

Abstract
There is increasing interest in the therapeutic targeting of proteases for the treatment of important diseases. Additionally new protein-based therapeutic strategies have the potential to widen the available treatments against these pathologies. In the last decade, accumulated evidence has confirmed that the family of proteases known as proprotein convertases (PCs) are potential targets for viral infections, osteoarthritis, cancer and cardiovascular disease, among others. Nevertheless, there are still many unanswered questions about the relevance of targeting PCs in a therapeutic context, especially regarding the anticipated secondary effects of treatment, considering the observed embryonic lethality of some PC knockout mice. In this review, the benefits of PCs as pharmacological targets will be discussed, with focus on concepts and strategies, as well as on the state of advancement of actual and future inhibitors.
Keywords: drug design, inhibitors, peptidomimetics, peptides as drugs, proprotein convertases, therapeutic applications, translational medicine
Introduction
After synthesis, proteins go through post-translational modification events that regulate their activity, function and localization within the cell. For many protein precursors, pro-teolysis at pairs of basic residues is one of these primordial modifications. Consequently, different compounds with very divergent activities can arise from the processing of a single protein precursor. In the secretory pathway of eukaryotic cells, these processing events are catalyzed by enzymes known as proprotein convertases (PCs). PCs are a family of serine proteases that, due to their endoproteolytic activity on precursors, generate bioactive proteins and peptides. Because these proteolytic events occur at specific sites, PCs are distinct from other serine proteases that are implicated in degradation events like trypsins and elastases. Indeed, PCs are characterized by their highly selective cleavage activity at pairs of basic residues, such as RR↓ or KR↓. Moreover, an additional arginine residue upstream of the cleavage site leads to the consensus recognition sequence R-X-K/R-R↓ and the minimal recognition sequence R-X-X-R↓ found in the majority of PC substrates.
PCs are encoded by nine different genes derived from yeast kexin and bacterial subtilisin homologues, and they maintain cellular homeostasis by processing multiple vital proteins that require a proteolytic activation step (1). Of the nine known PC enzymes, eight belong to the S8A family of serine proteases, according to the MEROPS peptidase database (http://merops.sanger.ac.uk). The enzymes furin, PC1/3, PC2, PACE4, PC4, PC5/6, PC7 (Figure 1), cleave secretory precursors at single or paired basic amino acids within the consensus cleavage site R-X-X-R↓ (2). The non-kexin like PCSK9 does not have any known in trans substrates identified so far, and seems to cleave only its own propeptide. However, PCSK9 also functions as a binding protein targeting the low density lipoprotein receptor (LDLR), as will be discussed later. In contrast to the other members of the PC family, the enzyme SKI-1/S1P, belongs to the S8B family, and does not require a basic amino acid at the cleavage site and has a consensus cleavage sequence R-X-V-R↓ (3). SKI-1/S1P plays a role in the regulation of lipid metabolism and cholesterol homeostasis (4).
Figure 1. Proprotein convertases structural domains.

Structurally, each PC possess five characteristic domains which are; (i) the signal peptide at the N-terminal extremity, responsible for the entry of the enzyme into the secretory pathway; (ii) the pro-domain which directs the suitable folding of the enzyme (chaperone), but is also an intramolecular inhibitor keeping the enzyme in its inactive form until it reaches a compartment with the appropriate calcium concentration and pH to be autoprocessed, allowing its removal; (iii) the catalytic domain, with high homology among the PC family, containing the catalytic triad composed of an Asp, His and a Ser, responsible for the interaction and the cleavage of the substrate’s multi-basic residues; (iv) the P-domain plays a key role in enzyme stability, calcium and pH dependence; and finally (v) a C-terminal domain that varies for each PC and may include transmembrane, cytoplasmic, amphipathic, serine/threonine or cysteine-histidine rich domains, contributing to each PCs unique cellular localization features.
PC cleavages result in a diversity of bioactive products, zymogen activation and sometimes inactivation of key proteins. Some PCs are expressed ubiquitously in the organism, such as furin or are widespread like PC5/6 and PC7, while others, such as PC1/3 and PC2 are more organ or system specific. Basic amino acid cleaving PCs process proteins of various categories including various receptors, growth factors, matrix-associated enzymes and components and, of course, peptidic hormone precursors, see (5) for a complete list. In addition to their endogenous functions, many studies have shown a relationship between PCs and certain pathologies due to their actions on exogenous molecules such as viral-coat proteins and bacterial toxins. Even so, heterozygous mutations in the PC1/3 gene (PCSK1) were correlated to monogenic and also polygenic obesity rodent and human phenotypes (6). Moreover, PC disorders may also lead to the development of several cellular/physiopathological states such as cancer, arthritis and dyslipidemia, which increases interest in the use of PCs as promising pharmacological targets (7).
The processing of intracellular precursors is essential in both regulated and constitutive secretory pathways. As illustrated in Figure 1, PCs are calcium-dependent serine proteases that contain multiple domains, see reviews (1, 8). Some PCs, such as PC1/3 and PC2, are mainly associated with proteolytic events that occur in the regulated secretory pathway of endocrine cells, but the activity of other PCs takes place in the constitutive secretion pathway. Furthermore, recent data show that some PCs can also be active at the cell surface, associated with heparan sulfate proteoglycans (HSPGs), or even within endosomal pathways (9). For all PCs, the catalytic site is highly conserved (50%–70%), despite the fact that they are considerably different at the C-terminal extremity (10). Indeed, these structural differences, for example differing in a transmembrane instead of a cysteine-rich domain, are responsible for their contrasting cellular localizations, as shown in Figure 2.
Figure 2. Proprotein convertases cellular localizations.

Structural characteristics, mainly in the C-terminal domain of each PC are responsible for the different cellular localizations of each enzyme. Furin, S1P/SKI-1 and PC7 are membrane associated PCs cycling between the TGN and the cell membrane PC5/6, is found in two isoforms; PC5/6A and B, originating from alternate splicing PC5/6A has been associated with the regulated secretory pathway (11), while PC5/6B is associated with the plasma membrane by its transmembrane domain (12) PACE4 and PC5/6 can also be associated with heparin sulfate proteoglycans by cysteine-rich domains (9) Both PC1/3 and PC2 are localized within the regulated secretory pathway.
Once a PC has cleaved a proprotein, carboxypeptidases are responsible for removing the newly generated C-terminal basic residues. Carboxypeptidase E (CPE) is responsible for the processing of target proteins within secretory granules, whereas the homologous carboxypeptidase D (CPD) is implicated in the same process but in the constitutive secretory pathway (13). Also, during these processes, other post-translational modifications of the proteins occur within the trans-Golgi network (TGN) and the endoplasmic reticulum including amidation, sulfation, glycosylation and/or acetylation of protein residues, which imposes a second level of regulation on them (14).
The identification of specific substrates attributable to each PC remains a real challenge because these enzymes share a highly homologous catalytic domain. An important consideration remains as to the overlapping cleavage specificity between PCs, especially if we are to establish potential strategies for therapeutic applications. While two or more PCs may cleave a precursor protein at the same cleavage site under ideal in vitro conditions, it is possible that only one of those PCs is actually carrying out that function in vivo, due to the intracellular localization of each of those PCs. In the opposite manner, cleavage redundancy also appears to be very common in the intracellular context. Although the exact nature of the specific PC(s) associated with these processing events is still difficult to predict, recent studies have investigated the proteolytic activity of each PC against multiple peptidic motifs in an attempt to identify these cleavage preferences (15, 16, 17) and produce predictive algorithms. However, even if these algorithms were fully predictive, it remains that within a cellular context the situation could be different and would require direct testing, for example using highly specific silencing technologies, such as interference RNA and mouse knockout models, in order to reveal which PC or PCs are processing that precursor in cell physiological or even pathophysiological context.
PC associated disorders can originate from either exogenous entities (e.g., bacterial or viral pathogens) requiring PC proteolysis to infect cells or, erroneous intracellular processing events that alter cellular phenotypes. It is easy to speculate that PCs are implicated in a plethora of diseases simply by the fact that they are expressed in all cells and many reviews on the subject have done just that. However, the realistic targeting of PCs in disease to obtain a beneficial therapeutic application certainly requires various levels of evidence-based validation. We will therefore limit our discussion only to those PCs and disease indications that have already provided significant proofs of concept, namely PCs in viral infections, cancer and cardiovascular disease.
Making the case for the implication of PCs in pathologies
Viral infections
Many viruses employ membrane fusion proteins to penetrate the cell membrane. To allow membrane fusion, these proteins must first be processed from their precursor form to generate the fully active form of the fusion peptide. This cleavage is necessary to induce the characteristic conformational changes required for membrane fusion, which is triggered either by an acidic pH, endosomal internalization or by receptor binding (18). For many viruses, the proteolytic events required to gain fusion activity have been linked to PCs, and this has generated interest in the development of potent antiviral therapies based on PC inhibition.
Transmembrane PCs such as furin, PC5/6, PC7 and S1P/SKI-1 (19) have been identified as being responsible for these processing events for many viral envelope proteins. One of the first identified viral fusion proteins that requires PC cleavage was the human immunodeficiency virus (HIV) envelope protein gp160, which matures into gp41 and gp120 isoforms, the latter of which is responsible for binding to the T lymphocyte CD4 receptor (20). Furin, and to a lesser extent PACE4, PC5/6 and PC1/3, have been suggested to be candidate proteases for HIV gp160 processing (21). Recent work has also linked extracellular PCs to the maturation of other HIV proteins such as viral protein R (Vpr), a lentiviral protein that facilitates HIV pre-integration to the nuclear core (22). This protein is characterized by the processing of its extracellular C-terminal arginine-rich motif (RQRR88↓) by PCs, as demonstrated by the use of the general irreversible PC inhibitor decanoyl-RVKR-chloromethylketone (dec-RVKR-cmk). The transient co-expression of PCs with this viral substrate has suggested that PC5/6 and PACE4 are the most likely candidates for Vpr cleavage, and therefore, this increases their relevance for antiviral strategies.
Influenza A viruses mediate their fusion with host cells by the binding of their hemagglutinin protein (HA0), which once processed correctly, is divided into two fragments: HA1, which contains the receptor binding domain, and HA2, which is membrane-anchored and is required for membrane fusion. These fragments are internalized by a receptor-induced endocytosis mechanism (18). Depending on the viral strain, the nature of the HA0 cleavage site can be either a single or multiple basic residues. The viral strains where HA cleavage occurs at single basic sites are generally less virulent than those with multiple basic residues. This difference can be explained by the proteases responsible for their processing. HA0 proteins from less virulent strains are mainly cleaved by trypsin-like proteases. However, PCs ensure efficient cleavage of HA0 at multiple basic residues of highly pathogenic strains, which facilitates the uptake of viral particles (23). The nature of the cellular protease is not the only factor taken into account for virulence analysis because contextual elements around the cleavage site, such as masking carbohydrates and the length of the cleavage site loop, may also interfere (18).
In addition to HIV and influenza viruses, other viruses, including papillomavirus (24), Semliki Forest virus (25), chikungunya virus (26) and arenavirus (27) also require PC cleavage of their coat proteins to become fully virulent. Considering this widespread mechanism of action, it is exciting to think that PC inhibitors could impair viral protein maturation and become a promising therapeutic approach for multiple viral infections. Furthermore, since PC are enzymes of the host cell, targeting them to block viral entry as opposed to targeting viral proteins directly, would have the tremendous advantage of limiting viral resistance, for example due to mutations or antigenic drift.
Cancer
Initially, it was speculated that PCs play an important role in neoplasia by promoting cell proliferation, migration and metastasis, mainly due to the nature of a variety of endogenous substrates which include MMPs, adhesion molecules, growth factors and their receptors. Although much remains to be defined, there is growing evidence of the role of PCs in cancer. (28–31). More recently, it has been shown that PACE4 overexpression was sufficient to induce the malignant conversion of non-tumorigenic mouse keratinocytes, leading to an increase of the basement membrane disruption. This conversion is attributed to the enhanced processing of MMPs and membrane-type MMP (MT-MMPs) to their active forms by the overexpressed PACE4, which leads to the highly invasive and metastatic phenotype that is observed (31, 32). However, the complexity of PC roles in cancer soon became apparent as variations are observed in different tumor types. For example, furin overexpression and its role in malignant phenotype has been observed in many cancers, such as head and neck (33), ovarian (34) and breast cancers (35). Other studies have reported that the role of PC1/3 and PC2 as they are overexpressed in neuroendocrine tumors such as pheochromocytomas and pituitary adenomas (36, 37). More recently, our group demonstrated the overexpression of PACE4 in human prostate cancer tissues and has provided strong evidence that PACE4 holds a central role for disease progression (38). PC expression levels have been monitored in breast cancer biopsies and cultured cell lines, showing that furin, PC1/3, PACE4 and PC7 mRNA are only detected in neoplastic tissues (35), with furin expression restricted to tumor cells. In contrast, other studies have reported a drastic reduction of PC expression in some cancers, such as PACE4 in ovarian cancer cells, but a protective role of PC5/6 against intestinal tumorigenesis has been suggested in mouse models (39, 40).
The complexity of cancer cell regulation is well documented (41), but we cannot overlook the important role in which PCs take part in the development and progression of this complex disease, potentially in key locations of convergent pathways. While indicative, it is not clear that the importance of one or more PCs in a cancer type or even in the many steps of cancer progression can be established only using over or under-expression data. A complete understanding of their role requires research on the genetic and molecular regulation of PC activity to clearly elucidate the specific contribution of PC-selective substrates in neoplastic states. Interestingly, recent studies have begun to link pathways and signal cascades to the expression of PCs, thereby increasing knowledge of the crosstalk between PCs and cellular signal transduction pathways. As an example, osteopontin, a multifaceted molecule that controls multiple carcinogenesis steps [see (42) for a complete review], regulates CD44-mediated p38 phosphorylation to induce furin expression by an NF-κB-dependent process that enhances cervical cancer cell motility (43).
We can conclude that the determination of which PCs are critical in cancer will be increasingly difficult to predict if based simply on not fully understood cellular mechanisms. This is due to the ever-increasing pathway crosstalk implicating PCs and their substrates being uncovered. A more direct approach is to directly and specifically inhibit specific PCs either in cellular or animal models to assess potential benefits. In principal, this could be achieved with exogenously applied pharmacological compounds that inhibit PCs. However, due to the lack of specificity and generally poor pharmacokinetics, these studies have not yet had the desired impact. We are left with molecular approaches that are highly specific, such as small interfering RNAs. While this molecular approach has begun to provide much needed target validation, it remains to be seen if these molecular tools can actually be used as drugs. Taken as a whole, direct PC inhibition is the best way to confirm the therapeutic potential of these proteases in the repression of malignant proliferation, invasiveness and metastatic potential (44, 45).
Osteoarthritis
Osteoarthritis (OA) is another pathophysiological state where PCs inhibitors could have a beneficial therapeutic impact. Osteoarthritis is a degenerative joint disease that is characterized by the degradation of the flexible and compression-resistant matrix known as the aggrecan, which is mainly composed of proteoglycan (PG) produced by chondrocytes (46). One of the processes known to be a major contributor to cartilage damage is the proteolytic degradation of aggrecan by enzymes such as aggrecanases, also known as ADAMTS (A disintegrin-like and metalloprotease with thrombospondin type 1 repeats). In situ studies indicate that ADAMTS-4 and -5 are the major aggrecanases involved in the pathogenesis of OA (47). These enzymes are initially synthesized as zymogens and are subsequently processed by PCs, especially PACE4, into their mature forms (48). PC cleavage, which occurs in the cartilage, removes the prodomains of both ADAMTS-4 and ADAMTS-5 at the PC recognition motifs RAKR212↓ and RRRR261↓, respectively. Considering the unique upstream position of PCs in the molecular cascade that leads to OA, PACE4 appears to be an attractive, yet challenging target for the treatment of this disease. For example, the exact distribution and targeting of the active inhibitors will be critical particularly in cartilage uptake (49). Should these obstacles be overcome, the therapeutic use of PACE4 inhibitors for OA will become more promising.
Cardiovascular diseases
The major discovery of PCSK9 in the cholesterol homeostasis has generated intense activity in the area of cardiovascular intervention. The role of the non-kexin-like PC, namely PCSK9 in the regulation of the blood low density lipoprotein cholestrol (LDL-c) concentration, has been exhaustively studied in the last few years (50, 51) and is responsible for a major push by the pharmaceutical industry as means to prevent and treat hypercholesterolemia, atherosclerosis and cardiovascular diseases. Briefly, as illustrated in Figure 3, PCSK9 enhances the degradation of the LDL-c receptor (LDLR), which is an important participant in plasma cholesterol level regulation, by inducing its endocytosis subsequent to binding or by intercepting de novo synthesized LDLRs before they even reach the plasma membrane (54). Most importantly, gain- and loss-of-function mutations found within the PCSK9 gene correlate with clinical observations of significant cholesterol level variations, making PCSK9 an important target for PC inhibitor development as a cutting edge approach to cardiovascular diseases (56).
Figure 3. PCSK9 pharmacological inhibition strategy.

From the blood circulation, cholesterol cellular uptake of LDL-c particles within the liver is mediated by the recognition of ApoB100 by the hepatocyte LDLR. This receptor-mediated endocytosis of the LDL-c leads to a decrease in plasma concentrations PCSK9, which is a secreted protein, is capable of binding LDLR (and VLDLR) (52) by its EGF-A domain (53) and mediates its degradation through late endosomal or lysosomal digestion PCSK9 can also mediate the degradation of de novo synthesized receptors by intercepting them before they even reach the plasma membrane (54) PCSK9 in circulation can be inactivated by furin or PC5/6A cleavage, which indicates a new regulation mechanism to control levels of active PCKS9. Interesting findings also show that some PCSK9 gain-of-function mutation are due to the removal of such inactivating cleavage site, leading to hypercholesterolemia (55).
Previous to the discovery of PCSK9’s role in lipid metabolism, another PC family member, SKI-1/S1P had been associated with sterol metabolic disorders. This association was based on the knowledge that SKI-1/S1P substrates; sterol regulatory element-binding proteins (SREBP-1/-2), are important transcription factors implicated in cholesterol uptake and synthesis regulation (50). The SREBP-1 precursor is an endoplasmic reticulum membrane-linked protein that is brought to the Golgi apparatus only after the binding of the sterol level responsive SREBP-cleavage-activating protein (SCAP). When low cellular sterol/lipid levels occur, SREBPs are brought within range of SKI-1/S1P in the TGN where they lose their cytosolic tail upon cleavage and can translocate into the nucleus. Genes under SREBP control mainly encode proteins that regulate cholesterol biosynthesis and uptake, but they also include PCSK9 itself (50, 57, 58).
Because of their high expression levels in macrophages and vascular smooth muscle cells that compose some human atherosclerotic plaques, furin, PACE4 and PC5/6A have also been implicated in atherosclerosis and restenosis (59). Moreover, recent evidence has linked PCs to lipid metabolism. Angiopoietin-like protein 3 (ANGPTL3), a modulator of plasma triglyceride levels, promotes the PC-mediated cleavage of lipoprotein lipase (LPL) (60). This inactivation process issued from the catalytic activity of furin and PACE4 increases plasma triglyceride levels and, consequently, increases risks of cardiovascular diseases such as atherosclerosis. To our knowledge, ANGPTL3 is the first PC activator identified so far. ANGPLT4, another member of the ANGPTL family, is known as an inhibitor of LPL activity by promoting its degradation by PCs. Moreover, this inhibitory effect is enhanced by the PC cleavage of ANGPTL4 itself, which generates an even more potent inhibitor of LPL (61).
PC inhibitors as potential therapeutic agents
As molecular evidences build for the implication of PCs in various pathologies, the need for the development of potent and specific PC inhibitors becomes more pressing. Needless to say that this enthusiasm is tempered by the fact that PCs function in multiple cellular pathways raise some doubts about targeting these enzymes in a therapeutic approach, particularly regarding potential side effects. In this section we will attempt to build the case for PCs as excellent therapeutic targets, in spite of potential perceived obstacles.
Protease inhibitors have previously been proposed to treat pathologies such as cancer because they play a major role in the progression of these diseases (62). As examples of drug-targeted proteases, cystatins and MMPs are potential therapeutic targets in cancer (63). Moreover, MMP inhibitors such as hydroxamate compounds have been tested in clinical trials after in vitro and in vivo validation (64). Unfortunately, unexpected side effects associated with the use of these compounds have overshadowed their therapeutic potential, even though the aberrant activity of MMPs is clearly associated with cancer progression. These side effects are due to a lack of specificity against the particular MMPs implicated in cancer. In fact, some MMPs that ensure physiological homeostasis, and also some non-MMPs, were presumed to be affected by the drug, which led to the serious side effects (63, 64). However, these results do not exclude these proteases as potential therapeutic targets because these enzymes have critical signaling and subcellular activity in addition to their extracellular and membrane-associated degradative functions (64). Therefore, the side effects observed with MMP inhibitors may not be applicable to all protease targets, or they may be avoided by using more selective compounds. In the case of PCs, the redundancy observed among this enzyme family may actually constitute an advantage over other protease families because the presence of alternative processing capabilities might attenuates the side effects caused by the specific inhibition of one member of the family. This redundancy concept has been extensively validated in vitro, and recent studies have explored it in animal models (65).
There has been skepticism about the use of PC inhibitors as therapeutics since lethality and serious phenotypic defects have been reported in some PC null mouse models (65, 66). Indeed, furin, PC5/6 and SKI-1/S1P null mice die during embryogenesis. However, PACE4 knockout mice are viable (75% of newborns) but show some severe craniofacial malformations. Recent findings also suggest that mouse background strains can impact the observations of these phenotypes, as PACE4 knockout mice in the C57BL/6 background are less affected (personal communication A.M. Malfait, Rush University, Chicago). Significant phenotypic modifications like hyperproinsulinemia and retarded growth are observed in PC1/3 and PC2 null mice, respectively. PC7 knockout mice do not show severe phenotypic symptoms but show anxiolytic attributes (67). These striking phenotypes have been used to argue for the fact that PC inhibition in a clinical context would result in severe side effects.
However, the distinction between the role of PCs in embryogenesis and development and their cellular functions in the adult animal needs to be closely examined. Knockout models allow a better understanding of the unique role of PCs in development, such as that of furin in the axial development triggered by the TGF-β family members such as Nodal, whose processing defects imply a limited redundancy of events in a spatio-temporal context during development (68, 69). The true impact of PCs silencing has remained unclear until a liver conditional knockout mouse model was obtained using an interferon-inducible Cre/lox knockout system. Using this animal model, it was observed that a liver-specific furin knockout after developmental maturity did not compromise the liver. Moreover, even though furin expression was abolished, the processing of various proproteins that were suspected to be solely processed by furin based on in vitro and cellular studies (such as the insulin receptor) showed almost complete processing without any observable up-regulation of the other co-expressed PCs (70). Thus, the liver-specific furin knockouts reveal the redundancy concept in vivo.
In addition, similar observations were obtained for furin and PC5/6 conditional knockout mice for salivary glands and enterocytes, respectively (40). Salivary glands were slightly smaller but showed normal histological structures in MCre+-furflox/flox mice after MMTV-Cre mediated fur inactivation. The PC5/6 gene knockout was performed by Pcsk5flox/flox gene with the Cre recombinase gene under control of the intestine-specific villin promoter. In this case, no malformations were observed. Unfortunately, in contrast with the liver-specific knockout, substrate processing was not measured to quantify the redundancy level between PCs in these two unique mouse models. However, due to the minimal phenotypic effects observed, it can be assumed that compensation by other co-expressed enzymes may have reduced the overall physiological consequences of the specific PC knockout. Other in vivo studies have evaluated redundancy on substrate processing in brain extracts from PC1/3 and PC2 null mice using a quantitative peptidomic strategy (72, 73). This quantitative approach has been particularly revealing to evaluate the extent of redundancy. As expected, some unique processing events were observed in both mouse models, but a majority of substrates still showed some level of processing. Those observations are in agreement with a study on the human obesity striking phenotype caused by PC1/3 missense mutations (6). Indeed, as mentioned by the authors, the absence of a more severe phenotype associated to the sequestration of the PC1/3 enzyme in the endoplasmic reticulum is suggested to be due to the redundant activities of other co-expressed PCs.
Considering the high sequence homology of the PC catalytic domains and the characterization of their preferred cleavage sites (16), favorable and unfavorable residues within the cleavage sequence are slightly variable between PCs and thus can account for differences in processing effectiveness. Overall, if we take into consideration the high redundancy observed in organ-specific knockout mice and overlapping PC processing events observed in cell-based experimentation, the redundancy hypothesis leads us to further suggest that redundancy could be an important factor to limit toxic effects of PC inhibition in a pharmacological intervention. As cellular localization and expression levels are variable, they will certainly have an impact within the physiological and pathophysiological context. Non-PC redundancy mechanisms can also intervene since enzymes such as cathepsin L are reported to process some neuropeptides, normally considered as PC1/3 and PC2 substrates in secretory granule pathways (74).
If PCs are revealed to be major therapeutic targets, the sum of the above studies suggests that minimal side effects are entirely possible. However, the selectivity of the developed PC inhibitors remains a critical factor that ensures that redundancy will be achievable by uninhibited co-expressed PCs. For this reason, it will be essential to assess the inhibitory effect of a given inhibitor for each PC. Figure 4 presents an example of how this could play out in a cellular pathway targeted by PC inhibitors wherein selective inhibition is used to treat a pathological state.
Figure 4. Hypothetical scheme of pharmacological effects of PC inhibition in cancer cells.

Various examples have been shown where one or more PCs are dysregulated in cancer cells. In the case where a particular PC is overexpressed, to maintain the cell’s need for activated growth factors (GF) and to sustain tumor progression, the inhibition of that PC represents an obvious target. The application of an exogenous and selective PC inhibitor may not discriminate between the cancer cell and a normal cell, in the vicinity of the target area. Thus the target PC would also be inhibited in normal adjacent cells, which could result in unwanted effects. Nonetheless, it is hypothesized that the cancer cells would be more highly affected as their needs for the required growth factors are essential for progression, whereas in the normal cells, inhibitory effects of would be minimized by the redundant functions of other PCs.
As an example, we will describe the potential therapeutic use of a hypothetical furin inhibitor against breast cancer tumors, because this single PC is specifically overexpressed within these tumor cells (35). Considering the reported up-regulation of the PC substrate IGFR in cancer cells (75) and the abnormal expression of furin in breast cancer (35), this cellular pathway could be part of a key element that supports a neoplastic phenotype. Also, expression levels of IGFR are proportionally associated with the gradation of breast cancer tumors (75, 76) while higher IGFR expression levels are related to both proliferation and the induction of the pro-invasive protease MT1-MMP (77). A recent study has demonstrated the partial relocalization of furin from the TGN to the plasma membrane of cancer cells under hypoxic conditions, supporting the role of PCs in cancer progression (78). Moreover, this study suggests that this pharmacological target could be accessible from the extracellular media, reducing potential concerns associated to cell permeability of candidate drugs. Systemic administration of furin-selective inhibitors could thus lead to antineoplastic effects through a reduction of active IGFR in breast cancer tumors, but also by the reduction of the tumoral enhanced processing of proinvasive mediators such as MT1-MMP and integrins. The side effects may be minimal as other cells with normal furin and IGFR expression levels would not be radically affected due to the redundant effects from other co-expressed PCs that would sustain the processing of a majority of the furin usual substrates. As shown in the liver-specific furin knockout mouse model, none of the substrates expected to be processed by furin in the knockout liver were impaired by furin removal (70). An in vivo study that supports this idea was carried out using a double transgenic mouse model overexpressing the proto-oncogene PLAG1, a transcription factor known to regulate IGF-2 expression, combined with a furin knockout in the salivary gland (71). This animal model showed a highly reduced tumor onset suggesting a role of furin in the pathways initiated by its two known in vitro substrates IGFR and IGF-2 leading to tumor progression (71). This model of possible therapeutic application is based on the example that one PC is aberrantly overexpressed in a given pathological state, such as cancer. However, the expression of PCs in various cancers seems to be heterogeneous (30). In fact, multiple PCs appear to be overexpressed in human tumors and tumor cell lines originating from the same cancer type. Therefore, in parallel with the development of PC inhibitors of variable specificity, it is also critical to completely characterize PC expression patterns in each type of cancer. Some cancers are known to regularly overexpress the same PC, for example, furin in breast (35), head and neck (33) and ovarian cancers (34). Interestingly, it has also been suggested that the level of a given PC in tissues could be used as a prognostic marker because it would reflect the aggressiveness of cancer cells, which could be applicable to tumor biopsies (33, 34). In pathologies such as osteoarthritis, PACE4 has been singled out in association with cartilage degradation and an adaptive therapy is not necessarily as relevant as it is for cancer therapy. However, our recent data show that PACE4 is a proven target in prostate cancer, as it is the sole PC to be overexpressed and its specific inhibition results in significant decreased tumor progression in vivo (38).
Overview of existing PC inhibitors and their development
Protease inhibitor development is among the most promising experimental avenues because, beyond their therapeutic potential, they are useful tools for observing protein folding and mechanisms of action in cellular pathways. Indeed, because of the many protease-associated pathologies such as cancer, inflammation, hypertension, neurodegeneration, viral infections and coagulation disorders, protease-targeting drug development is clearly an important research and development sector (79). As extensively reviewed by Drag and Salvesen, peptidic and non-peptidic protease inhibitors have been marketed as drugs for many therapeutic purposes, which range from hypertension (e.g., captopril, which targets angiotensin converting enzyme) to cancer (e.g., bortezomib, which targets the 26S proteasome subunit) and coagulation defects (e.g., desirudin, which targets thrombin).
In the case of PCs, many promising inhibitors have been developed. The multiple approaches used to generate these inhibitors have been reviewed previously (1). We will focus on the validated inhibitors and thus on their effectiveness in an in vivo context and, secondly, on the latest progress in their development. The validated approaches are classified into four inhibitor subclasses: peptidic, small non-peptidic molecules, antibodies and even siRNAs.
PC inhibitors validated in vivo
The therapeutic utility of peptidic furin inhibitors for cellular protection against infections in mice has been recently demonstrated for various diseases. The H5N1 influenza A PC cleavage sequence was used as a scaffold for the development of a selective furin peptidic inhibitor by improving the sequence length and amino acid composition. The nanomolar range obtained by the inhibitor, namely TPRARRRKKRT (Table 1), showed a protective effect in vivo against Pseudomonas exotoxin and anthrax infection (80). In subsequent studies (81) also presented in Table 1, the same sequence was adjusted to a core active sequence RARRRKKRT and used to generate a peptide with an N-terminal non-peptidic variant (8-amino-octanoyl group) to increase cell permeability. This peptide also offered protection against anthrax infection in vivo. In both cases, the peptidic inhibitors were synergistic when combined with actual antibiotics such as ciprofloxacin. Therefore, these inhibitors could be used in new co-treatment approaches to fight bacterial resistance. Of greater interest is their potential for therapeutic intervention in viral infections that utilize a furin-based entry mechanism such as various subtypes of influenza A. As influenza A infects humans through the airways, an easily delivered inhaled furin inhibitor could be effective in limiting infection.
Table 1.
Validated PC inhibitors.
| Molecules | Type | In vivo validated uses | Hits by | Selectivity indexa (nM) | |
|---|---|---|---|---|---|
| TPRARRRKKRT-NH2 (80) | Peptidic | Protects against Pseudomonas exotoxin and anthrax infection Synergism with antibiotics | Extension of the Furin cleaving sequence in the HA of avian influenza A H5N1 | 23 | furinb |
| 162 | PACE4 | ||||
| 441 | PC4 | ||||
| 232 | PC5/6 | ||||
| 152 | PC7 | ||||
| 8-Amino-octanoyl-RARRRKKRT-NH2 (81) | Tailed peptide | Protect from anthrax infection in vivo Synergism with antibiotics | Derivation of TPRARRRKKRT peptide with lipid complexion to provide cellular entry | 8 | furin |
| 3 | PC5/6 | ||||
| 430 | PC7 | ||||
| 3 | PACE4 | ||||
| Hexa-D-arginine (82, 83) | Peptidic | Prevents and treats PEA sepsis in mice | PS-SPCLS | 265 | furin |
| 106 | |||||
| 13 000 | PC1b | ||||
| 1875 | PC7 | ||||
| 206 | PC5/6 | ||||
| 580 | PACE4 | ||||
| Nona-D-arginine peptide (82, 84, 85) | Peptidic | Reduces considerably the corneal damages of the keratitis caused by Pseudomonas aeruginosa and prophylaxis uses | Levoratory modification of the nona-L-arginine version previously identified by PS-SPCLs of L- and D-hexapeptides | 1 | furinb |
| 19 | PC5/6 | ||||
| 81 | PC7 | ||||
| CMGTINTRTKKC (86) | Peptidic | Limits rhabdomyosarcoma tumors progression Synergism with antineoplasic agent | Screening for phage-displayed peptides binding to rhabdomysarcoma cells | N/A | N/A |
| Decanoyl-RVKR-CMK (45, 87–89) | CMK-peptide | Reduces considerably squamous cell carcinomas (~30%) in TPA induced epidermal proliferation | Coupling CMK group used to study other proteinases on Arg and Lys peptidic sequences | 1 | furin |
| 0.12 | PC5/6 | ||||
| 0.12 | PC7 | ||||
| 3.6 | PACE4 | ||||
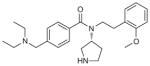
Aminopyrrolidineamide (19, 90, 91) |
Small molecule | Downregulates (~70% reduction) mice liver HMG-CoA reductase and fatty acid synthase and decrease cholesterol synthesis. Also, in vitro arenavirus’ glycoprotein processing reduction | High-throughput screening | 170 | SKI-1/S1P |
| No significant inhibition for furin, trypsin, elastase, Xia factor, thrombin, kallikrein | |||||
Based on Ki values against recombinant PC (human if not mentioned) and values marked with (b) give emphasis to the selectivity.
The treatment of Pseudomonas aeruginosa keratitis with PC inhibitors has also been tested. First, as shown in Table 1, the non-selective inhibitor nona-D-arginine peptide (82) provides efficient treatment of severe keratitis caused by bacteria, particularly when combined with ciprofloxacin (84). This treatment association is obviously required in these clinical circumstances because PC inhibitors are not used for bacterial eradication but rather to minimize tissue damage after the infection (85). The curative effects observed were not solely associated with the reduced activation of the Pseudomonas aeruginosa exotoxin A (PEA) but were also due to immunomodulating effects such as a deficient IL-1β response. This response led to a reduced inflammatory reaction related to attenuated MMP-9 expression, which in turn minimized corneal damage. Further studies from the same group showed that the nona-D-arginine peptide was also suitable for prophylactic keratitis treatment in vivo (85). This study is a significant extension of previous works that demonstrated the use of hexa-D-arginine peptide (Table 1) to prevent, and to a lesser extent treat, PEA-induced sepsis (92). These studies show that this inhibitory peptide, although non-selective against a specific PC (82), limits mouse death from PEA toxicosis. It is suggested that increased survival rate is due to hexa-D-arginine minimizing the consequences of sepsis by limiting PEA activation and thereby reducing the responses of TNF-α and other cytokines.
Two other groups have used exogenous PCs inhibitors in an attempt to limit tumoral cell progression. In the first study, an uncommon peptide inhibiting furin was recently developed using phage-display of cyclic random peptides in rhabdomyosarcoma cells and tumors (86). Using this method, a peptide with presumed affinity for furin was found (CMGTINTRTKKC, Table 1), although no kinetic constants have been calculated. In vivo, the systemic administration of the peptide in nude mice limited the progression of tumor xenografts and also had a significant synergistic action with the antineoplastic agent doxorubicin. However, a better pharmacological characterization, including the measurement of inhibition constants for all PCs, still remains to be done. In the second study, the peptide-based compound, dec-RVKR-CMK (Table 1) which has been mainly use to assess PC implication in cell based assays, was used as a topical therapeutic to retard skin tumor progression (45). The dec-RVKR-CMK compound is a non-selective, irreversible inhibitor of all individual PCs (87, 88, 89). Therefore, not surprisingly, it has been reported to exhibit some cytotoxic effects (81). For this reason, topical application may be the only use of this compound, in order to avoiding potential drawbacks of this suicide inhibitor.
An impressive demonstration of the efficacy of targeting a PC for inhibition is use of a human monoclonal antibody targeting human PCKS9 generated by Amgen, Inc. (93). When injected into either wild-type mice or mice expressing human PCSK9, the antibody considerably lowers (up to 66%) blood cholesterol in a dose-dependent manner. In cynomolgus monkeys, after a single injection, these non-human primates showed a reduction of nearly 50% in their blood cholesterol and a very significant (80%) and rapid decrease in circulating LDL cholesterol levels. In vitro assays performed also revealed that the PCSK9 antibody in combination with statins results in synergism, which suggests that PCSK9 targeting drugs could be used in combination with actual hypercholesterolemia treatments. Another group has used a completely different pharmacological approach, namely siRNA targeting of PCSK9 mRNA transcripts, to achieve similar blood cholesterol reductions. In these studies, a dose-response reduction of cholesterol levels was obtained of up to 30% in mice and 60% in rats. In cynomolgus monkeys, the treatment reduced LDL cholesterol levels more than 56% (94).
In another in vivo validation, SKI-1/S1P aminopyrrolidineamide inhibitors (Table 1) were developed using high-throughput screening by Pfizer Global Research and Development (90). The best small molecule hit was a selective inhibitor against SKI-1/S1P that did not significantly inhibit furin or other non-PC proteases. When administered to mice at high doses (30 mg/kg) to compensate for pharmacokinetic issues, the inhibitor showed short-term effectiveness for the reduction of HMG-CoA synthase and fatty acid synthase transcription (about 75%), and it also reduced the rates of cholesterol and fatty acid synthesis in the liver (91). These effects are explainable by the reduced processing of SREBP and therefore the reduced translation of multiple genes involved in sterol and cholesterol metabolism.
New approaches in PC inhibitor development
Presently, most described PC inhibitors are proteins derived or bioengineered from known or suspected PC/furin substrates. The development of these polypeptides has already been exhaustively described (89, 95, 96). The PC prodomains have also been shown to be potent PC inhibitors. Prodomains act as in cis inhibitors, as well as intramolecular chaperones in the zymogen forms of the enzymes, but have been proposed as selective inhibitors due to their considerably low homology from one PC to another. Unfortunately this is not the case, as prodomains used in trans do not show any preference for a single PC, sometimes surpassing their inhibitory potency on other PCs as compared to their cognate PC (89).
As powerful some of these protein-based PC inhibitors may be, they have the major inconvenience of being hard to apply in a therapeutic context compared to peptide-based and small molecule inhibitors. As reviewed previously (97), proteins used as drugs still have some issues to overcome. Indeed, these drugs are difficult and expensive to produce because they require mammalian cell culture to acquire their optimal post-translational modifications, which can lead to a heterogeneous population of molecules in the final product. Moreover, these molecules often require subsequent long and complex purification steps, which increases the final cost of production. In clinical use, protein-based drugs also need to be injected and, after administration, these compounds are generally unstable and present short plasmatic half-lives and, sometimes, immunogenic side effects if administered systemically. Taken together, these issues may compromise clinical trials. One possible solution may come from gene therapy approaches, although they are still not conclusively demonstrated at the present time. If this can be achieved, it is clear that protein-based inhibitors have undeniable potency. In light of the remaining hurdles, we will focus now on the more readily usable compounds for therapeutic applications: including peptide-based inhibitors, small molecules, antibodies and small interfering RNAs.
Peptide-based inhibitors
The peptide scaffold is an emerging development platform for obtaining powerful inhibitory compounds. Many new emerging therapeutics are generated as peptide-based molecules, such as exenatide (an incretin mimetic used in diabetes treatment) and icatibant (a bradykinin receptor antagonist) (98). These molecules are flexible and allow high selectivity and efficiency against their target, which is exactly what is required in cancer therapies and other life-threatening diseases (99). These molecules are attractive because they generally trigger low toxicological responses, mainly because of their high selectivity but also because of their low accumulation in organs, and they are of great importance for active inhibitory protein replacement (100). Indeed, inhibitory proteins are large moieties that display many drawbacks, such as low bioavailability, poor cellular penetration and fast degradation. However, protein-protein interactions are generally based on a few amino acid patterns called pharmacophores. Peptides can thereby be derived from these sequences to generate active peptides with the same capabilities as complete proteins but with the multiple advantages associated with these shorter molecules.
Nonetheless, peptide-based inhibitors also have some inconveniences associated with their use, such as poor cell membrane penetration because of their size or charge, short half-life due to proteolytic degradation, instability, aggregation, adsorption and infrequently will display immunogenicity. Despite these drawbacks, recent developments in peptide synthesis and peptide derivatives have given rise to new strategies to overcome these difficulties (101). Indeed, even though peptides are susceptible to degradation in physiological conditions, mainly by peptidases, structural modifications such as lipidization and PEGylation allow a considerable gain in stability and favor cell penetration. Additionally, there are multiple promising peptidomimetic variants that can substitute for the standard amino acids within the peptide backbone, which results in a higher resistance to degradative peptidases.
Many of the PC inhibitors that have been validated in physiological contexts are standard peptides often directly derived from PC substrates or natural inhibitory proteins. However, the attractive side of peptide development resides in the specific modifications that can be made once a peptidic backbone has proven its efficiency. The derived molecules that carry such optimized modifications may even have enhanced specific capabilities that result in a more selective and powerful inhibitor.
While the majority of peptide-based inhibitors that target PCs are derived from the sequences of either endogenous substrates or inhibitors, alternative strategies for development are based on the positional scanning of synthetic peptide combinatorial libraries (PS-SPCLs). PC-SPCLs which allows the identification of optimal sequences capable of PC inhibition from a degenerate mixture of peptides (95, 101). Because of the high homology within PC catalytic sites, the design of a selective inhibitor for a single PC constitutes a great challenge. Hopefully, PS-SPCLs combined with molecular modeling will give rise to new possibilities and a better understanding of the PC catalytic pocket sub-sites that interact with potential inhibitory peptides. These new data should lead to the design of more selective inhibitors. For example, a recent study addressed the preferred residues for each sub-site of the catalytic pockets of PC5/6 and PC7, and this strategy should lead to the optimization of novel inhibitory peptide sequences (83).
Using molecular modeling to observe the structural differences in different PC pockets combined with substrate cleavage analysis to monitor the cleavage efficiency of peptide sequences, it has been possible to identify slight differences between PC sub-sites (16, 102). Even if the only crystal actually available is for furin, homology models for all other PCs are available and could be used to improve inhibitors selectivity (10, 103). Using such data, it may soon be possible to select specific residues that are better embedded in or removed from inhibitory peptide sequences to facilitate the binding of a single PC and thereby create new selective inhibitors.
The incorporation of peptidomimetic variants into known peptide-based inhibitor scaffolds is a recent strategy that has been used to generate very powerful inhibitors. For example, an unnatural amino acid derivative, namely aromatic enediyne as presented on Table 2, was incorporated into the furin prodomain cleavage site to generate an enediynyl peptide with a low nanomolar range Ki against furin (Table 2) (104). Moreover, the substitution of the C-terminal arginine residue with amino acid mimetics led to a better inhibitory effect (109). More precisely, this group replaced the C-terminal arginylketones in the RVKR peptide with a series of decarboxylated arginine mimetics. As presented on Table 2, the substitution of arginine for the 4-amidinobenzylamide group generated high potency inhibitors against PC members and resulted in low nanomolar inhibitors, which also theoretically are less susceptible to carboxypeptidase degradation.
Table 2.
Promising PC inhibitor scaffolds allowing a strong or selective inhibition. Molecules were selected by their Ki values in the low micromolar range and/or demonstrating significant PC selectivity. Only peptides and small molecules are shown.
| Molecules/scaffolds | Type | Validation | Hits by | Selectivity indexa (nM) | |
|---|---|---|---|---|---|
|
Furin-Enedyinyl peptide (104) |
Peptidomimetic | Blocks pro-PDGF-A, pro-VEGF-C and pro-PDGF-B processing in cellular context | Furin prodomain cleaving site modification with enedyinyl unnatural group | 40 | furin |
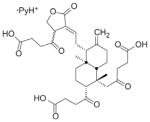 Diterpines of the labdane family (105) |
Small molecules | N/A | Scan of Andrographis paniculata (a widespread used plant to treat some human illness) compound | 2600 | furinb |
| 21 900 | PC1/3 | ||||
| 26 000 | PC7 | ||||
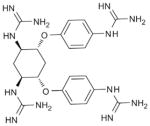 Guanidinylated Aryl 2,5-dideoxystreptamine derivatives (106) |
Small molecules | Compounds are able to limit anthrax protective antigen processing (EC50 in the low micromolar) and have low cytotoxicity | Examination of molecules with positively charged group derived from the ones capable of anthrax LF inhibition available in a small molecule collection | 12 | furin |
| 4 | PC5/6B | ||||
| 41 | PACE4 | ||||
| 595 | PC7 | ||||
| 200 | trypsin | ||||
| 200 | MT1-MMP | ||||
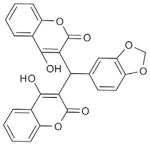 Dicoumarol derivatives (107) |
Small molecules | Nearly total protection from anthrax-toxin mediated death in the low micromolar without significant toxicity on murine macrophages | High-throughput screening of chemical diversity libraries with both enzyme-based and cell-based assays for furin and furin-like activities | 6100 | furinb |
| 14 000 | rPACE4 | ||||
| 16 000 | PC5/6 | ||||
| 91 000 | PC7 | ||||
| 15 000 | α-thrombin | ||||
 Pyrrolidine bis-piperazine compounds (108) |
Small molecules | Slightly limits recombinant mPOMC and human proglucagon processing at high micromolar concentrations | Screening of a positional scanning N-acylated bicyclic guanidine library | 660 | mPC2b |
| >40 000 | mPC1/3 | ||||
| >25 000 | furin | ||||
|
Phenylacetyl-RVR-4-amidino-benzylamide (109) |
Peptidomimetic | Limit seriously (IC50:10 μM) HA0 cleavage in Fowl Plague Virus infected cells | Incorporation of some peptidomimetic groups used for inhibition of trypsin-like serine proteases containing decarboxylated P1 Arg mimetics | 0.8 | Furin |
| 0.6 | PACE4 | ||||
| 1.6 | PC5/6 | ||||
| 0.8 | PC1/3 | ||||
| 312 | PC2 | ||||
| >600 000 | PC7 | ||||
Based on Ki values against recombinant PC (human if not mentioned) and values marked with (b) give emphasis to the potent selectivity.
Small molecule inhibitors
Small molecules have been the favored molecular forms used by the pharmaceutical industry for the generation of drug inhibitors because they are easily screenable and can be synthesized at a relatively low cost. However, this drug pipeline appears to be running dry as these small molecules have a high propensity for toxicological effects that are often the cause of trial failures. This is one of the main reasons why peptides are one of the new avenues for novel drug designs that can be ‘translated’ back into small molecules (110).
The number of small molecules that target PC family members is increasing. Some groups have previously investigated the inhibitory effects of some molecules that could be used as lead compounds for the molecular scaffolds that are required for further inhibitor development. A number of natural compounds derived from plants called andrographolides have shown weak potency inhibition (low micromolar inhibition) against furin, PC1/3 and PC7 (105). All of the effective constituents of the plant Andrographis paniculata have a diterpine labdane skeleton in common, shown in Table 2, that could be exploited to generate inhibitors as good as peptide-based compounds. Until such chemistries are developed, these classes of compounds remain a simple observation and are of no practical therapeutic potential application. In contrast, and as outlined in Table 2, 2,5-dideoxystreptamine-derived molecules also have inhibitory activity against most of the PCs with the best inhibitory constants in the low nanomolar range (106). These molecules were identified by testing positively charged molecules (such as 2,5-dideoxystreptamine guanidinylated analogs) to assess PC inhibition based on the hypothesis that basic residues have a high propensity to interact with furin and other PCs. As expected, these small molecules were highly effective against furin, PACE4, PC5/6 and PC7, but they had a much lower affinity for other proteases such as trypsin and MT1-MMP. It remains to be seen if these compounds can be effectively used in vivo. Finally, it should be noted that some copper and zinc complexes irreversibly inhibit kex2 and furin with an EC50 mostly in the low micromolar range (111). However, none of these chelates were tested against other types of proteases, which often require metal ions to catalyze their reactions. It is unlikely that these molecules will have therapeutic or even research based applications.
Recent studies have demonstrated the inhibitory properties of dicoumarol derivatives against PCs. One group screened 30 000 small molecules using a high-throughput real-time cellular assay employing a chimeric alkaline phosphatase fused to a TGN protease recognition domain and a Golgi retention signal to monitor the activity of TGN resident proteases (112). Using this process, followed by a cellular characterization assay, this group demonstrated that the dicoumarol derivative compounds shown in Table 2 have considerable abilities to inhibit furin with inhibitory constants of 6 μM and are at least two times more selective for furin than for the other PCs (107). Also using high-throughput screening, groups from Pfizer Global Research and Development have found the aminopyrrolidineamide small molecule inhibitor of SKI-1/S1P shown in Table 1 (19, 90). This compound is thought to be a competitive and selective nanomolar inhibitor of SKI-1/S1P within the PC family because it does not alter furin activity. Finally, for the first time, small molecule inhibitors of PC2 have been described (108). Positional scanning of small molecule library revealed that pyrrolidine bis-piperazine scaffolds (Table 2) and bicyclic guanidines inhibited PC2 in the mid-nanomolar and in the micromolar range respectively, while cross-reactivity with PC1/3 and furin was limited. Although these molecules seem to offer a good selectivity against the secretory pathway-related enzyme PC2, complete inhibition studies over the other PCs are still lacking to better evaluate their selectivity. At this point a selective PC2 inhibitor may be usefulness in basic research, in addition to RNA-silencing, but from a therapeutic point of view, inhibiting this secretory PC appears to have limited usefulness.
Antibodies
Since the demonstration in 1975 that murine monoclonal antibodies have significant potency for clinical uses, the development of novel antibodies with more and more designable capabilities has accelerated until these drugs took the lead of the pharmaceutical industry’s new therapeutics entering clinical trials (113). Antibody therapeutics have been successful in clinical use because they have a potent ability to target proteins that are hard to attain with other drugs, and because they can be genetically humanized to reduce immunotoxic reactions. However, these proteins are difficult and very expensive to synthesize, which compromises easily dispensable treatments and, to a lesser extent, basic research applications.
Over the past few years, antibodies have reached a new summit as therapeutics with the emergence of single-domain antibodies (sdAbs) that are produced by camelids such as llamas, camels and dromedaries [see (114) for a complete review]. This class of antibodies appears very promising because they offer unique features compared to other antibodies. Indeed, sdAbs consist of only a single protein, which dramatically facilitates genetic manipulation and production because they can be easily produced in biofermentors with simple microorganisms such as E. coli, thus considerably reducing their costs. Moreover, sdAbs are much more capable of reaching hidden epitopes (such as an enzyme’s catalytic site) due to their smaller size. Their high hydrophilicity and physicochemical stability also give them significant benefits in physiological contexts. No sdAbs targeting PCs have yet been reported in the literature, but this promising class of molecules will soon be of considerable value in in vivo studies and therapeutically-oriented applications.
PCSK9 is the only PC family member that has been pharmacologically approached with monoclonal antibodies. The choice of this strategy is reasonable considering the absence of enzymatic activity of this protein, which radically changes the strategic approach to inhibition of PCSK9. Still small molecules or peptide-based inhibitors of PCSK9 remain a distinct possibility. Because this protein attracted much attention from pharmaceutical companies for its dyslipidemia potential, a large number of patents were obtained for antibodies and short antisense RNAs that target PCSK9 (56).
siRNA
siRNAs are a very promising pharmacological avenue that have been used to target PCSK9 with high efficiency. This future class of potential pharmaceutical drugs has many advantages compared to synthetic molecules and antibodies (115). In contrast to large proteins or antibodies, siRNAs have the advantage of being able to target the mRNA issued from the transcription of virtually any gene within the genome with high selectivity. These molecules can also be synthesized at low cost because of industry’s investment in high performance nucleotide synthesis. However, it is important to note that siRNAs as drugs still need to surmount a number of hurdles. The development of selective siRNAs is critical for preventing off-target toxicity, which could be substantial, particularly in a systemic administration context (115). Moreover, the interferon cellular response needs to be fully understood and controlled to produce safer drugs. Another issue is the very low pharmacokinetic properties of these molecules due to quick elimination by the kidneys and degradation by serum RNases. A group targeting the PCSK9 gene with siRNA has reported a persistent effect of more than three weeks before levels returned to their baseline values, which is a substantial result in the preliminary development of a pharmacological agent (94). Nonetheless, chemical modifications and lipid/peptide complexes are potent avenues that need to be explored to increase the physiological half-life and cell delivery properties of siRNA drugs.
Expert opinion
We can easily conclude that PCs contribute to many disease states due both to the nature and known functions of their endogenous substrates (e.g., growth factors, signal molecules, etc.) and also due to their key roles in the proteolytic activation of infectious microorganisms and/or toxins. It is less obvious to conclude that targeting these PCs (one or many) will result in a beneficial clinical application. Taking into consideration the high redundancy level already observed and the capability of living organisms to survive local specific PC withdrawal without significant substrate misprocessing, the inhibition of PCs with considerable selectivity index appears to be a feasible treatment for preventing pathological states without the suspected severe drawbacks. Many powerful and selective inhibitors have already been discovered, and they should serve as a point of departure for the generation of molecules with enhanced and optimized properties, such as in vivo stability, advantageous pharmacokinetic parameters, and low toxicity that could be exploited as potent new drugs. We conclude that the described biomolecular data lead us to the concept that PCs have their place in the list of drug-targeted proteases at the cutting edge of pharmacological research.
Outlook
We are reaching an exciting crossroads in the field of PCs pharmacological research wherein a few specific indications are closer than ever to a significant clinical application. It is predicted that within the next decade, inhibition of PCSK9 will result in an entirely new therapeutic approach to hypercholesterolemia. Whether the approach will be used alone or in combination with statin treatment remains to be seen. While other indications are not as close to the clinical setting, they nonetheless have a large potential for the treatment of major diseases, including cancer and viral/bacterial infections. While we anticipate major clinical applications, it is also possible that the drive to better define PC mechanisms, substrates and pathways, will also lead to other developments such as the identification of specific biomarkers in pathophysiological progression.
Highlights.
PCs constitute effective therapeutic targets in multiple pathologies, including cancer, osteoarthritis, infectious and cardiovascular diseases.
The proof of concept for the PCs as therapeutic targets comes from the pharmacological and/or molecular inhibition of PCs in various cell based and animal models of bacterial infections, cancer and hypercholesterolemia.
Although not fully defined, the PC family of enzymes exerts important distinct and redundant functions.
While the development of more potent and selective PC inhibitors is regarded as important to reduce potential side effects, it is apparent that the redundant actions of co-expressed PCs can also reduce these unwanted effects.
Multiple design strategies of PCs inhibitors have been explored, but peptide-based inhibitors and antibodies remain the most promising molecular scaffolds for future developments in terms of potency, selectivity and innocuity.
Recent developments of medicinal chemistry adapted to peptide pharmaceuticals has overcome particular issues associated to their use, including stability, bioavailability, and toxicity.
Acknowledgments
This work was funded by grants from the Canadian Institutes of Health Research (CIHR) and the Ministère du Développement Économique de l’Innovation et de l’Exportation (MDEIE) to R.D. R.D. is a member of the Centre de Recherche Clinique Etienne-Le Bel (Sherbrooke, QC, Canada).
Biographies

Frédéric Couture is a promising graduate student under the mentorship of Robert Day at the Université de Sherbrooke. His research project tests the molecular and pharmacological targeting of proprotein convertases. Structure activity studies leading to novel inhibitors are optimized for improved pharmacodynamic and pharmacokinetic properties. His long term goal is to establish novel bioconcepts in the pharmacological development of inhibitor molecules.

François D’Anjou has a PhD in Pharmacology from the Université de Sherbrooke and is currently a senior research scientist in the Day lab. His key findings of the role of PACE4 in prostate cancer are providing novel insights in the complex pathways involving proprotein convertases in disease.

Robert Day is a full professor and pharmacologist at the Institut de pharmacologie de Sherbrooke (IPS) of the University of Sherbrooke. He is recognized for his participation in the co-discovery and study of the family of proprotein convertases in the last 20 years. His focus is now on establishing these enzymes as drug targets in cancer, infectious and cardiovascular disease.
References
- 1.Fugère M, Day R. Cutting back on pro-protein convertases: the latest approaches to pharmacological inhibition. Trends Pharmacol Sci. 2005;26:294–301. doi: 10.1016/j.tips.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hosaka M, Nagahama M, Kim WS, Watanabe T, Hatsuzawa K, Ikemizu J, Murakami K, Nakayama K. Arg-X-Lys/Arg-Arg motif as a signal for precursor cleavage catalyzed by furin within the constitutive secretory pathway. J Biol Chem. 1991;266:12127–30. [PubMed] [Google Scholar]
- 3.Pasquato A, Pullikotil P, Asselin MC, Vacatello M, Paolillo L, Ghezzo F, Basso F, Di Bello C, Dettin M, Seidah NG. The pro-protein convertase SKI-1/S1P In vitro analysis of Lassa virus glycoprotein-derived substrates and ex vivo validation of irreversible peptide inhibitors. J Biol Chem. 2006;281:23471–81. doi: 10.1074/jbc.M513675200. [DOI] [PubMed] [Google Scholar]
- 4.Seidah NG, Prat A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med. 2007;85:685–96. doi: 10.1007/s00109-007-0172-7. [DOI] [PubMed] [Google Scholar]
- 5.Seidah NG, Chrétien M. Interactive report Proprotein and pro-hormone convertases: a family of subtilases generating dive rse bioactive polypeptides 1. Brain Res. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- 6.Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–6. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 7.Taylor NA, Van De Ven WJM, Creemers JWM. Curbing activation: proprotein convertases in homeostasis and pathology. FASEB J. 2003;17:1215–27. doi: 10.1096/fj.02-0831rev. [DOI] [PubMed] [Google Scholar]
- 8.Bergeron F, Leduc R, Day R, et al. Subtilase-like pro-protein convertases: from molecular specificity to therapeutic applications. J Mol Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- 9.Nour N, Mort JS, Salvas A, Mbikay M, Morrison CJ, Overall CM, Seidah NG. The cysteine-rich domain of the secreted proprotein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metallo-proteinases. Mol Biol Cell. 2005;16:5215–26. doi: 10.1091/mbc.E05-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henrich S, Lindberg I, Bode W, Than ME. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J Mol Biol. 2005;345:211–27. doi: 10.1016/j.jmb.2004.10.050. [DOI] [PubMed] [Google Scholar]
- 11.De Bie I, Marcinkiewicz M, Malide D, Lazure C, Nakayama K, Bendayan M, Seidah NG. The isoforms of proprotein convertase PC5 are sorted to different subcellular compartments. J Cell Biol. 1996;135:1261–75. doi: 10.1083/jcb.135.5.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Essalmani R, Marcinkiewicz J, Chamberland A, Mbikay M, Chrétien M, Seidah NG, Prat A. Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol Cell Biol. 2006;26:354–61. doi: 10.1128/MCB.26.1.354-361.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arolas JL, Vendrell J, Aviles FX, Fricker LD. Metallocarboxy-peptidases: emerging drug targets in biomedicine. Curr Pharm Des. 2007;13:349–66. doi: 10.2174/138161207780162980. [DOI] [PubMed] [Google Scholar]
- 14.Maeda Y, Kinoshita T. The acidic environment of the golgi is critical for glycosylation and transport. Methods Enzymol. 2010;480:495–510. doi: 10.1016/S0076-6879(10)80022-9. [DOI] [PubMed] [Google Scholar]
- 15.Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17:107–12. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- 16.Remacle AG, Shiryaev SA, Oh E-S, Cieplak P, Srinivasan A, Wei G, Liddington RC, Ratnikov BI, Parent A, Desjardins R, Day R, Smith JW, Lebl M, Strongin AY. Substrate cleavage analysis of furin and related proprotein convertases. A comparative study. J Biol Chem. 2008;283:20897–906. doi: 10.1074/jbc.M803762200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian S, Huang Q, Fang Y, Wu J. Furin DB: a database of 20-residue furin cleavage site motifs, substrates and their associated drugs. Int J Mol Sci. 2011;12:1060–5. doi: 10.3390/ijms12021060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garten W, Hans-Dieter K. Cleavage activation of the influenza virus hemagglutinin and its role in pathogenesis. Trends Microbiol. 2008;27:156–67. [Google Scholar]
- 19.Urata S, Yun N, Pasquato A, Paessler S, Kunz S, de la Torre JC. Antiviral activity of a small-molecule inhibitor of arenavirus glycoprotein processing by the cellular site 1 protease. J Virol. 2011;85:795–803. doi: 10.1128/JVI.02019-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360:358–61. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- 21.Vollenweider F, Benjannet S, Decroly E, Savaria D, Lazure C, Thomas G, Chrétien M, Seidah NG. Comparative cellular processing of the human immunodeficiency virus (HIV-1) envelope glycoprotein gp160 by the mammalian subtilisin/kexin-like convertases. Biochem J. 1996;314:521–32. doi: 10.1042/bj3140521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Y, Chen G, Richard J, Rougeau N, Li H, Seidah NG, Cohen EA. Cell-surface processing of extracellular human immunodeficiency virus type 1 Vpr by proprotein convertases. Virology. 2008;372:384–97. doi: 10.1016/j.virol.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo X-L, Li L, Wei D-Q, Zhu Y-S, Chou K-C. Cleavage mechanism of the H5N1 hemagglutinin by trypsin and furin. Amino Acids. 2008;35:375–82. doi: 10.1007/s00726-007-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci USA. 2006;103:1522–7. doi: 10.1073/pnas.0508815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Fugère M, Day R, Kielian M. Furin processing and proteolytic activation of semliki forest virus. J Virol. 2003;77:2981–9. doi: 10.1128/JVI.77.5.2981-2989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozden S, Lucas-Hourani M, Ceccaldi P-E, Basak A, Valentine M, Benjannet S, Hamelin J, Jacob Y, Mamchaoui K, Mouly V, Desprès P, Gessain A, Butler-Browne G, Chrétien M, Tangy F, Vidalain PO, Seidah NG. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors: impairment of the maturation of the E2 surface glycoprotein. J Biol Chem. 2008;283:21899–908. doi: 10.1074/jbc.M802444200. [DOI] [PubMed] [Google Scholar]
- 27.Rojek JM, Pasqual G, Sanchez AB, Nguyen N-T, de la Torre J-C, Kunz S. Targeting the proteolytic processing of the viral glycoprotein precursor is a promising novel antiviral strategy against arenaviruses. J Virol. 2010;84:573–84. doi: 10.1128/JVI.01697-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creemers JW, Roebroek AJ, Van de Ven WJ. Expression in human lung tumor cells of the proprotein processing enzyme PC1/PC3 Cloning and primary sequence of a 5 kb cDNA. FEBS Lett. 1992;300:82–8. doi: 10.1016/0014-5793(92)80169-h. [DOI] [PubMed] [Google Scholar]
- 29.Clark DA, Day R, Seidah N, Moody TW, Cuttitta F, Davis TP. Protease inhibitors suppress in vitro growth of human small cell lung cancer. Peptides. 1993;14:1021–8. doi: 10.1016/0196-9781(93)90081-q. [DOI] [PubMed] [Google Scholar]
- 30.Khatib A-M, Siegfried G, Chrétien M, Metrakos P, Seidah NG. Proprotein convertases in tumor progression and malignancy: novel targets in cancer therapy. Am J Pathol. 2002;160:1921–35. doi: 10.1016/S0002-9440(10)61140-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bassi DE, Cicco RLD, Cenna J, Litwin S, Cukierman E, Klein-Szanto AJP. PACE4 expression in mouse basal keratinocytes results in basement membrane disruption and acceleration of tumor progression. Cancer Res. 2005;65:7310–9. doi: 10.1158/0008-5472.CAN-05-1213. [DOI] [PubMed] [Google Scholar]
- 32.Mahloogi H, Bassi DE, Klein-Szanto AJP. Malignant conversion of non-tumorigenic murine skin keratinocytes overexpressing PACE4. Carcinogenesis. 2002;23:565–72. doi: 10.1093/carcin/23.4.565. [DOI] [PubMed] [Google Scholar]
- 33.Bassi DE, Mahloogi H, Al-Saleem L, Lopez De Cicco R, Ridge JA, Klein-Szanto AJ. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol Carcinog. 2001;31:224–32. doi: 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- 34.Page RE, Klein-Szanto AJP, Litwin S, Nicolas E, Al-Jumaily R, Alexander P, Godwin AK, Ross EA, Schilder RJ, Bassi DE. Increased expression of the pro-protein convertase furin predicts decreased survival in ovarian cancer. Cell Oncol. 2007;29:289–99. doi: 10.1155/2007/930321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng M, Watson PH, Paterson JA, Seidah N, Chrétien M, Shiu RP. Pro–protein convertase gene expression in human breast cancer. Int J Cancer. 1997;71:966–71. doi: 10.1002/(sici)1097-0215(19970611)71:6<966::aid-ijc10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 36.Konoshita T, Gasc J, Villard E, Takeda R, Seidah N, Corvol P, Pinet F. Expression of PC2 and PC1/PC3 in human pheochromocytomas. Mol Cell Endocrinol. 1994;99:307–14. doi: 10.1016/0303-7207(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 37.Jin L, Kulig E, Qian X, Scheithauer BW, Young WF, Davis DH, Seidah NG, Chrétien M, Lloyd RV. Distribution and regulation of proconvertases PC1 and PC2 in human pituitary adenomas. Pituitary. 1999;1:187–95. doi: 10.1023/a:1009909232243. [DOI] [PubMed] [Google Scholar]
- 38.D’Anjou F, Routhier S, Perreault JP, Latil A, Bonnel D, Fournier I, Salzet M, Day R. Molecular validation of PACE4 as a target in prostate cancer. Transl Oncol. 2011;4:157–72. doi: 10.1593/tlo.10295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu Y, Campbell EJ, Shepherd TG, Nachtigal MW. Epigenetic regulation of proprotein convertase PACE4 gene expression in human ovarian cancer cells. Mol Cancer Res. 2003;1:569–76. [PubMed] [Google Scholar]
- 40.Sun X, Essalmani R, Seidah NG, Prat A. The proprotein convertase PC5/6 is protective against intestinal tumorigenesis: in vivo mouse model. Mol Cancer. 2009;8:73. doi: 10.1186/1476-4598-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Chakraborty G, Jain S, Behera R, Ahmed M, Sharma P, Kumar V, Kundu GC. The multifaceted roles of osteopontin in cell signaling, tumor progression and angiogenesis. Curr Mol Med. 2006;6:819–30. doi: 10.2174/156652406779010803. [DOI] [PubMed] [Google Scholar]
- 43.Kumar V, Behera R, Lohite K, Karnik S, Kundu GC. p38 kinase is crucial for osteopontin-induced furin expression that supports cervical cancer progression. Cancer Res. 2010;70:10381–91. doi: 10.1158/0008-5472.CAN-10-1470. [DOI] [PubMed] [Google Scholar]
- 44.Scamuffa N, Siegfried G, Bontemps Y, Ma L, Basak A, Cherel G, Calvo F, Seidah NG, Khatib A-M. Selective inhibition of proprotein convertases represses the metastatic potential of human colorectal tumor cells. J Clin Invest. 2008;118:352–63. doi: 10.1172/JCI32040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bassi DE, Zhang J, Cenna J, Litwin S, Cukierman E, Kleinszanto AJP. Proprotein convertase inhibition results in decreased skin cell proliferation, tumorigenesis, and metastasis. Neoplasia. 2010;12:516–26. doi: 10.1593/neo.92030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grushko G, Schneiderman R, Maroudas A. Some biochemical and biophysical parameters for the study of the pathogenesis of osteoarthritis: a comparison between the processes of ageing and degeneration in human hip cartilage. Conect Tissue Res. 1989;19:149–76. doi: 10.3109/03008208909043895. [DOI] [PubMed] [Google Scholar]
- 47.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma H-L, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK, Morris EA. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434:644–8. doi: 10.1038/nature03369. [DOI] [PubMed] [Google Scholar]
- 48.Malfait M, Arner EC, Song RH, Alston JT, Markosyan S, Staten N, Yang Z, Griggs DW, Tortorella MD. Proprotein convertase activation of aggrecanases in cartilage in situ. Arch Biochem Biophys. 2008;478:43–51. doi: 10.1016/j.abb.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 49.Byun S, Tortorella MD, Malfait A-M, Fok K, Frank EH, Grodzinsky AJ. Transport and equilibrium uptake of a peptide inhibitor of PACE4 into articular cartilage is dominated by electrostatic interactions. Arch Biochem Biophys. 2010;499:32–9. doi: 10.1016/j.abb.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seidah NG, Khatib AM, Prat A. The proprotein convertases and their implication in sterol and/or lipid metabolism. Biol Chem. 2006;387:871–7. doi: 10.1515/BC.2006.110. [DOI] [PubMed] [Google Scholar]
- 51.Davignon J, Dubuc G, Seidah NG. The influence of PCSK9 polymorphisms on serum low-density lipoprotein cholesterol and risk of atherosclerosis. Curr Atheroscler Rep. 2010;12:308–15. doi: 10.1007/s11883-010-0123-6. [DOI] [PubMed] [Google Scholar]
- 52.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem. 2008;283:2363–72. doi: 10.1074/jbc.M708098200. [DOI] [PubMed] [Google Scholar]
- 53.Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis of LDL receptor recognition by PCSK9. Proc Natl Acad Sci USA. 2008;105:1820–5. doi: 10.1073/pnas.0712064105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poirier S, Mayer G, Poupon V, Mcpherson PS, Desjardins R, Ly K, Asselin M-C, Day R, Duclos FJ, Witmer M, Parker R, Prat A, Seidah NG. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation. J Biol Chem. 2009;284:28856–64. doi: 10.1074/jbc.M109.037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benjannet S, Rhainds D, Hamelin J, Nassoury N, Seidah NG. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem. 2006;281:30561–72. doi: 10.1074/jbc.M606495200. [DOI] [PubMed] [Google Scholar]
- 56.Abifadel M, Pakradouni J, Collin M, Samson-Bouma M-E, Varret M, Rabès J-P, Boileau C. Strategies for proprotein convertase subtilisin kexin 9 modulation: a perspective on recent patents. Expert Opin Ther Pat. 2010;20:1547–71. doi: 10.1517/13543776.2010.518615. [DOI] [PubMed] [Google Scholar]
- 57.Sakai J, Rawson RB, Espenshade PJ, Cheng D, Seegmiller AC, Goldstein JL, Brown MS. Molecular identification of the sterol-regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Mol Cell Biol. 1998;2:505–14. doi: 10.1016/s1097-2765(00)80150-1. [DOI] [PubMed] [Google Scholar]
- 58.Rawson RB. Control of lipid metabolism by regulated intra-membrane proteolysis of sterol regulatory element binding proteins (SREBPs) Biochem Soc Symp. 2003;70:221–31. doi: 10.1042/bss0700221. [DOI] [PubMed] [Google Scholar]
- 59.Stawowy P, Fleck E. Proprotein convertases furin and PC5: targeting atherosclerosis and restenosis at multiple levels. J Mol Med. 2005;83:865–75. doi: 10.1007/s00109-005-0723-8. [DOI] [PubMed] [Google Scholar]
- 60.Liu J, Afroza H, Rader DJ, Jin W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J Biol Chem. 2010;285:27561–70. doi: 10.1074/jbc.M110.144279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lei X, Shi F, Basu D, Huq A, Routhier S, Day R, Jin W. Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J Biol Chem. 2011;286:15747–56. doi: 10.1074/jbc.M110.217638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lah TT, Duran Alonso MB, Van Noorden CJ. Antiprotease therapy in cancer: hot or not? Expert Opin Biol Ther. 2006;6:257–79. doi: 10.1517/14712598.6.3.257. [DOI] [PubMed] [Google Scholar]
- 63.Vihinen P, Ala-aho R, Kähäri V-M. Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets. 2005;5:203–20. doi: 10.2174/1568009053765799. [DOI] [PubMed] [Google Scholar]
- 64.Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–50. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 65.Creemers JW, Khatib AM. Knock-out mouse models of pro-protein convertases: unique functions or redundancy? Front Biosci. 2008;172:4960–71. doi: 10.2741/3055. [DOI] [PubMed] [Google Scholar]
- 66.Scamuffa N, Calvo F, Chrétien M, Seidah NG, Khatib A-M. Proprotein convertases: lessons from knockouts. FASEB J. 2006;20:1954–63. doi: 10.1096/fj.05-5491rev. [DOI] [PubMed] [Google Scholar]
- 67.Seidah NG. What lies ahead for the proprotein convertases? Ann NY Acad Sci. 2011;1220:149–161. doi: 10.1111/j.1749-6632.2010.05883.x. [DOI] [PubMed] [Google Scholar]
- 68.Chitramuthu BP, Baranowski DC, Cadieux B, Rousselet E, Seidah NG, Bennett HPJ. Molecular cloning and embryonic expression of zebrafish PCSK5 co-orthologues: functional assessment during lateral line development. Dev Dyn. 2010;239:2933–46. doi: 10.1002/dvdy.22426. [DOI] [PubMed] [Google Scholar]
- 69.Mesnard D, Constam DB. Imaging proprotein convertase activities and their regulation in the implanting mouse blastocyst. J Cell Biol. 2010;191:129–39. doi: 10.1083/jcb.201005026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, Lauwers A, Van de Ven WJ, Hartmann D, Creemers JW. Limited redundancy of the proprotein convertase furin in mouse liver. J Biol Chem. 2004;279:53442–50. doi: 10.1074/jbc.M407152200. [DOI] [PubMed] [Google Scholar]
- 71.De Vos L, Declercq J, Rosas GG, Van Damme B, Roebroek A, Vermorken F, Ceuppens J, van de Ven W, Creemers J. MMTV-cre–mediated fur inactivation concomitant with PLAG1 proto-oncogene activation delays salivary gland tumorigenesis in mice. Int J Oncol. 2008;32:1073–83. [PubMed] [Google Scholar]
- 72.Pan H, Che F-Y, Peng B, Steiner DF, Pintar JE, Fricker LD. The role of prohormone convertase-2 in hypothalamic neuropeptide processing: a quantitative neuropeptidomic study. J Neurochem. 2006;98:1763–77. doi: 10.1111/j.1471-4159.2006.04067.x. [DOI] [PubMed] [Google Scholar]
- 73.Wardman JH, Zhang X, Gagnon S, Castro LM, Zhu X, Steiner DF, Day R, Fricker LD. Analysis of peptides in prohormone convertase 1/3 null mouse brain using quantitative peptidomics. J Neurochem. 2010;114:215–25. doi: 10.1111/j.1471-4159.2010.06760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Funkelstein L, Beinfeld M, Minokadeh A, Zadina J, Hook V. Unique biological function of cathepsin L in secretory vesicles for biosynthesis of neuropeptides. Neuropeptides. 2010;44:457–66. doi: 10.1016/j.npep.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonneterre J, Peyrat JP, Beuscart R, Dã A. Prognostic significance of insulin-like growth factor 1 receptors in human breast cancer. Cancer Res. 1990;50:6931–5. [PubMed] [Google Scholar]
- 76.Happerfield LC, Miles DW, Barnes DM, Thomsen LL, Smith P, Hanby A. The localization of the insulin-like growth factor receptor 1 (IGFR-1) in benign and malignant breast tissue. J Pathol. 1997;183:412–7. doi: 10.1002/(SICI)1096-9896(199712)183:4<412::AID-PATH944>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 77.Stawowy P, Kallisch H, Kilimnik A, Margeta C, Seidah NG, Chrétien M, Fleck E, Graf K. Proprotein convertases regulate insulin-like growth factor 1-induced membrane-type 1 matrix metalloproteinase in VSMCs via endoproteolytic activation of the insulin-like growth factor-1 receptor. Biochem Biophys Res Commun. 2004;321:531–8. doi: 10.1016/j.bbrc.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 78.Arsenault D, Lucien F, Dubois CM. Hypoxia enhances cancer cell invasion through relocalization of the proprotein convertase furin from the trans-Golgi network to the cell surface. J Cell Physiol. 2011 doi: 10.1002/jcp.22792. [DOI] [PubMed] [Google Scholar]
- 79.Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9:690–671. doi: 10.1038/nrd3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shiryaev SA, Remacle G, Ratnikov BI, Nelson NA, Savinov AY, Wei G, Bottini M, Rega MF, Parent A, Desjardins R, Fugere M, Day R, Sabet M, Pellecchia M, Liddington RC, Smith JW, Mustelin T, Guiney DG, Lebl M, Strongin AY. Targeting host cell furin proprotein convertases as a therapeutic strategy against bacterial toxins and viral pathogens. J Biol Chem. 2007;282:20847–53. doi: 10.1074/jbc.M703847200. [DOI] [PubMed] [Google Scholar]
- 81.Remacle AG, Gawlik K, Golubkov VS, Cadwell GW, Liddington RC, Cieplak P, Millis SZ, Desjardins R, Routhier S, Yuan XW, Neugebauer WA, Day R, Strongin AY. Selective and potent furin inhibitors protect cells from anthrax without significant toxicity. Int J Biochem Cell Biol. 2010;42:987–95. doi: 10.1016/j.biocel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cameron A, Appel J, Houghten RA, Lindberg I. Polyarginines are potent furin inhibitors. J Biol Chem. 2000;275:36741–9. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- 83.Fugère M, Appel J, Houghten RA, Lindberg I, Day R. Short polybasic peptide sequences are potent inhibitors of PC5/6 and PC7: use of positional scanning-synthetic peptide combinatorial libraries as a tool for the optimization of inhibitory sequences. Mol Pharmacol. 2007;71:323–32. doi: 10.1124/mol.106.027946. [DOI] [PubMed] [Google Scholar]
- 84.Karicherla P, Hobden JA. Nona-D-arginine therapy for Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2009;50:256–62. doi: 10.1167/iovs.08-2344. [DOI] [PubMed] [Google Scholar]
- 85.Karicherla P, Hobden JA. Nona-D-arginine amide for prophylaxis and treatment of experimental Pseudomonas aeruginosa keratitis. Curr Eye Res. 2010;35:220–4. doi: 10.3109/02713680903487609. [DOI] [PubMed] [Google Scholar]
- 86.Hajdin K, D’Alessandro V, Niggli FK, Schäfer BW, Bernasconi M. Furin targeted drug delivery for treatment of rhabdomyosarcoma in a mouse model. PLoS One. 2010;5:e10445. doi: 10.1371/journal.pone.0010445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Angliker H, Wikstrom P, Shaw E, Brenner C, Fuller RS. The synthesis of inhibitors for processing proteinases and their action on the Kex2 proteinase of yeast. Biochem J. 1993;293(Pt 1):75–81. doi: 10.1042/bj2930075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jean F, Stella K, Thomas L, Liu G, Xiang Y, Reason AJ, Thomas G. alpha1-Antitrypsin Portland, a bioengineered serpin highly selective for furin: application as an antipathogenic. Proc Natl Acad Sci USA. 1998;95:7293–8. doi: 10.1073/pnas.95.13.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fugère M, Limperis PC, Beaulieu-Audy V, Gagnon F, Lavigne P, Klarskov K, Leduc R, Day R. Inhibitory potency and specificity of subtilase-like pro-protein convertase (SPC) prodomains. J Biol Chem. 2002;277:7648–56. doi: 10.1074/jbc.M107467200. [DOI] [PubMed] [Google Scholar]
- 90.Hay BA, Abrams B, Zumbrunn AY, Valentine JJ, Warren LC, Petras SF, Shelly LD, Xia A, Varghese AH, Hawkins JL, Van Campb JA, Robbinsa MD, Landschulza K, Harwood HJ., Jr Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg Med Chem Lett. 2007;17:4411–4. doi: 10.1016/j.bmcl.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 91.Hawkins JL, Robbins MD, Warren LC, Xia D, Petras SF, Valentine JJ, Varghese AH, Wang I–K, Subashi TA, Shelly LD, Hay BA, Landschulz KT, Geoghegan KF, Harwood HJ., Jr Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J Pharmacol Exp Ther. 2008;326:801–8. doi: 10.1124/jpet.108.139626. [DOI] [PubMed] [Google Scholar]
- 92.Sarac MS, Cameron A, Lindberg I. The furin inhibitor hexa-D-arginine blocks the activation of Pseudomonas aeruginosa exotoxin A in vivo. Infect Immun. 2002;70:7136–9. doi: 10.1128/IAI.70.12.7136-7139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chan JCY, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, Dia Y, Shetterlya S, Arimuraa Z, Salomonisa H, Romanowb WG, Thibaultb ST, Zhange R, Caoe P, Yange X-P, Yue T, Lue M, Retterf MW, Kwonf G, Henneg K, Panc O, Tsaid M-M, Fuchslocherd B, Yangd E, Zhouh L, Leed KJ, Darisd M, Shengd J, Wange Y, Shene WD, Yeha W-C, Emeryf M, Walkerb NPC, Shana B, Schwarza M, Jacksona SM. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci USA. 2009;106:9820–5. doi: 10.1073/pnas.0903849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frank-Kamenetsky M, Grefhorst A, Anderson NN, Racie TS, Bramlage B, Akinc A, Butler D, Charisse K, Dorkin R, Fan Y, Gamba-Vitalo C, Hadwiger P, Jayaraman M, John M, Jayaprakash KN, Maier M, Nechev L, Rajeev KG, Read T, Röhl I, Soutschek J, Tan P, Wong J, Wang G, Zimmermann T, de Fougerolles A, Vornlocher H-P, Langer R, Anderson DG, Manoharan M, Koteliansky V, Horton JD, Fitzgerald K. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci USA. 2008;105:11915–20. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fugère M, Day R. Inhibitors of the subtilase-like pro-protein convertases (SPCs) Curr Pharm Des. 2002;8:549–62. doi: 10.2174/1381612023395736. [DOI] [PubMed] [Google Scholar]
- 96.Chrétien M, Seidah NG, Basak A, Mbikay M. Proprotein convertases as therapeutic targets. Expert Opin Ther Targets. 2008;12:1289–300. doi: 10.1517/14728222.12.10.1289. [DOI] [PubMed] [Google Scholar]
- 97.Szymkowski DE. Rational optimization of proteins as drugs: a new era of ‘medicinal biology’. Drug Discov Today. 2004;9:381–3. doi: 10.1016/S1359-6446(03)02957-X. [DOI] [PubMed] [Google Scholar]
- 98.Stevenson CL. Advances in peptide pharmaceuticals. Curr Pharm Biotechnol. 2009;10:122–37. doi: 10.2174/138920109787048634. [DOI] [PubMed] [Google Scholar]
- 99.Bellmann-Sickert K, Beck-Sickinger AG. Peptide drugs to target G protein-coupled receptors. Trends Pharmacol Sci. 2010;31:434–41. doi: 10.1016/j.tips.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 100.Pillarisetti S. Are peptides the future? Small molecule versus peptides. Curr Pharm Biotechnol. 2006;7:225–7. doi: 10.2174/138920106777950799. [DOI] [PubMed] [Google Scholar]
- 101.Kumar TR, Soppimath K, Nachaegari SK. Novel delivery technologies for protein and peptide therapeutics. Curr Pharm Biotechnol. 2006;7:261–76. doi: 10.2174/138920106777950852. [DOI] [PubMed] [Google Scholar]
- 102.Tian S, Jianhua W. Comparative study of the binding pockets of mammalian proprotein convertases and its implications for the design of specific small molecule inhibitors. Int J Biol Sci. 2010;6:89–95. doi: 10.7150/ijbs.6.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol. 2003;10:520–6. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- 104.Basak A, Khatib A-M, Mohottalage D, Basak S, Kolajova M, Bag SS, Basak A. A novel enediynyl peptide inhibitor of furin that blocks processing of proPDGF-A, B and proVEGF-C. PLoS One. 2009;4:e7700. doi: 10.1371/journal.pone.0007700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Basak A, Cooper S, Roberge AG, Banik UK, Chrétien M, Seidah NG. Andrographis paniculata and their succinoyl esters. Biochem J. 1999;338:107–13. [PMC free article] [PubMed] [Google Scholar]
- 106.Jiao G-S, Cregar L, Wang J, Millis SZ, Tang C, O’Malley S, Johnson AT, Sareth S, Larson J, Thomas G. Synthetic small molecule furin inhibitors derived from 2,5-dideoxystreptamine. Proc Natl Acad Sci USA. 2006;103:19707–12. doi: 10.1073/pnas.0606555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Komiyama T, Coppola JM, Larsen MJ, van Dort ME, Ross BD, Day R, Rehemtulla A, Fuller RS. Inhibition of furin/proprotein convertase-catalyzed surface and intracellular processing by small molecules. J Biol Chem. 2009;284:15729–38. doi: 10.1074/jbc.M901540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kowalska D, Liu J, Appel JR, Ozawa A, Nefzi A, Mackin B, Houghten RA, Lindberg I. Synthetic small-molecule prohormone convertase 2 inhibitors. Mol Pharmacol. 2009;75:617–25. doi: 10.1124/mol.108.051334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Becker GL, Sielaff F, Than ME, Lindberg I, Routhier S, Day R, Lu Y, Garten W, Steinmetzer T. Potent inhibitors of furin and furin–like proprotein convertases containing decarboxylated P1 arginine mimetics. J Med Chem. 2010;53:1067–75. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hummel G, Reineke U, Reimer U. Translating peptides into small molecules. Mol Biosyst. 2006;2:499–508. doi: 10.1039/b611791k. [DOI] [PubMed] [Google Scholar]
- 111.Podsiadlo P, Komiyama T, Fuller RS, Blum O. Furin inhibition by compounds of copper and zinc. J Biol Chem. 2004;279:36219–27. doi: 10.1074/jbc.M400338200. [DOI] [PubMed] [Google Scholar]
- 112.Coppola JM, Hamilton CA, Bhojani S, Larsen MJ, Ross BD, Rehemtulla A. Identification of inhibitors using a cell based assay for monitoring golgi-resident protease activity. Anal Biochem. 2007;364:19–29. doi: 10.1016/j.ab.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 114.Harmsen MM, De Haard HJ. Properties, production, and applications of camelid single-domain antibody fragments. Appl Microbiol Biotechnol. 2007;77:13–22. doi: 10.1007/s00253-007-1142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dykxhoorn DM, Palliser D, Lieberman J. The silent treatment: siRNAs as small molecule drugs. Gene Ther. 2006;13:541–52. doi: 10.1038/sj.gt.3302703. [DOI] [PubMed] [Google Scholar]