

Abstract
Metal ions are an important part of many natural proteins, providing structural, catalytic and electron transfer functions. Reproducing these functions in a designed protein is the ultimate challenge to our understanding of them. Here, we present an artificial metallohydrolase, which has been shown by X-ray crystallography to contain two different metal ions – a Zn(II) ion which is important for catalytic activity and a Hg(II) ion which provides structural stability. This metallohydrolase displays catalytic activity that compares well with several characteristic reactions of natural enzymes. It catalyses p-nitrophenyl acetate hydrolysis (pNPA) to within ~100-fold of the efficiency of human carbonic anhydrase (CA)II and is at least 550-fold better than comparable synthetic complexes. Similarly, CO2 hydration occurs with an efficiency within ~500-fold of CAII. While histidine residues in the absence of Zn(II) exhibit pNPA hydrolysis, miniscule apopeptide activity is observed for CO2 hydration. The kinetic and structural analysis of this first de novo designed hydrolytic metalloenzyme uncovers necessary design features for future metalloenzymes containing one or more metals.
Metal ions are found in over one-third of natural proteins and are essential to many biological processes, including hydrolytic chemistry, respiration, and photosynthesis. These elements make up various sites, one or more in a single protein, with catalytic, structural, and electron transfer functions.1 One of the most challenging approaches to understanding how metals function in proteins is to reproduce these sites using de novo protein design. The successful preparation of artificial metalloenzymes not only allows one to reveal previously unappreciated features defining metal behavior in biology, but also inspires confidence in the ability to design novel systems with catalytic reactivity not naturally observed that could support a wide range of biotechnological and pharmaceutical applications.2,3 While considerable progress in enhancing the catalytic properties of natural enzymes through protein redesign methods has occurred, there are few examples of successful de novo designed metalloenzymes.2–5 Our “bottom-up” approach is to design a three-stranded coiled coil (3SCC) containing both a catalytic 6,7 metal site, ZnN3O, as found in CA and a separate HgS3 site for structural stabilization. Our 3SCCs are simplified constructs which retain sufficient complexity to resemble a native protein environment.
There has been significant interest recently in the design of artificial metalloenzymes, with two fundamentally different approaches being utilized. The first involves introducing a metal site into a preexisting, stable natural protein fold. The second requires both the design of a catalytic metal site and a sequence which folds into a defined three-dimensional structure.8 This approach exploits and tests first principle understanding of biophysics and metallobiochemistry with the ultimate goal of establishing criteria that allows for generation of stable new catalysts presently not found in biology. Unfortunately, there are few reports of true de novo metalloenzymes based on this second approach, none of which are hydrolytic.2–5 One elegant example has been reported in which the active site consists of a di-iron centre in the interior of a four-helix bundle that is capable of catalyzing the O2-dependent phenol oxidase reaction.4,5 The authors remarked on the important balance between conformational stability and catalytic activity.
Our designed metallopeptides represent a “middle ground” between studying the native metalloprotein and preparing small-molecule synthetic models. In fact, they offer a number of advantages not easily employed in small molecules, including the ability to work under aqueous conditions, use the natural amino acid ligands, and stabilize lower coordination numbers within a hydrophobic core. In comparison to natural proteins, our constructs are significantly simplified to retain only core elements, allowing us to isolate and establish different contributing factors to protein function. Further, these peptides can be designed for increased thermal stability and their facile synthesis allows incorporation of non-coding amino acids.9
Our objective was to incorporate a catalytically active mononuclear metal site into a de novo designed construct and, at the same time, incorporate a separate and distinct structural site for stabilization. This represents both the first example of a de novo designed mononuclear metalloenzyme and of a de novo metalloprotein containing two different metals in two different coordination environments with distinct functions. We targeted CA for our active site because it displays high catalytic activity. Conveniently, CA’s three-fold symmetric histidine (His) binding site fits well into the designed coiled coil scaffolds and its numerous studies provide us with a wealth of literature to which we can compare our results. Previous reports exist on the design of Zn(His)3O sites into various de novo protein folds10–12 and into stable natural protein structures,13,14 however, neither structural elucidation of these centers nor catalytic activity has been reported. Therefore, our catalytically active ZnN3O-type site represents both the first structurally characterized synthetic and active CA site within a “protein framework”.
Results and discussion
Design of stabilizing structural and catalytic sites
We have reported numerous studies investigating binding of metal ions such as Hg(II), Cd(II), As(III), Bi(III) and Pb(II) to 3SCC scaffolds based on the TRI family of peptides, Ac-G(LKALEEK)4G-NH2, which contain thiol substitutions for leucine (Leu) residues in the interior, and have observed that addition of metal ions to these destabilized constructs confers significant stability.6,7,15–17 The peptides used in this study (refer to Supplementary Table S1 for full sequences) each contain an additional His for Leu substitution in the interior of the 3SCC, which is expected to destabilize the peptide aggregate markedly. TRIL9CL23H contains both a His substitution in the 23rd position towards the C-terminus and a thiol substitution for preparation of a structural site in the 9th position, near the N-terminus (TRIL23H has only a Leu to His substitution at the 23rd position). When Hg(II) is bound to the three cysteine (Cys) residues, this peptide will be referred to as [Hg(II)S](TRIL9CL23H)3−, where ‘S’ represents binding to the sulfur site. When Zn(II) is bound to the His site, there are three coordinated nitrogen atoms from protein ligands and one exogenous water or hydroxide, depending on pH. We will use ZnN to indicate that the Zn(II) is bound at the His site and specify within parentheses the protonation state of the coordinated solvent. Under pH conditions where the solvent may occur both as the H2O and OH− forms, we will use H2O/OH−. [Hg(II)]S[Zn(II)(H2O)]N(TRIL9CL23H)3+ will then refer to a trigonally coordinated Hg(II) at the Cys site, and a psuedotetrahedral Zn(II) with three coordinated imidazoles and one water molecule.
Characterization of structural and catalytic sites
First we assessed the utility of the proposed structural site, HgS3, in TRIL9CL23H. To determine the relative folding and stability of the 3SCC in the presence and absence of Hg(II), we monitored the peptide region of the CD spectrum that yielded characteristic coiled coil peaks with negative minima at 208 and 222 nm. Molar ellipticity (θ) values obtained at 222 nm and pH 8.5 of −24,102 deg dmol−1 cm2 for (TRIL23H)3 and −25,704 deg dmol−1 cm2 for (TRIL9CL23H)3 are consistent with folded coiled coil structures. While this data cannot be quantitatively fit to a two-state model (Supplementary Fig. S1), it is apparent that the midpoint for unfolding of the doubly-substituted (TRIL9CL23H)3 is at a lower concentration of denaturant than for (TRIL23H)3. The lower stability of (TRIL9CL23H)3 is anticipated as an additional hydrophobic packing layer has been substituted. As expected, a significant shift of the midpoint to a higher concentration is observed in the denaturation for [Hg(II)S](TRIL9CL23H)3−, incorporating the structural HgS3 site, as compared to that for both apopeptides (Supplementary Fig. S1). This shift clearly demonstrates the benefit of incorporating a structural Hg(II)S3 site into the design. UV-visible spectra of [Hg(II)S](TRIL9CL23H)3− confirm that Hg(II) binds in a trigonal fashion in the sulfur site, as compared to previously reported values.18
Protein crystallography clarifies how Hg(II) and Zn(II) bind to the structural Cys and catalytic His sites, respectively. For these studies, a derivative of the analogous CoilSer (CS) peptide was utilized, CSL9PenL23H (Pen = penicillamine, sequence Ac-EWEALEKK PenAALESK LQALEKK HEALEHG NH2); CoilSer behaves very similarly to the TRI peptide family, but is more amenable for crystallographic characterization.9,19 Crystals in space group P21 were obtained and the structure was solved at 2.20 Å resolution using molecular replacement. The resulting model, [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ (pdb 3PBJ), contains two independent well folded, parallel 3SCCs in the asymmetric unit (Fig. 1A). One contains Hg(II) bound to the sulfurs of Pen in a trigonal planar structure (Fig. 1B) with Hg(II)-S distances 2.21, 2.07, 2.41 Å, and four-coordinate zinc bound to three His ligands and a chloride ion. The second 3SCC contains a T-shaped Hg(II) (Hg(II)-S distances 2.29, 2.13, 3.06 Å) and a four-coordinate Zn(II) with three His residues and a water/hydroxyl ligand (Fig. 1C). The Zn(II)-His distances in both 3SCCs are close to 2.0 Å. Excitingly, the Zn(II)His3X site closely resembles that found in CA (pdb 2CBA, Fig. 2) and is the best reported structural model for CA in a synthetic system.
Figure 1. Ribbon diagrams of the [Hg(II)]S[Zn(II)(H2O/OH )]N(CSL9PenL23H)3n+ parallel 3SCC (one of two different 3-helix bundles present in the asymmetric unit) at pH 8.5.

Shown are the main chain atoms represented as helical ribbons (cyan) and the Pen and His side chains in stick form (sulphur = yellow, nitrogen = blue, oxygen = red). a, One of two trimers found in the asymmetric unit of the crystal structure. b, a top down view of the structural trigonal thiolate site, Hg(II)S3, confirming the proposed structure of Hg(II) in Cys-containing TRI peptides.17 This metal site should mimic well the structural site in the metalloregulatory protein MerR.47 c, a side view of the tetrahedral catalytic site, Zn(II)N3O, which closely mimics carbonic anhydrase and matrix metalloproteinase active sites.1 All figures are shown with 2Fo-Fc electron density contoured at 1.5 σ overlaid.
Figure 2. Overlay of the Zn(II)N3O site in [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ with the active site of human CAII.
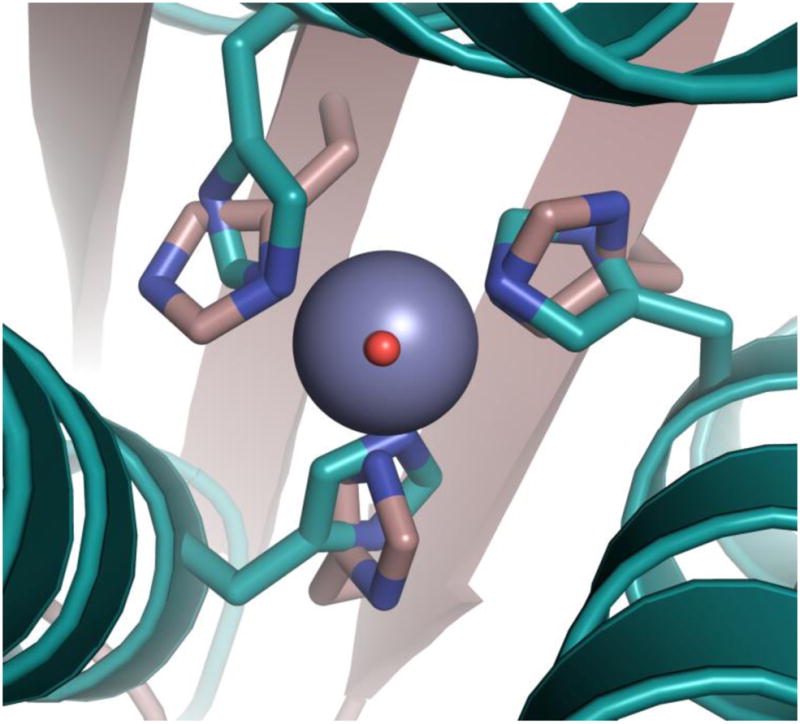
[Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ is shown in cyan (pdb 3PBJ) and CAII in tan (pdb 2CBA). The solvent molecule associated with [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ is shown in red and that associated with CAII lies underneath. The model displays an excellent structural overlay for the first coordination sphere atoms with CAII; however, the orientation of the imidazoles differs between the two proteins. Another subtle difference is that the present structure has three ε amino nitrogens bound to the Zn(II) ion whereas CAII has a mixed two ε and one δ coordination sphere. Hence, the present structure better mimics the MMP adamalysin II which also has three ε amino nitrogens bound to Zn(II).48 Overlay was performed manually in Pymol. See Supplementary Fig. S2 for a side-on view.
Characterization of ester hydrolysis
After demonstrating that we obtained the best structural model of CA, it was necessary to determine if this strictly first-coordination sphere structural relationship would be enough to confer catalytic activity. The α-CAs can hydrolyze esters such as p-nitrophenyl acetate (pNPA) by a similar mechanism (nucleophilic attack by Zn(II)-bound hydroxide) to the physiologically relevant CO2 hydration reaction.20,21 Modeling the structure and reactivity of this metalloenzyme has been the focus of many studies, however, relatively few active model complexes that can be considered structurally true to the active site of CA have been reported for pNPA hydrolysis.22–27 Of these, there are three notable small molecule examples of N3-type ligands reported to bind Zn(II) and facilitate the hydrolysis of pNPA with measurable second-order rate constants. These are the macrocyclic amine complexes [12]aneN3 and [15]aneN3O2, and a tris(imidazolyl)phosphine complex. A second-order rate constant of 0.041 M−1 s−1 was reported for [12]aneN3 at pH 8.2 and 25°C (pKa 7.3)22 and of 0.6 M−1 s−1 for [15]aneN3O 2, assuming 100% active hydroxide complex (pKa 8.8).25 The Zn(II) complex of tris(4,5-din-propyl-2-imidazolyl)phosphine is reported to catalyze this reaction in micelles with a second order rate constant of 0.86 M−1 s−1, however, the corresponding estimated value under aqueous (and more comparable) conditions is only 0.0186 M−1 s−1.24 There are also several relatively poor structural models, such as [12]aneN4 with four coordinating nitrogens, which have rate constants up to 0.1 M−1 s−1 at pH 9.3 (pKa 7.9) in 10% CH3CN28 and up to 5.0 M−1 s−1 under micellar conditions (for the 1–hexadecyl derivative of [12]aneN4).29 Notably, these synthetic complexes, in addition to having important drawbacks such as not being able to perform in 100% aqueous solution and often dimerizing, typically suffer from product inhibition.
We find that [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+ exhibits saturation kinetics for pNPA hydrolysis in buffered aqueous solution, with kcat 2.2 (±0.5) × 10−3 s−1 and kcat/KM 1.38 ± 0.04 M−1 s−1 at pH 7.5 (25 °C, 10 μM catalyst, turnover number (TON) > 10, Table 1). Compared to the best small-molecule models (having mixed solvent systems), this designed metallopeptide is over 33-fold superior to [12]aneN3 (pH 8.2, TON < 1 under the conditions at which this rate was measured) at its slowest (pH 7.5) and over 550-fold at pH 9.5.22 At pH 9.5, it is ~40-fold faster than the maximal rate for [15]aneN3O225 and 1,250-fold faster than Brown’s phosphine complex under comparable conditions.24 Additionally, designed proteins have been reported which can catalyze this reaction using His residues. Their maximal activities (at pH 7.0) are ten-fold lower than the maximal [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 activity at pH 9.5 (refer to Supplementary Table S4 and the Supplementary Discussion). 30–32 The observed increase in rate and catalytic efficiencies of [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+ as a function of increasing pH suggests a change in the active site species, presumably deprotonation of the Zn(II)-aqua ligand. kcat/KM of up to 23.3 ± 0.3 M−1 s−1 can be reached at pH 9.5, while the KM values remain essentially constant (~2 mM, within error) over the range. pH-dependency profiles can be fitted to yield pKa’s of 8.82 ± 0.11 for kcat/KM vs. pH and 8.77 ± 0.08 for kcat vs. pH, suggesting that the rate-limiting step is chemical in nature (Fig. 3). Notably, variation of the concentration of [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 relative to substrate at pH 9.5 confirms that the reaction is first order in Zn(II) protein concentration over the tested range of 10 to 60 μM catalyst. Further, we do not observe product inhibition under the conditions of our kinetic experiments (for p-nitrophenol or acetate up to 15 mM and at a single substrate concentration of 0.5 mM). A detailed analysis with potassium acetate (varying both pNPA and acetate concentrations) yields competitive inhibition with a rather high KI of 0.45 M. Refer to Supplementary Table S4 and the Supplementary Discussion for the above comparisons of catalytic efficiencies and second-order rate constants as well as those of the peptides in the absence of Zn(II). Given that imidazole is known to catalyze this reaction, the apopeptide (containing three imidazoles in the trimer) also facilitates hydrolysis with an efficiency at pH 9.0 that represents ~30% of that of the Zn(II)-bound trimer (this activity is accounted for in the values reported for Zn(II)-facilitated hydrolysis). Notably, little to no background peptide activity is observed for CO2 hydration (below).
Table 1.
Kinetics for hydrolysis of p-nitrophenyl acetate by [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+ compared to CAII.
| Designed metalloprotein (pKa = 8.8)a | CAII (pKa = 6.8) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| pH | kcat/KM (M−1 s−1) | kcat (s−1) | KM (mM) | kcat/KM (M−1 s−1) | kcat (s−1)b | KM (mM) |
| 7 | nd | nd | nd | 1,670b | 37 ± 10 | 22.1 ± 5.0b |
| 7.5 | 1.38 ± 0.04 | 2.2 (±0.5) × 10−3 | 1.6 ± 0.4 | 2,600 ± 5c | 30.53 ± 2.10c | |
| 8 | 3.1 ± 0.1 | 5.4 (±1.5) × 10−3 | 1.7 ± 0.5 | 2,550b | 53 ± 10 | 20.7b |
| 8.5 | 6.0 ± 0.1 | 12 (±4) × 10−3 | 1.9 ± 0.6 | |||
| 8.75 | 10.8 ± 0.3 | 21 (±1) × 10−3 | 2.0 ± 0.9 | |||
| 9 | 17.6 ± 0.3 | 38 (±1) × 10−3 | 2.1 ± 0.6 | 2,320b | 56 ± 10 | 23.9b |
| 9.5 | 23.3 ± 0.3 | 40 (±1) × 10−3 | 1.7 ± 0.5 | |||
Conditions: 10 μM [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+ in 50 mM buffer (HEPES or CHES). kcat, KM, and kcat/KM calculations and their error analysis are described in Supplementary Methods.
Taken from ref. 20.
Taken from ref. 33, at pH 7.4.
Figure 3. pH-dependency of pNPA hydrolysis by [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+.

Plots of a, kcat/KM vs. pH and b, kcat vs. pH for the hydrolysis of pNPA by [Hg(II)]S[Zn(II)(H2O/OH−)]N(TRIL9CL23H)3n+ (10 μM). pKa values of 8.82 ± 0.11 and 8.77 ± 0.08 for plots a and b, respectively, can be determined from the fittings and presumably represent the deprotonation of Zn-OH2 to form an active Zn-OH− nucleophile, as with CAII. See Supplementary Methods for a description of the fitting and error analysis.
The maximum kcat/KM achieved for pNPA hydrolysis by [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 is just over 100-fold less at pH 9.5 than that of CAII at pH 8 (maximum efficiency). Furthermore, it is only ~16-fold less efficient than CAI.20,33 Notably, secondary interactions in CA (as in many metalloenzymes) are very important and in particular, mutation of Thr199 (which forms a hydrogen bonding interaction with the hydroxide nucleophile of the Zn(II) center) in CAII results in a 100-fold loss in catalytic efficiency and a 1.5–2.5 unit increase in pKa from 6.8.34,35 These new kinetic parameters for T199A CAII would yield an enzyme possessing an efficiency within a factor of two of [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 and exhibiting an essentially identical pKa. Thus, the catalytic efficiency of our minimal first generation metallopeptide model towards pNPA hydrolysis, which has yet to take into account these secondary interactions that significantly contribute to optimal activity in CA, is essentially mimicking one of the fastest known enzymes, CAII. It should be noted that the relative kcat/KM values for [Zn(II)]N(TRIL23H)32+ are similar/identical from pH 7.5–8.75, suggesting that the structural site enhances stability with no expense to catalytic activity. Furthermore, above pH 9.0, [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 exhibits ~20% enhanced catalytic efficiency compared to the complex lacking the structural site, suggesting the added stability conferred by Hg(II) may be important at higher pH.
Evaluation of CO2 hydration activity
Of course, CO2 hydration is the native reaction catalyzed by CA so we have evaluated whether our complex can perform this chemistry. This is a relatively facile reaction, especially for model complexes, because product inhibition is less of a problem than for pNPA hydrolysis. While there are many examples of Zn(II) complexes which can catalyze CO2 hydration36–43, the macrocyclic amine [12]aneN4 and Brown’s phosphine complex (described earlier in terms of pNPA hydrolysis) are among the fastest models.40,42 The fastest N3-type model, therefore, is tris(4,5-di-n-propyl-2-imidazolyl)phosphine with a reported second order rate constant 2,480 M−1 s−1. CAII can catalyze this reaction with a pH-independent turnover per second of approximately one million (8.2 × 105 s−1) and a catalytic efficiency of 9.2 × 107 M−1 s−1, which is nearly 30,000-fold faster than even [12]aneN4 and 37,000-fold over the more structurally faithful tris(4,5-di-n-propyl-2-imidazolyl)phosphine complex.40,42,44 We have performed CO2 hydration studies at pH 9.5 and 25 °C. We find that [Hg(II)]S[Zn(II)(OH−)]N(TRIL9CL23H)3 attains a kcat of 1806 ± 434 s−1, a kcat/KM of 180,430 ± 26,240 M−1 s−1 and a KM of 10.0 ± 2.4 mM. This catalytic efficiency is over 70-fold faster than any previously reported model and within ~500-fold of CA II. Future work will involve not only completing the determination of this activity over the full pH range (7.5–9.5) but also incorporating potential hydrogen bonding residues in an attempt to improve the properties of our model.
Conclusions
The design reported here takes advantage of our ability to generate peptides which contain adjacent metal ion sites with differing affinities, properties, and functions.45,46 We have prepared two spatially removed sites within the interior of a coiled coil, in which very different coordinating ligands are presented to the metal ions. These sites differ greatly and as such we are now capable for the first time of building an artificial protein that contains both a structural and catalytic site. Not only is this the first example of a true de novo designed protein containing two separate metal sites with different functions, but it is also the first hydrolytic metalloenzyme designed from scratch and competitive with one of the fastest known natural metalloenzymes, CA. Furthermore, it represents a unique synthetic model with rates higher than any previously reported nitrogenous catalytic Zn(II) complex for either pNPA hydrolysis or CO2 hydration. An important question in protein design that is difficult, if not impossible, to address with mutagenesis studies on the natural protein in question, is what the minimal unit required for catalytic activity is. We have begun to answer this question with our first coordination sphere-only model and hope to gradually build in secondary interactions to gain further insight. The versatility of the TRI system will allow us to readily make changes to incorporate residues capable of secondary interactions like hydrogen bonding and to study the resulting effects on both catalytic activity and pKa. These results also inspire confidence that more economically important processes may be developed within a biomolecular scaffold for future biotechnological and pharmaceutical applications.
Methods
See the supplementary information for additional experimental details.
Supplementary Material
Acknowledgments
V.L.P. thanks the National Institutes of Health for support of this research (Grant R01 ES0 12236). M.L.Z. thanks the National Institutes of Health Chemistry-Biology Interface Training Program for support of this research. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). J.A.S. is supported by the University of Michigan Center for Structural Biology.
Footnotes
Author Contributions
M.L.Z. carried out the experimental work and completed crystal structure refinement. A.F.A.P. obtained and solved the crystal structure. M.L.Z., A.F.A.P., J.A.S., and V.L.P. contributed to the design of the experiments and analysis of the data. All of the authors contributed to writing the paper.
The authors declare no competing financial interests. Supplementary information and chemical compound information accompany this paper at www.nature.com/naturechemistry. Reprints and permission information is available online at http:npg.nature.com/reprintsandpermissions/.
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data Bank under accession code 3PBJ.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Structural and functional aspects of metal sites in biology. Chem Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nanda V, Koder R. Designing artificial enzymes by intuition and computation. Nature Chem. 2010;2:15–24. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan J, DeGrado W. F De novo design of catalytic proteins. Proc Natl Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faiella M, et al. An artificial di-iron oxo-protein with phenol oxidase activity. Nature Chem Biol. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieckmann GR, et al. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J Mol Biol. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 7.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Thermodynamic model for the stabilization of trigonal thiolato mercury(II) in designed three-stranded coiled coils. Biochemistry. 2001;40:14696–14705. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 8.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. De novo design and structural characterization of proteins and metalloproteins. Annu Rev Biochem. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 9.Peacock AFA, Stuckey JA, Pecoraro VL. Chirally switching metal coordination environments in designed peptides. Angew Chem Int Ed. 2009;48:7371–7374. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handel T, DeGrado WF. De novo design of a Zn2+-binding protein. J Am Chem Soc. 1990;112:6710–6711. [Google Scholar]
- 11.Pessi A, et al. A designed metal-binding protein with a novel fold. Nature. 1993;362:367–369. doi: 10.1038/362367a0. [DOI] [PubMed] [Google Scholar]
- 12.Kiyokawa T, et al. Binding of Cu(II) or Zn(II) in a de novo designed triple-stranded α-helical coiled-coil toward a prototype for a metalloenzyme. J Pept Res. 2004;63:347–353. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 13.Müller HN, Skerra A. Grafting of a high-affinity Zn(II)-binding site on the β-barrel of retinol-binding protein results in enhanced folding stability and enables simplified purification. Biochem. 1994;33:14126–14135. doi: 10.1021/bi00251a023. [DOI] [PubMed] [Google Scholar]
- 14.Vita C, Roumestand C, Toma F, Ménez A. Scorpion toxins as natural scaffolds for protein engineering. Proc Natl Acad Sci, USA. 1995;92:6404–6408. doi: 10.1073/pnas.92.14.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrer BT, McClure CP, Penner-Hahn JE, Pecoraro VL. Arsenic(III)-cysteine interactions stabilize three-helix bundles in aqueous solution. Inorg Chem. 2000;39:5422–5423. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 16.Matzapetakis M, Ghosh D, Weng TC, Penner-Hahn JE, Pecoraro VL. Peptidic models for the binding of Pb(II), Bi(III) and Cd(II) to mononuclear thiolate binding sites. J Biol Inorg Chem. 2006;11:876–890. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 17.Dieckmann GR, et al. De novo design of mercury-binding two- and three-helical bundles. J Am Chem Soc. 1997;11:876–890. [Google Scholar]
- 18.Pecoraro VL, Peacock AFA, Iranzo O, Luczkowski M. Understanding the biological chemistry of mercury using a de novo protein design strategy. Bioinorganic Chemistry ACS Symposium Series. 2009:183–197. [Google Scholar]
- 19.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Identifying important structural characteristics of arsenic resistance proteins by using designed three-stranded coiled coils. Proc Nat Acad Sci, USA. 2007;104:11969–11974. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem. 1967;242:4221–4229. [PubMed] [Google Scholar]
- 21.Gould SM, Tawfik DS. Directed Evolution of the promiscuous esterase activity of carbonic anhydrase II. Biochemistry. 2005;44:5444–5452. doi: 10.1021/bi0475471. [DOI] [PubMed] [Google Scholar]
- 22.Kimura E, Shiota T, Koike T, Shiro M, Kodama M. A Zinc(II) Complex of 1,5,9-triazacyclododecane ([12]aneN3) as a model for carbonic anhydrase. J Am Chem Soc. 1990;112:5805–5811. [Google Scholar]
- 23.Olmo CP, Bohmerle K, Vahrenkamp H. Zinc enzyme modeling with zinc complexes of polar pyrazolylborate ligands. Inorg Chim Acta. 2007;360:1510–1516. [Google Scholar]
- 24.Koerner TB, Brown RS. The hydrolysis of an activated ester by a tris(4,5-di-n-propyl-2-imidazolyl)phosphine-Zn2+ complex in neutral micellar medium as a model for carbonic anhydrase. Can J Chem. 2002;80:183–191. [Google Scholar]
- 25.Bazzicalupi C, et al. Carboxy and phosphate esters cleavage with mono- and dinuclear zinc(II) macrocyclic complexes in aqueous solution. Crystal structure of [Zn2L1(μ-PP)2(MeOH)2](ClO4)2 (L1 = [30]aneN6O4, PP− = diphenyl phosphate) Inorg Chem. 1997;36:2784–2790. doi: 10.1021/ic961521j. [DOI] [PubMed] [Google Scholar]
- 26.Sprigings TG, Hall DC. A simple carbonic anhydrase model which achieves catalytic hydrolysis by the formation of an ‘enzyme-substrate’-like complex. J Chem Soc, Perkin Trans. 2001;2:2063–2067. [Google Scholar]
- 27.Jairam R, Potvin PG, Balsky S. Zn2+ inclusion complexes of endodentate tripodands as carbonic anhydrase-inspired artificial esterases. Part 21 Micellar systems. J Chem Soc, Perkin Trans. 1999;2:363–367. [Google Scholar]
- 28.Koike T, Takamura M, Kimura E. Role of zinc(II) in β-lactamase II: a model study with a zinc(II)-macrocyclic tetraamine (1,4,7,10-tetraazacyclododecane, cyclen) complex. J Am Chem Soc. 1994;116:8443–8449. [Google Scholar]
- 29.Kimura E, Hashimoto H, Koike T. Hydrolysis of lipophilic esters catalyzed by a zinc(II) complex of a long alkyl-pendant macrocyclic tetraamine in micellar solution. J Am Chem Soc. 1996;118:10963–10970. [Google Scholar]
- 30.Broo K, Brive L, Ahlberg P, Baltzer L. Catalysis of hydrolysis and transesterification reactions of p-nitrophenyl esters by a designed helix-loop-helix dimer. J Am Chem Soc. 1997;119:11362–11372. [Google Scholar]
- 31.Nilsson J, Baltzer L. Reactive-site design in folded-polypeptide catalysts-the leaving group pKa of reactive esters sets the stage for cooperativity in nucleophilic and general-acid catalysis. Chem Eur J. 2000;6:2214–2220. doi: 10.1002/1521-3765(20000616)6:12<2214::aid-chem2214>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 32.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Innocenti A, et al. Investigations of the esterase, phosphatase, and sulfatase activities of the cytosolic mammalian carbonic anhydrase isoforms I, II, and XIII with 4-nitrophenyl esters as substrates. Bioorg Med Chem Lett. 2008;18:2267–2271. doi: 10.1016/j.bmcl.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Liang Z, Xue Y, Behravan G, Jonsson BH, Lindskog S. Importance of the conserved active-site residues Tyr7, Glu106, Thr199 for the catalytic function of human carbonic anhydrase II. Eur J Biochem. 1993;211:821–827. doi: 10.1111/j.1432-1033.1993.tb17614.x. [DOI] [PubMed] [Google Scholar]
- 35.Krebs JF, Ippolito JA, Christianson DW, Fierke CA. Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II. J Biol Chem. 1993;268:27458–27466. [PubMed] [Google Scholar]
- 36.Woolley P. Models for metal ion function in carbonic anhydrase. Nature. 1975;258:677–682. doi: 10.1038/258677a0. [DOI] [PubMed] [Google Scholar]
- 37.Huguet J, Brown RS. Catalytically active models for the active site in carbonic anhydrase. J Am Chem Soc. 1980;102:7571–7572. [Google Scholar]
- 38.Brown RS, Curtis NJ, Huguet J. Tris(4,5-diisopropylimidazol-2-yl)phosphine:Zinc(2+). A catalytically active model for carbonic anhydrase. J Am Chem Soc. 1981;103:6953–6959. [Google Scholar]
- 39.Brown RS, Salmon D, Curtis NJ, Kusuma S. Carbonic anhydrase models. 4 [Tris[(4,5-dimethyl-2-imidazolyl)methyl]phosphineoxide]cobalt(2+); A small-molecule mimic of the spectroscopic properties of Co(II) carbonic anhydrase. J Am Chem Soc. 1982;104:3188–3194. [Google Scholar]
- 40.Slebocka-Tilk H, Cocho JL, Frakman Z, Brown RS. Carbonic anhydrase models. 5 Tris(4,5-di-n-propyl-2-imidazolyl)phosphine-zinc(2+) and bis(4,5-diisopropyl-2-imidazolyl)-2-imidazolylphosphine-zinc(2+) Catalysts facilitating HCO3 − ↔ CO2 interconversion. J Am Chem Soc. 1984;106:2421–2431. [Google Scholar]
- 41.Zhang X, van Eldik R, Koike T, Kimura E. Kinetics and mechanism of the hydration of CO2 and dehydration of HCO3− catalyzed by a Zn(II) complex of 1,5,9-triazacyclododecane as a model for carbonic anhydrase. Inorg Chem. 1993;32:5749–5755. [Google Scholar]
- 42.Zhang X, van Eldik R. A functional model for carbonic anhydrase: Thermodynamic and kinetic study of a tetraazacyclododecane complex of zinc(II) Inorg Chem. 1995;34:5606–5614. [Google Scholar]
- 43.Nakata K, et al. Kinetic study of catalytic CO2 hydration by water-soluble model compound of carbonic anhydrase and anion inhibition effect on CO2 hydration. J Inorg Biochem. 2002;89:255–266. doi: 10.1016/s0162-0134(01)00419-6. [DOI] [PubMed] [Google Scholar]
- 44.Jackman JE, Merz KM, Jr, Fierke CA. Disruption of the active site solvent network in carbonic anhydrase II decreases the efficiency of proton transfer. Biochemistry. 1996;35:16421–16428. doi: 10.1021/bi961786+. [DOI] [PubMed] [Google Scholar]
- 45.Iranzo O, Cabello C, Pecoraro VL. Heterochromia in designed metallopeptides: geometry-selective binding of CdII in a de novo peptide. Angew Chem Int Ed. 2007;46:6688–6691. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peacock AFA, Hemmingsen L, Pecoraro VL. Using diastereopeptides to control metal ion coordination in proteins. Proc Natl Acad Sci USA. 2008;105:16566–16571. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright JG, Tsang HT, Penner-Hahn JE, O’Halloran TV. Coordination chemistry of the Hg-MerR metalloregulatory protein: Evidence for a novel tridentate Hg-cysteine receptor site. J Am Chem Soc. 1990;112:2434–2435. [Google Scholar]
- 48.Gomis-Rüth FX, Kress LF, Bode W. First structure of a snake venom metalloproteinase: a prototype for matrix metalloproteinases/collagenases. EMBO J. 1993;12:4151–4157. doi: 10.1002/j.1460-2075.1993.tb06099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.