reactive oxygen and nitrogen species, which are critical for killing microbes engulfed by phagocytes. MAPKs also regulate gene expression through promoting chromatin remodeling [64] and participate in the transport, stabilization, and translation of cytokine mRNA transcripts that contain AU-rich elements [33]. It is well known that p38 MAPK activates MAP kinase-activated protein kinase (MK)-2 through phosphorylation. MK-2, in turn, inactivates transacting RNA-binding protein tristetraprolin (TTP) by phosphorylation at serine 52 and 178. This phosphorylation sequesters TTP with 14 : 3 : 3 and inhibits the binding of TTP with ARE mRNA. Thus, ARE mRNAs are spared from TTP shuttling to degradation machinery, and TTP-mediated deadenylation and destabilization of ARE-containing transcripts are inhibited, giving mRNAs an opportunity for translation [66–68].
Once the inflammatory stimulus is resolved, negative regulators modulate the strength and duration of the activated MAPK signaling pathway and control the production of inflammatory cytokines, thus restraining the potentially devastating actions of the immunological system on the host and preventing self-destruction. These negative regulators include tyrosine, serine/threonine, and dual specificity phosphatases. A group of dual specificity protein phosphatases (often referred to as MAPK phosphatases (MKPs)) [69] are the primary phosphatases responsible for dephosphorylation/deactivation of MAP kinases (see Figure 2). MKP-1, an archetypal member of the MKP family, is essential for the dephosphorylation/deactivation of MAPKs p38 and JNK. Additionally, it serves as pivotal feedback control regulator in the innate immune response during microbial infection and thus, plays a significant role in the progression of periodontitis.
Figure 2.
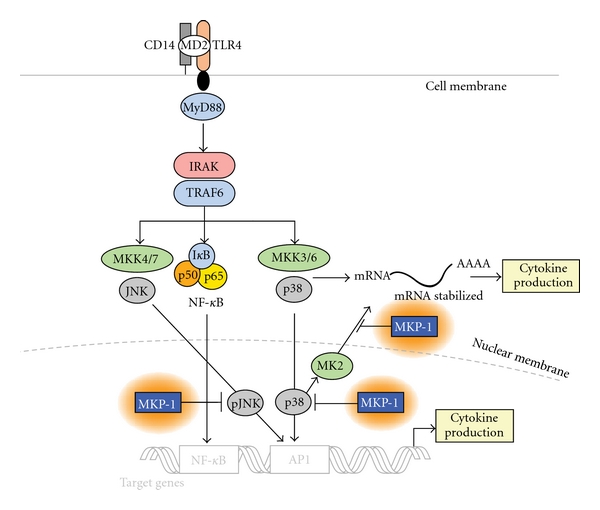
MAPK activation and regulation via MKP-1. Bacterial LPS induces MAPK signaling as described in Figure 1. MAP Kinase Phosphatase-1 (MKP-1) negatively regulates MAPK activation via dephosphorylation of target kinases at multiple points of kinase activation.
2.2. Expression of Immune Cytokines
Periodontal diseases are characterized by chronic inflammation due to the overproduction of inflammatory mediators such as cytokines, chemokines, nitric oxide, and reactive oxygen species by immune and nonimmune cells. In the progression of the disease, the extent and severity of tissue destruction are the result of cytokine overproduction, which is largely dependent on the nature of the host-microbial interactions.
Cytokines interact functionally in networks and integrate aspects of innate and adaptive immunity, mobilizing leukocytes to the infection site, initiating the adaptive immune response, and initiating the acute phase response [16]. To date, two proinflammatory cytokines, TNF-α and IL-1β, are the best understood cytokines that correlate significantly with periodontal diseases.
Of the IL-1 family, IL-1α and IL-1β have been recognized to be central proinflammatory cytokines with similar biological activity. IL-1ra is an endogenous receptor antagonist and an anti-inflammatory nonsignaling molecule that competes for receptor binding with IL-1α and IL-1β. The overall contribution of IL-1 to the proinflammatory response depends on the balance among these three molecules [70, 71]. In periodontal research, many studies are conducted to measure IL-1β in gingival tissue and gingival crevicular fluid (GCF) in patients with various periodontal conditions. Increased IL-1β expression has been consistently detected in both samples [72, 73] and was associated with periodontitis severity [74], and IL-1β decreased after treatment [75]. In periodontitis patients, IL-1β production increased via circulating monocytes or oral polymorphonuclear neutrophils (PMNs). Furthermore, blockage of IL-1β activity slowed progression of experimental periodontitis in primates [76]. In addition, IL-1β stimulates many cells to produce MMPs and prostaglandins, resulting in bone resorption and connective tissue degradation, all of which contribute to the pathogenesis of periodontitis [77].
TNF-α is released by activated monocytes, macrophages, and T-lymphocytes and promotes critical inflammatory responses. TNF-α induces bone resorption and upregulates PGE2 and MMP secretion during periodontal disease [56]. Clinical evidence indicates that patients with periodontal disease have increased TNF-α in the gingival crevice fluid [44], and TNF-α has been shown to be enhanced during destruction of periodontal tissues [49–51]. TNF-α binds to two distinct TNF receptors: TNF-R1 (p55) and TNF-R2 (p75), and most of TNF's inflammatory activities are transduced by TNF-R1. TNF-R2 is thought to enhance this activity by binding TNF and then passing it on to the TNF-R1 [78]. Blocking TNF-α has been proven to effectively inhibit osteoclast formation [79]. Indeed, the blockade of TNF can be used as a probe to understand the molecular basis of osteoclastogenesis and it may be a target for therapeutic agent development.
3. Cytokine Inhibition Strategies via MAPK Blockade
The contemporary concept of periodontal therapy focuses on eliminating bacteria through mechanical and chemotherapeutics means because the role of bacteria in the initiation and progress of periodontal disease is undisputed. Various therapeutic strategies aimed at the microorganisms, including local and systemic delivery of antimicrobial and antibiotic agents, have been studied over the years, but none of these methods has proven universally efficacious, particularly in the case of tissue-invasive species such as Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans). As we understand more about the immune response in the pathobiology of infectious diseases, including periodontitis, therapeutic strategies have focused more on host modulation. For example, therapeutic manipulation involving inhibition of TLR signaling using a small molecule inhibitor TAK-242 has been demonstrated to be beneficial to sepsis [80]. Specific soluble receptors (antagonists) of IL-1 and TNF-α inhibited the progression of bone loss by reducing the formation of osteoclast and recruitment of inflammatory cells in a nonhuman primate experimental periodontitis model [81]. Approaches to block the progression of inflammatory bone loss observed in periodontitis include host modulation of MMPs, COX2, and arachidonic acid metabolites. However, these therapies target singular mechanisms of alveolar bone destruction. Cytokines are well known to compensate for one another, thereby limiting the effect of cytokine-specific inhibitors. Alternatively, targeting a common regulatory mechanism for multiple cytokines may repress periodontal disease progression and improve treatment response. Also, inflammatory cell signaling pathways that generate inflammatory and tissue destruction proteins have become promising therapeutic targets. Therapeutic modulation of signaling pathways can affect various genes, depending not only on the pathway but also on the relative position targeted for inhibition in the signaling cascade. Because the MKK-MAPK-MK2 pathway phosphorylates downstream intermediates and regulates ARE proinflammatory cytokines, including IL-6, TNF-α, GM-CSF, IL-8, and iNOS, through mRNA stability, it and its pathway components could be excellent targets for therapeutic designs.
3.1. p38 Inhibition Studies
As described previously, p38 MAPK is an upstream effector common to many inflammatory cytokines. Activation of p38 MAPK signaling mediates inflammatory cytokine expressions such as IL-1β, IL-6, and TNF-α either directly or indirectly. These cytokines synergistically stimulate the production of other inflammatory cytokines, MMPs, and prostanoids [82–86]. p38 has also been implicated in the regulation of, IL-3, IL-8, macrophage inhibitory protein 1-α (MIP), GM-CSF, VEGF, urokinase-type plasminogen activator, and inducible NO synthase [87–90]. Evidence suggests that it is involved in rheumatoid arthritis, Alzheimer's disease, ischemic heart disease, asthma, dermatitis, inflammatory bowel disease, and periodontitis [86, 91–98].
Four members of the p38 MAPK family have been cloned: p38-α, -β, -γ, and -δ. All isoforms share conserved residues involved in ATP and ion binding and share significant sequence homology in their kinase domain and the 24- to 27 amino acid N-terminal to this domain. These regions are most likely to be involved in substrate specificity and activity. The α isoform is ubiquitously expressed to induce apoptosis, whereas the β isoforms is highly expressed in the brain and heart to promote cell survival in cardiac muscle cells. The γ isoform is chiefly expressed in muscle cells, and the δ isoform is expressed in the lung, kidney, gut, and salivary gland epithelium.
Bicyclic imidazole (compound SB203580) was initially identified to be an ATP-competitive inhibitor of p38 kinase [99], and later, more potent and specific inhibitors such as VX-745 and BIRB-796 were explored [100]. These developments lead to p38 kinase inhibition therapies which have been shown to be efficacious in several animal disease models, including rheumatoid arthritis (RA), psoriasis, Crohn's disease, stroke, asthma, chronic obstructive pulmonary disease (COPD), and periodontitis [99, 101–105]. For example, p38 inhibitors SC409 and SD282 have been shown to be efficacious in reducing and reversing bone and cartilage destruction in an experimental arthritis model [91]. In LPS-induced arthritis, mice treated with p38 MAPK inhibitors RO4399247 and AVE8677 diminished IL-6 to background levels [106]. Given that cytokines act synergistically, simultaneously blocking them is substantially more effective than blocking one alone. In the first study to test p38 inhibitors in humans, a single dose decreased TNF-α, IL-1, and -6 by 90%. Among inhibitors, an important distinction among isoforms is their ability to be inhibited by SB203580. Such inhibitors have 14.3 to >1,000 times greater activity against p38α than p38β, p38γ, or p38δ isoforms [91].
Research from our laboratory documents the relevance of p38 MAPK in the regulation of expression of IL-6, MMP-13, and receptor activator of NF-κB ligand in periodontally relevant resident cells, such as fibroblasts and osteoblasts, in vitro [17, 59, 107, 108]. In vivo, we have shown the significance of p38 MAPK signaling in periodontal disease progression, in which orally active p38 inhibitors reduced periopathogenic LPS-induced bone destruction in a rat model [105]. To study the preventive function of p38α inhibitors in periopathogenic LPS-induced experimental alveolar bone loss using a rat model, two simultaneous doses of SD-282 (15 or 45 mg/kg) were administered twice daily by oral gavage for 8 weeks. Bone area and volumetric analysis by μCT indicated significant bone volume loss with LPS treatment, but this was blocked with both doses of p38α inhibitor (see Figure 3). Histological examination indicated significantly fewer tartrate-resistant acid phosphatase-positive osteoclasts adjacent to the areas of active bone resorption, including the periodontal ligament area, and a significant decrease in IL-6, IL-1β, and TNF-α expression in p38 inhibitor-treated groups compared with LPS groups by immunostaining. This proof-of-principle study supports—for the first time—the role of an orally active p38α MAPK inhibitor (SD-282) to potentially benefit LPS-induced alveolar bone loss. These data also suggest that the α isoform is the predominate isoform expressed in LPS-induced periodontal disease pathology.
Figure 3.
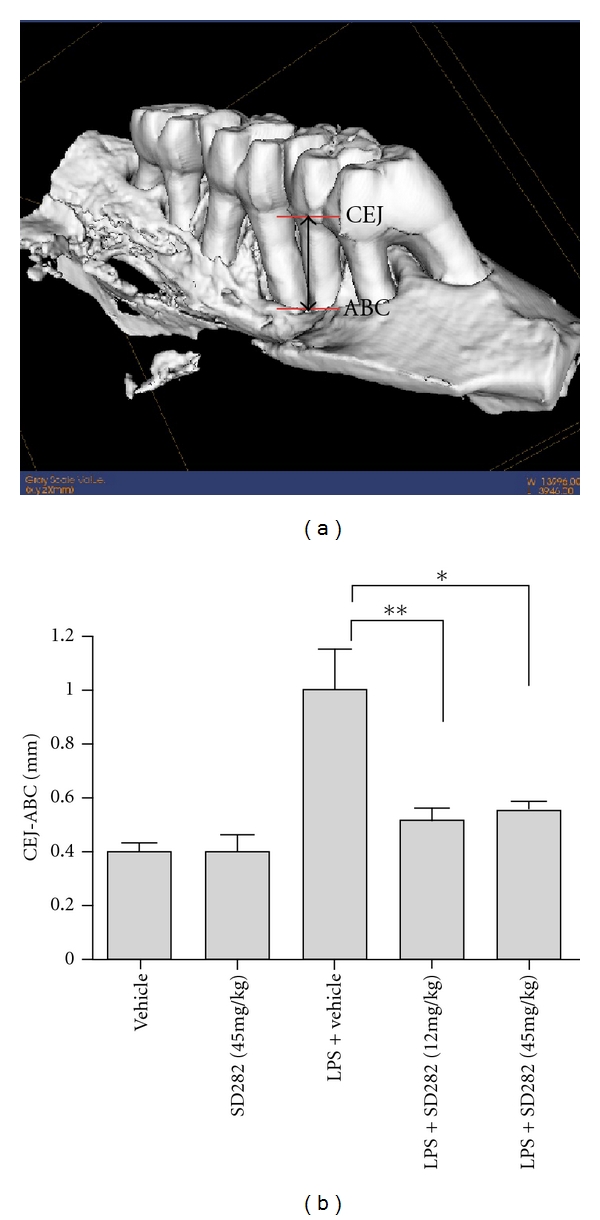
A. actinomycetemcomitans LPS induces significant linear bone loss which is blocked by SD-282. (a) Reformatted μCT isoform display from 8 weeks A. actinomycetemcomitans LPS-injected rat maxillae exhibits dramatic palatal and interproximal bone loss. Landmarks used for linear measurements were the cementoenamel junction (CEJ) to the alveolar bone crest (ABC). Differences between these anatomical locations using defined locations of 2D displays determined alveolar bone loss. (b) Linear bone loss as measured from the CEF to AB (Mean ± SEM). Significant bone loss (P < 0.01) was observed between control (n = 6) and A. actinomycetemcomitans LPS injected rats (n = 12). Significant reduction of LPS-induced periodontal bone loss (**P < 0.01 for SD-282 [15 mg/kg; n = 8] and *P < 0.05 for SD-282 [45 mg/kg; n = 8]) [105]. Reproduced with permission.
Clinically, the therapeutic goal is to prevent further advancement of alveolar bone loss. Thus, in a second therapeutic model, we investigated the effect of a p38 MAPK inhibitor on established periodontal disease [109]. The periodontal disease state was established by LPS injections to the palatal molar gingiva three times per week for 4 weeks. p38α MAPK inhibitor SD282 (45 mg/kg) was administrated from weeks 5 through 8 via oral gavage in addition to LPS injections. The data from this study revealed that treatment with an orally active p38 MAPK inhibitor stopped the established periodontal disease progression in vivo and decreased inflammatory cytokine (IL-1β, TNF-α) expression and osteoclastogenesis. Interestingly, in this study, SD282 had a slight anabolic effect; the difference between the 8-week LPS-only and LPS + SD282 groups was significant for increased bone volume. The reasons for this are unclear and may be due to a relatively high suppression of osteoclastogenesis without compensatory cessation of osteoblastic differentiation. Conceptually, this makes p38 inhibitor strategies appealing as a host-modulating agent for treatment of periodontitis because physiologic bone turnover (induced by PTH/PTHrP) would occur, but inflammatory bone loss (induced by LPS, IL-1β, and TNF-α) would be pharmacologically antagonized.
Collectively, these data highlight the therapeutic potential of this novel class of inhibitors in bacterial-induced alveolar bone loss—the hallmark of periodontitis, but developing p38 inhibitors as a therapeutics in clinical settings have failed due to unacceptable safety profiles, central role for activation of various downstream kinases and transcription factors, ubiquitous expression, toxicity, significant off-target effects, and lack of oral bioavailability [110]. Preclinical and clinical side effects include hepatotoxicity, cardiotoxicity, light-headedness, central nervous system toxicities, skin rash, gastrointestinal tract symptoms, and bacterial infections [88, 111–115]. In addition, p38 MAPK-knockout mouse is embryonic lethal [90, 116–118]. Until now none of these p38 inhibitors has been approved for clinical usage.
3.2. Intraoral Silencing of MK2 Signaling
Due to the concerns about p38 inhibitors indicated above, targeting downstream substrates of p38 MAPK and factors that regulate transcription, nuclear export, mRNA stability, and translation could be a promising therapeutic alternative for inhibiting inflammatory gene expression to treat various inflammatory diseases. As a direct substrate of the stress-activated MAPK p38α and β[119], MAPK-activated protein kinase 2 (MAPKAPK-2, MK2) is regulated exclusively by p38α/β [119].
Landmark studies performed with MK2-deficient cells in vitro or with MK2-deficient mice in vivo clearly revealed the physiological roles of MK2 activation. The studies performed with MK2-deficient cells in vitro demonstrated a central role of MK2 in the production of proinflammatory mediators such as TNF-α, IL-1β, MIP-1α, IL-8, IL-6, and INFγ [120]. MK2 is also involved in atherogenesis by promoting the recruitment of inflammatory monocytes/macrophages through increasing endothelial expression of VCAM-1 and MCP-1 [121]. MK2-deficient mice show an increased stress resistance to LPS-induced endotoxic shock, owing to a severely impaired inflammatory response leading to a 90% reduction in TNF-α production, as well as reduction of IFN-γ, IL-1β, IL-6, and nitric oxide [120]. The MK2 gene deletion protected DBA/1LacJ mice from collagen-induced arthritis due to a significantly lower LPS-induced TNF-α and IL-6 serum levels when compared with wild-type controls [122]. Deficiency of MK2 also protected against cerebral ischemic injury [123]. In addition, MK2 also participates in other diverse cellular processes such as cell division, apoptosis, cell differentiation, endocytosis, reorganization of the cytoskeleton, cell migration, cell cycle progression, chromatin remodeling, respiratory burst, and chemotaxis [124–126].
Targeting MK2 should be a more specific target than p38, with potentially fewer side effects, because MK2 acts on a more limited downstream substrate repertoire compared to p38. Importantly, MK2-deficient mice are viable with a normal phenotype [90, 120]. Therefore, there has been much research exploiting MK2 as molecular target for the development of experimental therapeutics for number of conditions such as RA, Alzheimer's disease, atherosclerosis, and cancer. As periodontal disease has remarkably similar inflammatory pathways and mediator profiles with other inflammatory diseases [6, 127, 128], it is reasonable to anticipate that MK2 would be an attractive and potentially selective target for the treatment of periodontitis. However, targeting MK2 with small molecular inhibitors is complex and difficult because of the relatively planar ATP-binding site of this critical MAPK.
In our study, we hypothesized that silencing MK2 through an RNAi strategy would provide a novel anti-inflammatory target that selectively blocks signaling mechanisms needed for enhanced cytokine mRNA stability/translation in periodontitis progression. First, we validated MK2 silencing in cytokine production in vitro to evaluate the feasibility of choosing MK2, instead of p38, as a highly specific and potent drug candidate. Our data clearly showed that LPS-induced IL-6 expression was significantly attenuated, both at the mRNA and protein levels, a result consistent with previous observations in MK2−/−mice [120, 122]. We observed that MK2 siRNA delivery significantly reduced TNF-α mRNA and protein expression. The role of MK2 in the regulation of LPS-induced inflammatory cytokine gene expression is further confirmed by significant reductions of mRNA expression for COX-2, IL-1β, and the chemokine CXCL1 in cells transfected with MK2 siRNA. MK2 siRNA gene knockdown changed the activation of JNK and ERK MAPKs without obvious phospho-p38 expression variation, suggesting the existence of crosstalk and compensatory mechanisms and underscoring the complexities of Toll-like receptor signaling pathways. Secondly, in vivo studies employed the rat LPS-induced experimental periodontitis model to further elucidate the role of MK2 in the pathogenesis of periodontitis and evaluate the therapeutic potential by targeting MK2 employing an RNAi strategy in periodontal disease. The protection of MK2 siRNA from alveolar bone loss in LPS-induced periodontitis model was further verified by μCT analysis (see Figure 4; [129]). Histological examination displayed that MK2 siRNA in vivo delivery attenuated the inflammatory infiltrate associated with A. actinomycetemcomitans LPS-induced bone loss. This is consistent with the decrease of osteoclast formation after MK2 silencing. In conclusion, with an RNAi strategy, our recent work validated that MK2 plays a role in a preventive model of experimental periodontitis, suggesting a novel target for controlling periodontal inflammation.
Figure 4.
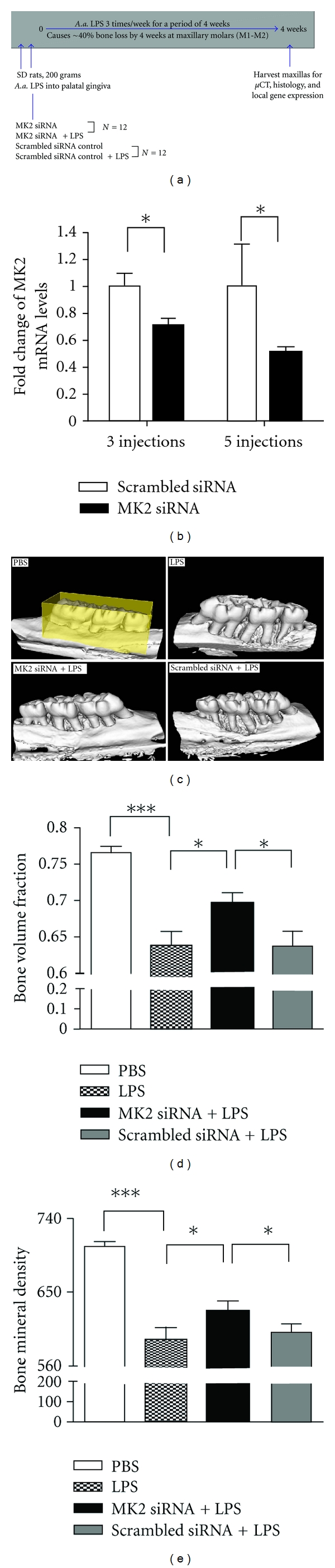
Specific MK2 siRNA in vivo delivery silenced target gene expression and reduced LPS-induced bone loss. (a) A schematic showing overall experimental protocol. (b) MK2 mRNA expression in palate gingiva after 3 and 5 times siRNA in vivo delivery. Results are expressed as mean ± SE (n = 5 or 6 rats/group, *P < 0.05). (c) Representative μCT images of rat maxillae from indicated treatment groups. ROI for quantitative analysis is highlighted. (d) Volumetric analysis of bone loss levels. (e) Bone mineral density (BMD) analysis of bone loss levels. (*P < 0.05, ***P < 0.001) [153]. Reproduced with permission.
3.3. MKP-1 Alters MAPK Signaling in Periodontal Disease Progression
MAPKs are activated by phosphorylation of critical tyrosine, serine, or threonine residues. Negative regulation of MAPK activity is mediated by the MAPK phosphatases (MKPs) that dephosphorylate these functional residues [130]. To date, this group of dual-specificity protein phosphatases (MKPs) includes 11 members in mammalian cells and the founding member is MKP-1. MKP-1 is localized to the nucleus, where it preferentially dephosphorylates activated p38 and JNK compared with ERK MAPK [131]. In vitro studies conducted using cultured immortalized macrophages provided compelling evidence that MKP-1 attenuates TNF-α and IL-6 after LPS stimulation [132–134]. MKP-1 functions as a feedback control mechanism, which governs the production of proinflammatory cytokines by deactivating p38 and JNK, thereby limiting proinflammatory cytokine biosynthesis in innate immune cells exposed to microbial components [132, 133, 135]. Consistent with in vitro data, MKP-1 null mice had markedly more production of proinflammatory cytokine TNF-α, IL-6, and an anti-inflammatory cytokine IL-10 compared with wild-type animals. Sustained p38 and JNK activity in response to stress support the central role of MKP-1 in the restraint of the innate immune response and in the prevention of endotoxemia, experimentally induced autoimmune arthritis, septic shock syndrome, and multiorgan dysfunction during pathogenic microbial infection [134–138].
The critical role of the p38/MKP-1 axis of regulation on the innate immune response and in maintaining bone homeostasis has been clearly demonstrated. Moreover, as MKP-1 not only regulates p38 MAPK, but also JNK and ERK activities, overexpression of MKP-1 has potent capacity to prevent an exuberate immune response and osteoclastogenesis in response to stimuli compared with p38 MAPK inhibitors. Using gain- and loss-of-function approaches, the role and potential therapeutic target of MKP-1 in inflammatory bone loss was explored.
First, decreased IL-6 expression was observed in murine macrophage cell line RAW264.7 transfected with expression vector containing MKP-1 cDNA in pSRII-Flag. This data provided in macrophages supports the role of MKP-1 in negative regulation of A. actinomycetemcomitans LPS-induced p38 activation and IL-6 production [139]. MKP-1 plays a crucial role in decreasing inflammatory cytokine biosynthesis. Then in our chronic periodontitis model, wild-type and MKP-1 null mice received A. actinomycetemcomitans LPS injection in the palatal region or PBS control 3 times/wk for 30 days. Results indicated that, in LPS injected MKP-1−/− mice, significantly greater bone loss occurred with more inflammatory infiltrate in the periodontal areas injected with LPS and a significant increase in osteoclastogenesis compared with MKP-1−/− control sites or either wild-type littermates. MKP-1 displayed a protective response in this more chronic model of inflammation and bone loss [139].
In gain-of-function experiments, MKP-1 was able to dephosphorylation all three MAPKs via MKP-1 gene transfer with recombinant adenovirus MKP-1 in rat macrophages. Ex vivo data indicated that MKP-1 gene transfer in bone marrow macrophages from MKP-1 KO mice significantly decreased IL-6, IL-10, TNF-α, and select chemokine levels compared with wild-type mice when stimulated by LPS [140]. In addition, bone marrow cultures from MKP-1 KO mice exhibited significantly more osteoclastogenesis induced by LPS than when compared with WT mice. This observation correlated with more osteoclasts seen in bone marrow cells of MKP-1 KO mice compared with osteoclasts from WT mice in response to LPS stimuli. Furthermore, in vivo MKP-1 gene transfer in an experimental periodontal disease model attenuated bone resorption induced by LPS (See Figure 5; [140]). Histological analysis confirmed that periodontal tissues transduced with Ad. MKP-1 exhibited less infiltrated inflammatory cells, less osteoclasts, and less IL-6 than compared with rats of control groups.
Figure 5.
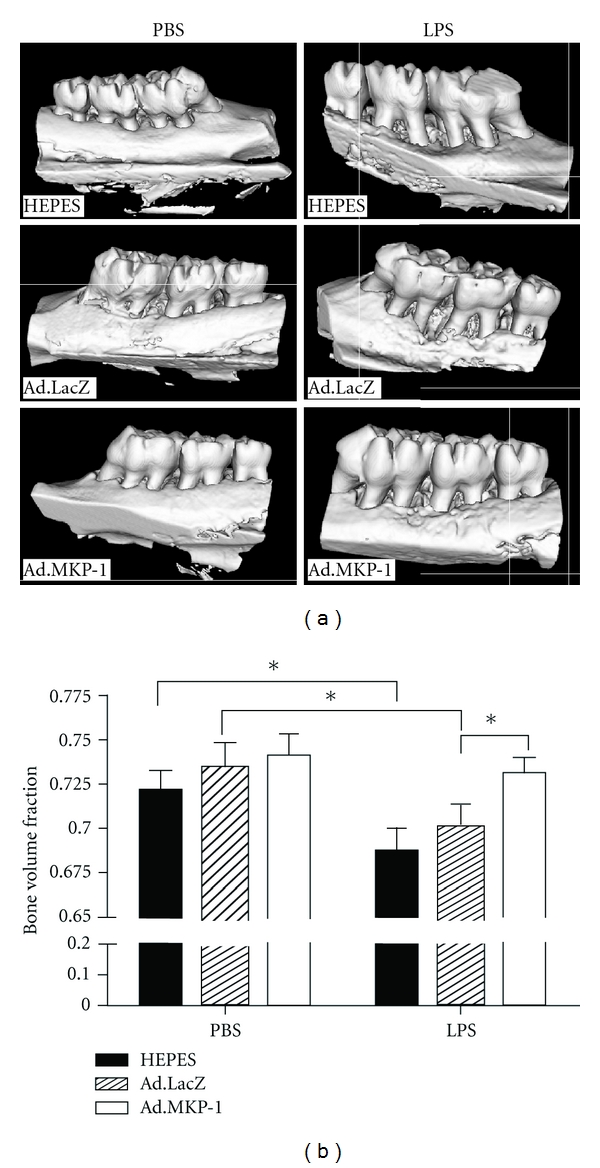
MKP-1 gene transfer alleviated bone resorption in rats after LPS challenge. Eight-week-old male Sprague-Dawley rats (17 rats/group) were injected either Ad.MKP-1, or Ad. LacZ (1×109 pfu in 4 μl), or HEPES buffered saline (4 μl). Forty-eight hours after the adenovirus injection, the rats were injected with 2 μl of either 20 μg of LPS (from A. actinomycetemcomitans) or PBS three times a week for four weeks. (a) Representative microcomputed tomography images of rat maxillae from indicated treatment groups. (b) Volumetric analysis of bone loss levels (n = 7 for PBS groups, n = 10 for LPS groups, *P < 0.05); [140]. Reproduced with permission.
Together, our studies indicate the importance of MKP-1 in the development of immune responses that contribute to LPS-induced alveolar bone loss. It can be used as a key therapeutic target to control of inflammation-induced bone loss associated with increased MAPK activation.
3.4. Local Gene Delivery of TTP as an Anti-Inflammatory Agent
Cells of the immune system tightly regulate the production of potentially harmful cytokines at many levels: transcriptional, posttranscriptional, translational, and posttranslational levels. Posttranscriptional regulation of cytokines occurs at different stages such as nuclear export, cytoplasmic localization, and stability/degradation. Central to the posttranscriptional regulatory events is the interaction of RNA with RNA-binding proteins that influence their splicing, localization, stability, and association with the translation machinery [141–143].
The cisacting elements, which interacted with RNA-binding protein, are AU-rich elements (AREs) [67, 144]. They are located in the 3′ untranslated regions (UTRs) of many cytokines (e.g., granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α, IL-2, IL-3, IL-6), and other proinflammatory factors (e.g., COX2 and MMP-13). A hallmark of AREs is the pentamer AUUUA that occurs either alone or clustered [145]. Recently, the structure and the functional significance of AREs have been clearly demonstrated by insertion strategy and deletion strategy.
At least 20 different proteins that can bind to ARE segments have been identified to date, including TTP, HuR, butyrate response factor (BRF)-1 and BRF-2, ARE/poly(U)-binding/degradation factor-1 (AUF-1), T-cell intracellular antigen-1 (TIA-1), and T-cell-restricted intracellular antigen-related protein (TIAR). However, only a subset of RNA-BPs has been shown to influence the stability or translational efficiency of target mRNAs. Some RNA-binding proteins, such as TTP, AU-binding factor 1, and K homology splicing-regulatory protein, promote mRNA decay, whereas others, like members of the Hu family, prevent mRNA degradation.
TTP is a well-characterized, zinc-finger-containing, RNA-binding protein. The function of TTP was elucidated through several studies with TTP-deficient mice. TTP deficiency is associated with cachexia, arthritis, autoimmunity, and myeloid hyperplasia, secondary to increased TNF-α and GM-CSF levels. In TTP−/− mice, the increased cytokine production was shown to be a result of increased mRNA stability [66, 146]. Many studies also have demonstrated that the overexpression of TTP promoted the decay of reporter transcripts that contained AU-rich sequences from TNF-α in vitro [147].
TTP binds to AREs located in 3′ untranslated regions of cytokine genes and targets them to the exosome for rapid degradation. In this context, TTP can be considered an anti-inflammatory protein. As a point of therapeutic potential, we hypothesized that targeting a common upstream RNA-binding protein, TTP, may inhibit periodontitis progression.
Using adenoviral-delivered TTP, TTP overexpression was evaluated in a preventive model of experimental periodontitis to determine whether altering cytokine mRNA stability affected pathological bone resorption (see Figure 6; [148]). In vivo analysis indicated a significant protective effect from inflammation-induced bone loss and inflammatory infiltrate in animals overexpressing TTP compared with reporter controls. In addition, significant reductions of IL-6, TNF-α, and PGE2 were observed after TTP overexpression in vitro through a mechanism consistent with targeting mRNA stability. Collectively, these findings provide experimental evidence that mRNA stability is a valid therapeutic target in inflammatory bone loss.
Figure 6.
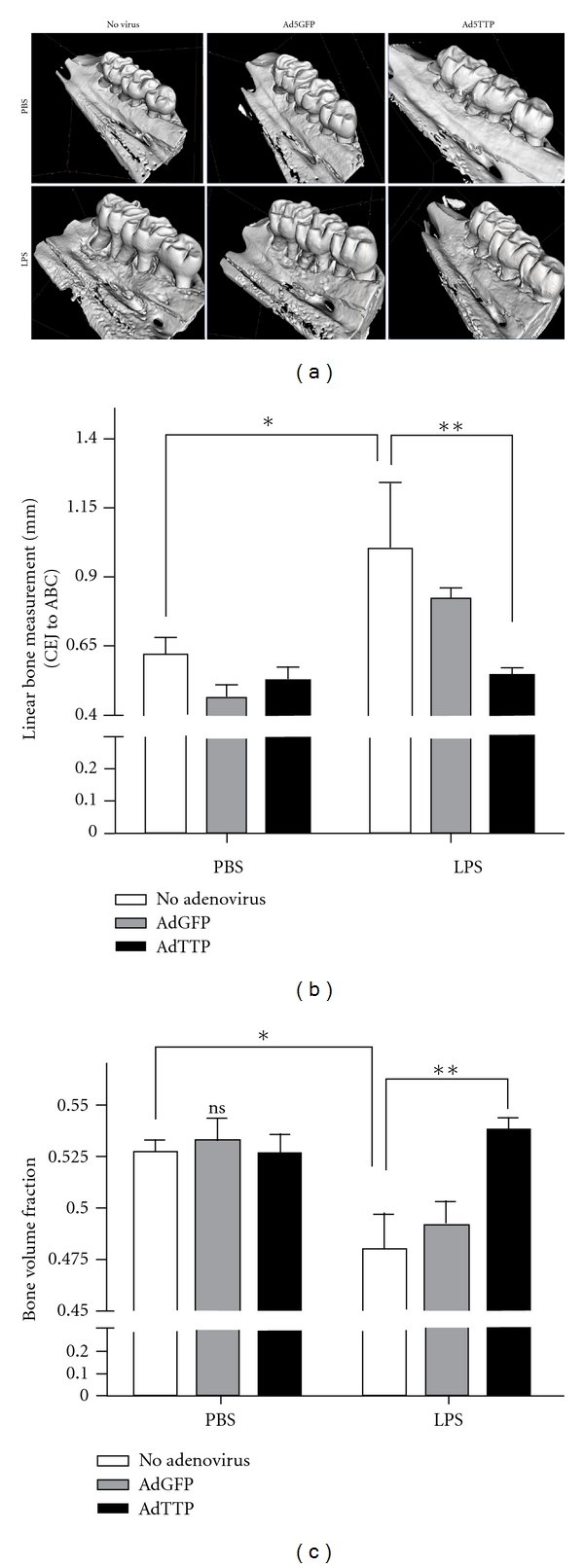
Ad5-TTP protects bone in experimental periodontal disease model. (a) representative microcomputed tomography images of rat maxillae from indicated treatment groups. (b) Linear measurement from cementoenamel junction (CEJ) to alveolar bone crest (ABC) (n = 8/group; *P < 0.05, **P < 0.01), (c) Volumetric analysis of bone loss levels (n = 8/group; *P < 0.05, **P < 0.01) [148]. Reproduced with permission.
In vitro, we focused on testing if p38/MK2 signaling is required for cytokines expression regulation via TTP activity. Using MC3T3-E1 cell line as an osteoblastic model, we observed that p38 MAPK regulates IL-1β-stimulated IL-6 at a posttranscriptional mechanism and one of the primary targets of IL-6 gene regulation is the 3′ UTR of IL-6 [107]. Using mouse embryonic fibroblasts (MEFs) derived from p38α −/− mice, we further confirmed the above conclusion and determined p38α as the critical isoform. We identified three ARE elements that require p38α signaling, and IL-6 3′-UTR-(56–173) is critical for p38α to promote mRNA stability [149]. After this, the role of TTP in posttranscriptional regulation of IL-6 was elucidated. Genetic and siRNA-mediated knockdown of TTP resulted in increased IL-6 production and overexpression of TTP had the reverse effect [150]. Significant IL-6 mRNA expression and a long half-life were observed in TTP−/− mice MEFs. Overexpression of TTP reduced IL-6 3′UTR luciferase reporter activity in an ARE-dependent manner. Mutation-based luciferase assays show that ARE2, ARE3, and ARE4 are required for TTP-mediated repression, and the constitutively activated p38-MK2 pathway abrogated TTP-mediated repression of IL-6 3′UTR reporter activity. An RNA immunoprecipitation assay indicated that the p38α deficiency resulted in increased affinity of TTP to IL-6 mRNA. Taken together, our serial studies showed that the RNA-binding protein, TTP, regulates IL-1β-induced IL-6 expression at the posttranscriptional level through an affinity shift for the transcript, which occurs in a p38 MAPK-dependent manner and involves specific AREs within the 3′UTR of the IL-6 mRNA.
4. Future Studies
Our research group has accumulated significant information that IL-6 regulation is highly dependent on p38 MAPK signaling in a variety of cell types [59, 105, 107, 108, 149, 151]. TTP directs mRNA stability of IL-6 largely in a p38 MAPK-dependent manner [150]. The clinical significance of this regulation was also demonstrated through inhibition of inflammation and bone loss via different strategies, such as small molecule inhibitors against p38 MAPK [105], MKP-1 overexpression, or TTP overexpression using gene transfer with recombinant adenovirus MKP-1 or TTP [140, 148, 149], and more recently using siRNA strategies targeting the p38 downstream kinase, MK2 [129]. Collectively, our series of studies targeting multiple molecules situated in inflammatory-related MAPK signaling cascade either to elucidate the functional mechanism or to verify the therapeutic potential for periodontal disease provide strong proof-of-principle evidence.
However, disease progression is complex with multiple aspects worthy of consideration. In human periodontal pathology, infectious bacteria are capable of interacting with the host. Although LPS is a potent inflammatory mediator, other components (e.g., cytoplasmic membranes, peptidoglycans, outer membrane proteins, capsules, and cell-surface fimbriae) within live organisms can induce apoptosis and modulate or evade the immune response. In addition, other classes of PRRs, such as Nod-like receptors (NLRs), have been recognized to have important roles in sensing intracellular infections [152]. NLRs have also been shown to modulate various signaling pathways, including p38 MAPK and NF-κB, underscoring the complexity of TLR signaling and the crosstalk with other signaling pathways involved in the pathobiology of periodontal disease. Cytokines in the periodontium integrate aspects of innate and adaptive immunity. It is becoming clear, however, that cytokines do not function in isolation, but form complex interactive networks involving both pro- and anti-inflammatory effects.
Future studies are being planned with large animals or nonhuman primates to further address the therapeutic effect of small molecule inhibitor strategies. In addition, elucidation of fundamental mechanisms, finding powerful target candidates, developing clinically viable delivery systems, and optimizing dose and delivery routes must be addressed, along with exploration of different infection models to elucidate the potential of these strategies to arrest other inflammatory diseases.
Acknowledgment
This study is supported by Grants from National Institutes of Health (NIH): 1R01DE021423 (KK), 1R01DE018290 (KK), 2P20 RR017696 (KK), and T32 DE017551 (MV).
References
- 1.Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. Journal of Bacteriology. 2001;183(12):3770–3783. doi: 10.1128/JB.183.12.3770-3783.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Socransky SS, Haffajee AD, Ximenez-Fyvie LA, Feres M, Mager D. Ecological considerations in the treatment of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis periodontal infections. Periodontology 2000. 1999;20(1):341–362. doi: 10.1111/j.1600-0757.1999.tb00165.x. [DOI] [PubMed] [Google Scholar]
- 3.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. Microbial complexes in subgingival plaque. Journal of Clinical Periodontology. 1998;25(2):134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 4.Savage A, Eaton KA, Moles DR, Needleman I. A systematic review of definitions of periodontitis and methods that have been used to identify this disease. Journal of Clinical Periodontology. 2009;36(6):458–467. doi: 10.1111/j.1600-051X.2009.01408.x. [DOI] [PubMed] [Google Scholar]
- 5.Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontology 2000. 1997;14:9–11. doi: 10.1111/j.1600-0757.1997.tb00189.x. [DOI] [PubMed] [Google Scholar]
- 6.Kirkwood KL, Cirelli JA, Rogers JE, Giannobile WV. Novel host response therapeutic approaches to treat periodontal diseases. Periodontology 2000. 2007;43(1):294–315. doi: 10.1111/j.1600-0757.2006.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Löe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. Journal of Clinical Periodontology. 1986;13(5):431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 8.Baelum V, Fejerskov O, Karring T. Oral hygiene, gingivitis and periodontal breakdown in adult Tanzanians. Journal of Periodontal Research. 1986;21(3):221–232. doi: 10.1111/j.1600-0765.1986.tb01454.x. [DOI] [PubMed] [Google Scholar]
- 9.Baelum V, Fejerskov O, Manji F. Periodontal diseases in adult Kenyans. Journal of Clinical Periodontology. 1988;15(7):445–452. doi: 10.1111/j.1600-051x.1988.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 10.Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Compendium of Continuing Education in Dentistry. 2008;29(7):402–413. [PubMed] [Google Scholar]
- 11.Feller L, Lemmer J. Necrotizing periodontal diseases in HIV-seropositive subjects: pathogenic mechanisms. Journal of the International Academy of Periodontology. 2008;10(1):10–15. [PubMed] [Google Scholar]
- 12.Handfield M, Baker HV, Lamont RJ. Beyond good and evil in the oral cavity: insights into host-microbe relationships derived from transcriptional profiling of gingival cells. Journal of Dental Research. 2008;87(3):203–223. doi: 10.1177/154405910808700302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor JJ. Cytokine regulation of immune responses to Porphyromonas gingivalis. Periodontology 2000. 2010;54(1):160–194. doi: 10.1111/j.1600-0757.2009.00344.x. [DOI] [PubMed] [Google Scholar]
- 14.Beutler B, Hoebe K, Du X, Ulevitch RJ. How we detect microbes and respond to them: the Toll-like receptors and their transducers. Journal of Leukocyte Biology. 2003;74(4):479–485. doi: 10.1189/jlb.0203082. [DOI] [PubMed] [Google Scholar]
- 15.Kirkwood KL, Rossa C. The potential of p38 MAPK inhibitors to modulate periodontal infections. Current Drug Metabolism. 2009;10(1):55–67. doi: 10.2174/138920009787048347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preshaw PM, Taylor JJ. How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? Journal of Clinical Periodontology. 2011;38(supplement 11):60–84. doi: 10.1111/j.1600-051X.2010.01671.x. [DOI] [PubMed] [Google Scholar]
- 17.Rossa C, Jr., Min L, Bronson P, Kirkwood KL. Transcriptional activation of MMP-13 by periodontal pathogenic LPS requires p38 MAP kinase. Journal of Endotoxin Research. 2007;13(2):85–93. doi: 10.1177/0968051907079118. [DOI] [PubMed] [Google Scholar]
- 18.Sugawara Y, Uehara A, Fujimoto Y, et al. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. Journal of Dental Research. 2006;85(6):524–529. doi: 10.1177/154405910608500609. [DOI] [PubMed] [Google Scholar]
- 19.Mori Y, Yoshimura A, Ukai T, Lien E, Espevik T, Hara Y. Immunohistochemical localization of Toll-like receptors 2 and 4 in gingival tissue from patients with periodontitis. Oral Microbiology and Immunology. 2003;18(1):54–58. doi: 10.1034/j.1399-302x.2003.180109.x. [DOI] [PubMed] [Google Scholar]
- 20.Ren L, Leung WK, Darveau RP, Jin L. The expression profile of lipopolysaccharide-binding protein, membrane-bound CD14, and toll-like receptors 2 and 4 in chronic periodontitis. Journal of Periodontology. 2005;76(11):1950–1959. doi: 10.1902/jop.2005.76.11.1950. [DOI] [PubMed] [Google Scholar]
- 21.Kajita K, Honda T, Amanuma R, et al. Quantitative messenger RNA expression of Toll-like receptors and interferon-α1 in gingivitis and periodontitis. Oral Microbiology and Immunology. 2007;22(6):398–402. doi: 10.1111/j.1399-302X.2007.00377.x. [DOI] [PubMed] [Google Scholar]
- 22.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Hemmi H, Kaisho T, Takeuchi O, et al. Small-antiviral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature Immunology. 2002;3(2):196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 24.Hoshino K, Kaisho T, Iwabe T, Takeuchi O, Akira S. Differential involvement of IFN-β in toll-like receptor-stimulated dendritic cell activation. International Immunology. 2002;14(10):1225–1231. doi: 10.1093/intimm/dxf089. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nature Immunology. 2003;4(11):1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420(6913):324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 28.O’Neill LAJ, Greene C. Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects, and plants. Journal of Leukocyte Biology. 1998;63(6):650–657. [PubMed] [Google Scholar]
- 29.An H, Yu Y, Zhang M, et al. Involvement of ERK, p38 and NF-κB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology. 2002;106(1):38–45. doi: 10.1046/j.1365-2567.2002.01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akira S. Mammalian Toll-like receptors. Current Opinion in Immunology. 2003;15(1):5–11. doi: 10.1016/s0952-7915(02)00013-4. [DOI] [PubMed] [Google Scholar]
- 31.Beutler B, Kruys V. Lipopolysaccharide signal transduction, regulation of tumor necrosis factor biosynthesis, and signaling by tumor necrosis factor itself. Journal of Cardiovascular Pharmacology. 1995;25(2, supplement):S1–S8. doi: 10.1097/00005344-199500252-00002. [DOI] [PubMed] [Google Scholar]
- 32.Ono K, Han J. The p38 signal transduction pathway Activation and function. Cellular Signalling. 2000;12(1):1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 33.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annual Review of Immunology. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 34.Bainbridge BW, Darveau RP. Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontologica Scandinavica. 2001;59(3):131–138. doi: 10.1080/000163501750266710. [DOI] [PubMed] [Google Scholar]
- 35.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. Journal of Immunology. 2000;165(2):618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 36.Madianos PN, Bobetsis YA, Kinane DF. Generation of inflammatory stimuli: how bacteria set up inflammatory responses in the gingiva. Journal of Clinical Periodontology. 2005;32(6):57–71. doi: 10.1111/j.1600-051X.2005.00821.x. [DOI] [PubMed] [Google Scholar]
- 37.Choi BK, Jung JH, Suh HY, et al. Activation of matrix metalloproteinase-2 by a novel oral spirochetal species Treponema lecithinolyticum. Journal of Periodontology. 2001;72(11):1594–1600. doi: 10.1902/jop.2001.72.11.1594. [DOI] [PubMed] [Google Scholar]
- 38.Jotwani R, Palucka AK, Al-Quotub M, et al. Mature dendritic cells infiltrate the T cell-rich region of oral mucosa in chronic periodontitis: in situ, in vivo, and in vitro studies. Journal of Immunology. 2001;167(8):4693–4700. doi: 10.4049/jimmunol.167.8.4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahanonda R, Pothiraksanon P, Sa-Ard-Iam N, et al. The effects of Porphyromonas gingivalis LPS and Actinobacillus actinomycetemcomitans LPS on human dendritic cells in vitro, and in a mouse model in vivo. Asian Pacific Journal of Allergy and Immunology. 2006;24(4):223–228. [PubMed] [Google Scholar]
- 40.Garlet GP, Martins W, Ferreira BR, Milanezi CM, Silva JS. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. Journal of Periodontal Research. 2003;38(2):210–217. doi: 10.1034/j.1600-0765.2003.02012.x. [DOI] [PubMed] [Google Scholar]
- 41.Garlet GP, Cardoso CR, Silva TA, et al. Cytokine pattern determines the progression of experimental periodontal disease induced by Actinobacillus actinomycetemcomitans through the modulation of MMPs, RANKL, and their physiological inhibitors. Oral Microbiology and Immunology. 2006;21(1):12–20. doi: 10.1111/j.1399-302X.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 42.Stashenko P, Fujiyoshi P, Obernesser MS, Prostak L, Haffajee AD, Socransky SS. Levels of interleukin 1 beta in tissue from sites of active periodontal disease. Journal of Clinical Periodontology. 1991;18(7):548–554. doi: 10.1111/j.1600-051x.1991.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 43.Stashenko P, Jandinski JJ, Fujiyoshi P, Rynar J, Socransky SS. Tissue levels of bone resorptive cytokines in periodontal disease. Journal of Periodontology. 1991;62(8):504–509. doi: 10.1902/jop.1991.62.8.504. [DOI] [PubMed] [Google Scholar]
- 44.Lee HJ, Kang IK, Chung CP, Choi SM. The subgingival microflora and gingival crevicular fluid cytokines in refractory periodontitis. Journal of Clinical Periodontology. 1995;22(11):885–890. doi: 10.1111/j.1600-051x.1995.tb01788.x. [DOI] [PubMed] [Google Scholar]
- 45.Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of a direct relationship between neutrophil collagenase activity and periodontal tissue destruction in vivo: role of active enzyme in human periodontitis. Journal of Periodontal Research. 1995;30(1):23–33. doi: 10.1111/j.1600-0765.1995.tb01249.x. [DOI] [PubMed] [Google Scholar]
- 46.Ejeil AL, Igondjo-Tchen S, Ghomrasseni S, Pellat B, Godeau G, Gogly B. Expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in healthy and diseased human gingiva. Journal of Periodontology. 2003;74(2):188–195. doi: 10.1902/jop.2003.74.2.188. [DOI] [PubMed] [Google Scholar]
- 47.Ejeil AL, Gaultier F, Igondjo-Tchen S, et al. Are cytokines linked to collagen breakdown during periodontal disease progression? Journal of Periodontology. 2003;74(2):196–201. doi: 10.1902/jop.2003.74.2.196. [DOI] [PubMed] [Google Scholar]
- 48.Reddi K, Nair SP, White PA, et al. Surface-associated material from the bacterium Actinobacillus actinomycetemcomitans contains a peptide which, in contrast to lipopolysaccharide, directly stimulates fibroblast interleukin-6 gene transcription. European Journal of Biochemistry. 1996;236(3):871–876. doi: 10.1111/j.1432-1033.1996.00871.x. [DOI] [PubMed] [Google Scholar]
- 49.Engebretson SP, Hey-Hadavi J, Ehrhardt FJ, et al. Gingival crevicular fluid levels of interleukin-1β and glycemic control in patients with chronic periodontitis and type 2 diabetes. Journal of Periodontology. 2004;75(9):1203–1208. doi: 10.1902/jop.2004.75.9.1203. [DOI] [PubMed] [Google Scholar]
- 50.Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Levels of interleukin-1β, -8, and -10 and RANTES in gingnival crevicular fluid and cell populations in adult periodontitis patients and the effect of periodontal treatment. Journal of Periodontology. 2000;71(10):1535–1545. doi: 10.1902/jop.2000.71.10.1535. [DOI] [PubMed] [Google Scholar]
- 51.Górska R, Gregorek H, Kowalski J, Laskus-Perendyk A, Syczewska M, Madaliński K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. Journal of Clinical Periodontology. 2003;30(12):1046–1052. doi: 10.1046/j.0303-6979.2003.00425.x. [DOI] [PubMed] [Google Scholar]
- 52.Geivelis M, Turner DW, Pederson ED, Lamberts BL. Measurements of interleukin-6 in gingival crevicular fluid from adults with destructive periodontal disease. Journal of Periodontology. 1993;64(10):980–983. doi: 10.1902/jop.1993.64.10.980. [DOI] [PubMed] [Google Scholar]
- 53.Hirose K, Isogai E, Miura H, Ueda I. Levels of Porphyromonas gingivalis fimbriae and inflammatory cytokines in gingival crevicular fluid from adult human subjects. Microbiology and Immunology. 1997;41(1):21–26. doi: 10.1111/j.1348-0421.1997.tb01168.x. [DOI] [PubMed] [Google Scholar]
- 54.Reinhardt RA, Masada MP, Kaldahl WB, et al. Gingival fluid IL-1 and IL-6 levels in refractory periodontitis. Journal of Clinical Periodontology. 1993;20(3):225–231. doi: 10.1111/j.1600-051x.1993.tb00348.x. [DOI] [PubMed] [Google Scholar]
- 55.Al-Shammari KF, Giannobile WV, Aldredge WA, et al. Effect of non-surgical periodontal therapy on C-telopeptide pyridinoline cross-links (ICTP) and interleukin-1 levels. Journal of Periodontology. 2001;72(8):1045–1051. doi: 10.1902/jop.2001.72.8.1045. [DOI] [PubMed] [Google Scholar]
- 56.Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. Journal of Periodontology. 2003;74(3):391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 57.Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. The Journal of Biological Chemistry. 2001;276(23):20659–20672. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- 58.Jiang Y, Mehta CK, Hsu TY, Alsulaimani FFH. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infection and Immunity. 2002;70(6):3143–3148. doi: 10.1128/IAI.70.6.3143-3148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rossa C, Ehmann K, Liu M, Patil C, Kirkwood KL. MKK3/6-p38 MAPK signaling is required for IL-1β and TNF-α-induced RANKL expression in bone marrow stromal cells. Journal of Interferon and Cytokine Research. 2006;26(10):719–729. doi: 10.1089/jir.2006.26.719. [DOI] [PubMed] [Google Scholar]
- 60.Teng YTA, Nguyen H, Gao X, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. Journal of Clinical Investigation. 2000;106(6):R59–R67. doi: 10.1172/jci10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taubman MA, Valverde P, Han X, Kawai T. Immune response: they key to bone resorption in periodontal disease. Journal of Periodontology. 2005;76(11, supplement):2033–2041. doi: 10.1902/jop.2005.76.11-S.2033. [DOI] [PubMed] [Google Scholar]
- 62.Mogi M, Otogoto J, Ota N, Togari A. Differential expression of RANKL and osteoprotegerin in gingival crevicular fluid of patients with periodontitis. Journal of Dental Research. 2004;83(2):166–169. doi: 10.1177/154405910408300216. [DOI] [PubMed] [Google Scholar]
- 63.Kawai T, Matsuyama T, Hosokawa Y, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. American Journal of Pathology. 2006;169(3):987–998. doi: 10.2353/ajpath.2006.060180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. Journal of Molecular Medicine. 1996;74(10):589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 65.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. The Journal of Biological Chemistry. 1993;268(20):14553–14556. [PubMed] [Google Scholar]
- 66.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science. 1998;281(5379):1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 67.Mahtani KR, Brook M, Dean JLE, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Molecular and Cellular Biology. 2001;21(19):6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stoecklin G, Stubbs T, Kedersha N, et al. MK2-induced tristetraprolin:14-3-3 Complexes prevent stress granule association and ARE-mRNA decay. EMBO Journal. 2004;23(6):1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Current Opinion in Cell Biology. 2000;12(2):186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 70.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–2147. [PubMed] [Google Scholar]
- 71.Giovine D. Cytokine Molecular Biology—A Practical Approach. 3rd edition. 2000. Detection and population analysis of IL-1 and TNF gene polymorphisms; pp. 21–46. [Google Scholar]
- 72.Rogers MA, Figliomeni L, Baluchova K, et al. Do interleukin-1 polymorphisms predict the development of periodontitis or the success of dental implants? Journal of Periodontal Research. 2002;37(1):37–41. doi: 10.1034/j.1600-0765.2002.00651.x. [DOI] [PubMed] [Google Scholar]
- 73.Yin L, Li L, Pan Y, Tan Y, He A. IL-1 beta mRNA and TNF-alpha mRNA expression in gingival tissues of patients with adult periodontitis. Hua xi Kou Qiang yi Xue za Zhi. 2001;19(5):318–321. [PubMed] [Google Scholar]
- 74.Ebersole JL, Singer RE, Steffensen B, Filloon T, Kornman KS. Inflammatory mediators and immunoglobulins in GCF from healthy, gingivitis and periodontitis sites. Journal of Periodontal Research. 1993;28(6):543–546. doi: 10.1111/j.1600-0765.1993.tb02121.x. [DOI] [PubMed] [Google Scholar]
- 75.Howells GL. Cytokine networks in destructive periodontal disease. Oral diseases. 1995;1(4):266–270. doi: 10.1111/j.1601-0825.1995.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 76.Assuma R, Oates T, Cochran D, Amar S, Graves DT. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. Journal of Immunology. 1998;160(1):403–409. [PubMed] [Google Scholar]
- 77.Birkedal-Hansen H. Role of matrix metalloproteinases in human periodontal diseases. Journal of Periodontology. 1993;64(5):474–484. doi: 10.1902/jop.1993.64.5s.474. [DOI] [PubMed] [Google Scholar]
- 78.Bouwmeester T, Bauch A, Ruffner H, et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nature Cell Biology. 2004;6(2):97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 79.Cheng X, Kinosaki M, Murali R, Greene MI. The TNF receptor superfamily: role in immune inflammation and bone formation. Immunologic Research. 2003;27(2-3):287–294. doi: 10.1385/IR:27:2-3:287. [DOI] [PubMed] [Google Scholar]
- 80.Ii M, Matsunaga N, Hazeki K, et al. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Molecular Pharmacology. 2006;69(4):1288–1295. doi: 10.1124/mol.105.019695. [DOI] [PubMed] [Google Scholar]
- 81.Graves DT. The potential role of chemokines and inflammatory cytokines in periodontal disease progression. Clinical Infectious Diseases. 1999;28(3):482–490. doi: 10.1086/515178. [DOI] [PubMed] [Google Scholar]
- 82.Ridley SH, Sarsfield SJ, Lee JC, et al. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. Journal of Immunology. 1997;158(7):3165–3173. [PubMed] [Google Scholar]
- 83.Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-γ . Journal of Infectious Diseases. 1999;179(4):939–944. doi: 10.1086/314659. [DOI] [PubMed] [Google Scholar]
- 84.Dean JLE, Brook M, Clark AR, Saklatvala J. p38 Mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. The Journal of Biological Chemistry. 1999;274(1):264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 85.Underwood DC, Osborn RR, Bochnowicz S, et al. SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. American Journal of Physiology. 2000;279(5):L895–L902. doi: 10.1152/ajplung.2000.279.5.L895. [DOI] [PubMed] [Google Scholar]
- 86.Mbalaviele G, Anderson G, Jones A, et al. Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. Journal of Pharmacology and Experimental Therapeutics. 2006;317(3):1044–1053. doi: 10.1124/jpet.105.100362. [DOI] [PubMed] [Google Scholar]
- 87.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. The Journal of Biological Chemistry. 1995;270(25):14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 88.Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Current Opinion in Pharmacology. 2004;4(4):372–377. doi: 10.1016/j.coph.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 89.Cuenda A, Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochimica et Biophysica Acta. 2007;1773(8):1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 90.Allen M, Svensson L, Roach M, Hambor J, McNeish J, Gabel CA. Deficiency of the stress kinase p38α results in embryonic lethality: characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. Journal of Experimental Medicine. 2000;191(5):859–869. doi: 10.1084/jem.191.5.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Medicherla S, Ma JY, Mangadu R, et al. A selective p38α mitogen-activated protein kinase inhibitor reverses cartilage and bone destruction in mice with collagen-induced arthritis. Journal of Pharmacology and Experimental Therapeutics. 2006;318(1):132–141. doi: 10.1124/jpet.105.098020. [DOI] [PubMed] [Google Scholar]
- 92.Wada Y, Nakajima-Yamada T, Yamada K, et al. R-130823, a novel inhibitor of p38 MAPK, ameliorates hyperalgesia and swelling in arthritis models. European Journal of Pharmacology. 2005;506(3):285–295. doi: 10.1016/j.ejphar.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 93.Culbert AA, Skaper SD, Howlett DR, et al. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity: relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. The Journal of Biological Chemistry. 2006;281(33):23658–23667. doi: 10.1074/jbc.M513646200. [DOI] [PubMed] [Google Scholar]
- 94.Takanami-Ohnishi Y, Amano S, Kimura S, et al. Essential role of p38 mitogen-activated protein kinase in contact hypersensitivity. The Journal of Biological Chemistry. 2002;277(40):37896–37903. doi: 10.1074/jbc.M207326200. [DOI] [PubMed] [Google Scholar]
- 95.Clark JE, Sarafraz N, Marber MS. Potential of p38-MAPK inhibitors in the treatment of ischaemic heart disease. Pharmacology and Therapeutics. 2007;116(2):192–206. doi: 10.1016/j.pharmthera.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 96.Duan W, Wong WSF. Targeting mitogen-activated protein kinases for asthma. Current Drug Targets. 2006;7(6):691–698. doi: 10.2174/138945006777435353. [DOI] [PubMed] [Google Scholar]
- 97.Sasaki Y, Aiba S. Dendritic cells and contact dermatitis. Clinical Reviews in Allergy and Immunology. 2007;33(1-2):27–34. doi: 10.1007/s12016-007-0034-7. [DOI] [PubMed] [Google Scholar]
- 98.Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. International Journal of Chronic Obstructive Pulmonary Disease. 2006;1(2):137–150. doi: 10.2147/copd.2006.1.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. Journal of Pharmacology and Experimental Therapeutics. 1996;279(3):1453–1461. [PubMed] [Google Scholar]
- 100.Gaestel M, Mengel A, Bothe U, Asadullah K. Protein kinases as small molecule inhibitor targets in inflammation. Current Medicinal Chemistry. 2007;14(21):2214–2234. doi: 10.2174/092986707781696636. [DOI] [PubMed] [Google Scholar]
- 101.Revesz L, Blum E, Di Padova FE, et al. Novel p38 inhibitors with potent oral efficacy in several models of rheumatoid arthritis. Bioorganic and Medicinal Chemistry Letters. 2004;14(13):3595–3599. doi: 10.1016/j.bmcl.2004.03.106. [DOI] [PubMed] [Google Scholar]
- 102.Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47(2-3):185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 103.Jackson JR, Bolognese B, Hillegass L, et al. Pharmacological effects of SB 220025, a selective inhibitor of p38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. Journal of Pharmacology and Experimental Therapeutics. 1998;284(2):687–692. [PubMed] [Google Scholar]
- 104.Peifer C, Wagner G, Laufer S. New approaches to the treatment of inflammatory disorders small molecule inhibitors of p38 MAP kinase. Current Topics in Medicinal Chemistry. 2006;6(2):113–149. doi: 10.2174/156802606775270323. [DOI] [PubMed] [Google Scholar]
- 105.Kirkwood KL, Li F, Rogers JE, et al. A p38α selective mitogen-activated protein kinase inhibitor prevents periodontal bone loss. Journal of Pharmacology and Experimental Therapeutics. 2007;320(1):56–63. doi: 10.1124/jpet.106.112466. [DOI] [PubMed] [Google Scholar]
- 106.Zwerina J, Hayer S, Redlich K, et al. Activation of p38 MAPK is a key step in tumor necrosis factor-mediated inflammatory bone destruction. Arthritis and Rheumatism. 2006;54(2):463–472. doi: 10.1002/art.21626. [DOI] [PubMed] [Google Scholar]
- 107.Patil C, Zhu X, Rossa C, Kim YJ, Kirkwood KL. p38 MAPK regulates IL-1β induced IL-6 expression through mRNA stability in osteoblasts. Immunological Investigations. 2004;33(2):213–233. doi: 10.1081/imm-120034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Patil C, Rossa C, Kirkwood KL. Actinobacillus actinomycetemcomitans lipopolysaccharide induces interleukin-6 expression through multiple mitogen-activated protein kinase pathways in periodontal ligament fibroblasts. Oral Microbiology and Immunology. 2006;21(6):392–398. doi: 10.1111/j.1399-302X.2006.00314.x. [DOI] [PubMed] [Google Scholar]
- 109.Rogers JE, Li F, Coatney DD, et al. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. Journal of Periodontology. 2007;78(10):1992–1998. doi: 10.1902/jop.2007.070101. [DOI] [PubMed] [Google Scholar]
- 110.Dominguez C, Powers DA, Tamayo N. p38 MAP kinase inhibitors: many are made, but few are chosen. Current Opinion in Drug Discovery and Development. 2005;8(4):421–430. [PubMed] [Google Scholar]
- 111.Dambach DM. Potential adverse effects associated with inhibition of p38α/β MAP kinases. Current Topics in Medicinal Chemistry. 2005;5(10):929–939. doi: 10.2174/1568026054985911. [DOI] [PubMed] [Google Scholar]
- 112.Rogers DF, Giembycz MA. Asthma therapy for the 21st century. Trends in Pharmacological Sciences. 1998;19(5):160–164. doi: 10.1016/s0165-6147(98)01198-5. [DOI] [PubMed] [Google Scholar]
- 113.Kumar S, Boehm J, Lee JC. P38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nature Reviews Drug Discovery. 2003;2(9):717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 114.Pargellis C, Regan J. Inhibitors of p38 mitogen-activated protein kinase for the treatment of rheumatoid arthritis. Current Opinion in Investigational Drugs. 2003;4(5):566–571. [PubMed] [Google Scholar]
- 115.van den Blink B, Juffermans NP, Ten Hove T, et al. p38 mitogen-activated protein kinase inhibition increases cytokine release by macrophages in vitro and during infection in vivo. Journal of Immunology. 2001;166(1):582–587. doi: 10.4049/jimmunol.166.1.582. [DOI] [PubMed] [Google Scholar]
- 116.Adams RH, Porras A, Alonso G, et al. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Molecular Cell. 2000;6(1):109–116. [PubMed] [Google Scholar]
- 117.Mudgett JS, Ding J, Guh-Siesel L, et al. Essential role for p38α mitogen-activated protein kinase in placental angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(19):10454–10459. doi: 10.1073/pnas.180316397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tamura K, Sudo T, Senftleben U, Dadak AM, Johnson R, Karin M. Requirement for p38α in erythropoietin expression: a role for stress kinases in erythropoiesis. Cell. 2000;102(2):221–231. doi: 10.1016/s0092-8674(00)00027-1. [DOI] [PubMed] [Google Scholar]
- 119.Stokoe D, Campbell DG, Nakielny S, et al. MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO Journal. 1992;11(11):3985–3994. doi: 10.1002/j.1460-2075.1992.tb05492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nature Cell Biology. 1999;1(2):94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 121.Jagavelu K, Tietge UJF, Gaestel M, Drexler H, Schieffer B, Bavendiek U. Systemic deficiency of the MAP kinase-activated protein kinase 2 reduces atherosclerosis in hypercholesterolemic mice. Circulation Research. 2007;101(11):1104–1112. doi: 10.1161/CIRCRESAHA.107.156075. [DOI] [PubMed] [Google Scholar]
- 122.Hegen M, Gaestel M, Nickerson-Nutter CL, Lin LL, Telliez JB. MAPKAP kinase 2-deficient mice are resistant to collagen-induced arthritis. Journal of Immunology. 2006;177(3):1913–1917. doi: 10.4049/jimmunol.177.3.1913. [DOI] [PubMed] [Google Scholar]
- 123.Wang X, Xu L, Wang H, Young PR, Gaestel M, Feuerstein GZ. Mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 deficiency protects brain from ischemic injury in mice. The Journal of Biological Chemistry. 2002;277(46):43968–43972. doi: 10.1074/jbc.M206837200. [DOI] [PubMed] [Google Scholar]
- 124.Martín-Blanco E. p38 MAPK signalling cascades: ancient roles new functions. BioEssays. 2000;22(7):637–645. doi: 10.1002/1521-1878(200007)22:7<637::AID-BIES6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 125.Hannigan MO, Zhan L, Ai Y, Kotlyarov A, Gaestel M, Huang CK. Abnormal migration phenotype of mitogen-activated protein kinase-activated protein kinase 2-/- neutrophils in zigmond chambers containing formyl-methionyl-leucyl-phenylalanine gradients. Journal of Immunology. 2001;167(7):3953–3961. doi: 10.4049/jimmunol.167.7.3953. [DOI] [PubMed] [Google Scholar]
- 126.Kotlyarov A, Yannoni Y, Fritz S, et al. Distinct cellular functions of MK2. Molecular and Cellular Biology. 2002;22(13):4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Greenwald RA, Kirkwood K. Adult periodontitis as a model for rheumatoid arthritis (with emphasis on treatment strategies) Journal of Rheumatology. 1999;26(8):1650–1653. [PubMed] [Google Scholar]
- 128.Mercado FB, Marshall RI, Bartold PM. Inter-relationships between rheumatoid arthritis and periodontal disease: a review. Journal of Clinical Periodontology. 2003;30(9):761–772. doi: 10.1034/j.1600-051x.2003.00371.x. [DOI] [PubMed] [Google Scholar]
- 129.Li Q, Yu H, Zinna R, et al. Silencing mitogen-activated protein kinase-activated protein kinase-2 arrests inflammatory bone loss. Journal of Pharmacology and Experimental Therapeutics. 2011;336(3):633–642. doi: 10.1124/jpet.110.172395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer and Metastasis Reviews. 2008;27(2):253–261. doi: 10.1007/s10555-008-9123-1. [DOI] [PubMed] [Google Scholar]
- 131.Wu JJ, Bennett AM. Essential role for mitogen-activated protein (MAP) kinase phosphatase-1 in stress-responsive MAP kinase and cell survival signaling. The Journal of Biological Chemistry. 2005;280(16):16461–16466. doi: 10.1074/jbc.M501762200. [DOI] [PubMed] [Google Scholar]
- 132.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. Journal of Immunology. 2002;169(11):6408–6416. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- 133.Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. The Journal of Biological Chemistry. 2004;279(52):54023–54031. doi: 10.1074/jbc.M408444200. [DOI] [PubMed] [Google Scholar]
- 134.Nimah M, Zhao B, Denenberg AG, et al. Contribution of MKP-1 regulation of p38 to endotoxin tolerance. Shock. 2005;23(1):80–87. doi: 10.1097/01.shk.0000145206.28812.60. [DOI] [PubMed] [Google Scholar]
- 135.Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. The Journal of Biological Chemistry. 2005;280(9):8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]
- 136.Chi H, Barry SP, Roth RJ, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hammer M, Mages J, Dietrich H, et al. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. Journal of Experimental Medicine. 2006;203(1):15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. Journal of Immunology. 2006;176(3):1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 139.Sartori R, Li F, Kirkwood KL. MAP kinase phosphatase-1 protects against inflammatory bone loss. Journal of Dental Research. 2009;88(12):1125–1130. doi: 10.1177/0022034509349306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yu H, Li Q, Herbert B, et al. Anti-inflammatory effect of MAPK phosphatase-1 local gene transfer in inflammatory bone loss. Gene Therapy. 2011;18(4):344– 353. doi: 10.1038/gt.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Patil CS, Kirkwood KL. P38 MAPK signaling in oral-related diseases. Journal of Dental Research. 2007;86(9):812–825. doi: 10.1177/154405910708600903. [DOI] [PubMed] [Google Scholar]
- 142.Hamilton TA, Novotny M, Datta S, et al. Chemokine and chemoattractant receptor expression: post-transcriptional regulation. Journal of Leukocyte Biology. 2007;82(2):213–219. doi: 10.1189/jlb.1206754. [DOI] [PubMed] [Google Scholar]
- 143.Hamilton TA, Ohmori Y, Tebo J. Regulation of chemokine expression by antiinflammatory cytokines. Immunologic Research. 2002;25(3):229–245. doi: 10.1385/IR:25:3:229. [DOI] [PubMed] [Google Scholar]
- 144.Stoecklin G, Stoeckle P, Lu M, Muehlemann O, Moroni C. Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA. 2001;7(11):1578–1588. [PMC free article] [PubMed] [Google Scholar]
- 145.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Molecular and Cellular Biology. 2006;26(24):9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95(6):1891–1899. [PubMed] [Google Scholar]
- 147.Lai WIS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Molecular and Cellular Biology. 1999;19(6):4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Patil CS, Liu M, Zhao W, et al. Targeting mRNA Stability Arrests Inflammatory Bone Loss. Molecular Therapy. 2008;16(10):1657–1664. doi: 10.1038/mt.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhao W, Liu M, Kirkwood KL. p38α stabilizes interleukin-6 mRNA via multiple AU-rich elements. The Journal of Biological Chemistry. 2008;283(4):1778–1785. doi: 10.1074/jbc.M707573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhao W, Liu M, D'Silva NJ, Kirkwood KL. Tristetraprolin regulates interleukin-6 expression through p38 MAPK-dependent affinity changes with mRNA 3′ untranslated region. Journal of Interferon and Cytokine Research. 2011;31(8):629–637. doi: 10.1089/jir.2010.0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rossa C, Jr., Liu M, Kirkwood KL. A dominant function of p38 mitogen-activated protein kinase signaling in receptor activator of nuclear factor-κB ligand expression and osteoclastogenesis induction by Aggregatibacter actinomycetemcomitans and Escherichia coli lipopolysaccharide. Journal of Periodontal Research. 2008;43(2):201–211. doi: 10.1111/j.1600-0765.2007.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Franchi L, Park JH, Shaw MH, et al. Intracellular NOD-like receptors in innate immunity, infection and disease. Cellular Microbiology. 2008;10(1):1–8. doi: 10.1111/j.1462-5822.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 153.Li Q, Yu H, Zinna R, et al. Silencing mitogen-activated protein kinase-activated protein kinase-2 arrests inflammatory bone loss. Journal of Pharmacology and Experimental Therapeutics. 2011;336(3):633–642. doi: 10.1124/jpet.110.172395. [DOI] [PMC free article] [PubMed] [Google Scholar]