

Background: Type I interferons are immunomodulatory cytokines that either prevent or promote apoptosis of T cells.
Results: Loss of Orai1-mediated Ca2+ entry sensitizes T cells to type I interferon-induced apoptosis via NF-κB activity.
Conclusion: A crosstalk between Ca2+ and type I interferon signaling dictates the survival of T cells.
Significance: Understanding how T cells escape apoptosis has clinical implications in many diseases.
Keywords: Apoptosis, Calcium, Cytokines/Interferon, Gene Transcription, NF-κB Transcription Factor, NFAT Transcription Factor, T Cell, Jurkat, Orai1, Stim1
Abstract
Store-operated Ca2+ entry (SOCE) is an essential process in T cell activation. SOCE is controlled by the Ca2+ release-activated Ca2+ (CRAC) channel encoded by the gene Orai1 that is expressed on the plasma membrane and activated by STIM1 when ER Ca2+ stores are depleted. Our earlier work showed that a somatic T-cell line Jurkat mutant H123 bearing a defect in Ca2+ signaling was susceptible to the apoptotic effects of type I interferons (IFN-α/β). The nature of the mutation and whether this mutation was linked to IFN-α/β apoptotic susceptibility was unknown. Here we show that H123 cells lacked Orai1 and exhibit reduced STIM1 protein. Reconstitution of both Orai1 and STIM1 in H123 cells rescued SOCE in response to thapsigargin and ionomycin and abrogated IFN-α/β-induced apoptosis. Reciprocally, overexpression of the dominant negative Orai1-E106A in either parental Jurkat cells or an unrelated human T cell line (CEM391) inhibited SOCE and led to sensitization to IFN-α/β-induced apoptosis. Furthermore, we showed that the Ca2+ response pathway antagonized the IFN-α/β -induced transcriptional responses; in the absence of SOCE, this negative regulatory effect was lost. However, the inhibitory effect of Ca2+ on type I IFN-induced gene transcription was diminished by pharmacological inhibition of NF-κB in cells with intact SOCE. Our findings reveal an unexpected and novel regulatory crosstalk mechanism between type I IFNs and store-operated Ca2+ signaling pathways mediated at least in part by NF-κB activity with significant clinical implications to both viral and tumor immunology.
Introduction
Type I interferons (IFN3-α/β) are family of cytokines that exert many pleiotropic effects on cells, including antiproliferative, antiviral, and immunomodulatory functions. Type I IFNs are notable in modulating T cell responses. One such effect is the direct inhibition of proliferation of mitogen and T cell receptor (TCR)-activated CD4+ T lymphocytes (1, 2). For instance, IFN-α can delay naïve T cells from entry into the cell cycle after TCR engagement and abrogate IL-2 production (2, 3). Another effect of IFN-α is to support and maintain the survival of memory CD8+ T cells (4) and rescue CD4+ T cells from undergoing apoptosis early upon TCR/CD28 activation (5). However, the anti-apoptotic effect of IFN-α is impaired in human immunodeficiency virus (HIV)-infected CD4+ T cells (6).
In our earlier studies, we reported the existence of a crosstalk between TCR and type I IFN signaling. The antiproliferative effects of IFN-α in T cells required Lck, Zap70, and CD45, all components of the TCR signal transduction pathway (7). These studies were conducted using both somatic mutants of human Jurkat T cells and primary murine T cells derived from knock-out mice missing individually these signaling components, all of which carried defects in the Ca2+ release from ER stores upon TCR activation. In a subsequent study, we examined type I IFN responses in a new somatic Jurkat mutant designated H123 (8). H123 is a clone of mutagenized Jurkat cells that were selected for a deficiency in Ca2+ signaling following pervanadate and TCR stimulation and this defect coincided with cells undergoing apoptosis in response to IFN-α stimulation (9). At that time the nature of the mutation that made H123 cells defective in the induction of Ca2+ transients had not been identified.
Recent investigations have led to the identification of members of the STIM and Orai protein families as the molecular mediators of store-operated Ca2+ entry (SOCE) (10, 11). Hence, STIM1 and STIM2, which are Ca2+ sensors on ER membrane, sense changes in ER Ca2+ content via their ER luminal EF hands (12, 13), while Orai1 is the store-operated Ca2+ channel located on the plasma membrane (14–19). When ER Ca2+ stores are depleted, STIM proteins aggregate and make contact with the plasma membrane. This in turn results in the opening of the Orai1 channel, leading to extended elevation of cytosolic Ca2+ levels, which triggers Ca2+-dependent signaling responses. In this study, we now report that impaired SOCE sensitizes T cells to type I IFN-induced apoptosis. Conversely, Ca2+ activated signals appear to be protective as they can inhibit T cells from undergoing apoptosis by IFN-α. Overall, our data indicate a novel role for SOCE in antagonizing the apoptotic and transcriptional activities of type I IFNs.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
The human leukemic T-cell line Jurkat (subclone E6), its somatic mutant H123, CEM391, and MOLT-4 cells were maintained in RPMI 1640 complete medium (Mediatech, Inc; Herndon, VA) containing 10% fetal calf serum (FCS), 2 mm l-glutamine, penicillin, and streptomycin (Invitrogen) at 37 °C and 5% CO2. H123 cells were isolated from Jurkat cells mutagenized with ICR-191 that failed to mobilize or sustain intracellular Ca2+ in response to pervanadate (8, 9). This approach facilitated the isolation of loss of function mutations in more distal components of the Ca2+ response pathway. Recombinant human IFN-α was purchased from Peprotech (Rocky Hill, NJ). Ionomycin (Iono), PMA, thapsigargin (Tg), EGTA, and CRAC channel inhibitor BTP2 were purchased from Sigma. NF-κB peptide inhibitor was a kind gift of Dr. Michael May (University of Pennsylvania).
Transfection
Human YFP-STIM1, Orai1-CFP, and Orai1 E106A were generated as previously described (17, 20). Parental and H123 Jurkat cells as well as CEM391 and MOLT-4 cells were transfected accordingly by electroporation using the Gene Pulser Xcell Electroporation system (Bio-Rad) at 280 V, 1000 μF (25 ms pulse length). Cells were also transfected with empty vector alone and used as control cells. Cells stably expressing Orai1 were selected in complete RPMI 1640 medium in the presence of 500 μg/ml of G418. The surviving populations were sorted two to three times for CFP fluorescence using a Becton Dickinson FACS-VantageSE/DiVa flow cytometer. >95% of the cells were positive for CFP.
Cytosolic Ca2+ Measurements
Ratiometric imaging of intracellular Ca2+ using fura-2 was performed as previously described (21). Cells were plated on coverslips coated with poly-l-lysine (Sigma) for 3 h before being placed in a cation-safe solution (107 mm NaCl, 7.2 mm KCl, 1.2 mm MgCl2, 11.5 mm glucose, 20 mm Hepes-NaOH, 1 mm CaCl2 pH 7.2) and loaded with fura-2/acetoxymethylester (2 μm) for 30 min at 24 °C. Cells were washed and dye was allowed to de-esterify for a minimum of 30 min at 24 °C. Ca2+ measurements were taken using a Leica DMI 6000B fluorescence microscope controlled by Slidebook Software (Intelligent Imaging Innovations; Denver, CO). Fluorescence emission at 505 nm was monitored while alternating between 340 and 380 nm excitation wavelengths at a frequency of 0.67 Hz. Intracellular Ca2+ measurements are shown as 340/380 nm ratios obtained from groups of 35 to 45 individual cells. Measurements shown are representative experiments of at least three that were performed.
Preparation of Protein Extracts and Western Blot Analysis
Cells were lysed in Nonidet P-40 lysis buffer (1% w/v Nonidet P-40, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0 with protease inhibitors), followed by centrifugation at 4 °C. The supernatant fraction was collected and protein concentration was measured using Bio-Rad protein assay. Proteins were resolved on 8% SDS-PAGE gels, transferred to PVDF membranes, and analyzed with the indicated antibodies. Peptide-affinity purified specific anti-STIM1 antibody was produced by 21st Century Biochemicals (Marlboro, MA), anti-STIM2 antibody was obtained from Cell Signaling (Danvers, MA), anti-Orai1 antibody was produced by Sigma and anti-actin antibody was purchased from Abcam (Cambridge, MA). Horseradish peroxidase-conjugated goat anti-rabbit and rabbit anti-mouse antibodies were purchased from Invitrogen and used as secondary antibodies. Blots were developed using ECL (GE Healthcare; Piscataway, NJ) and images captured using AlphaInnotech HD2 image system (San Leandro, CA).
Quantitative PCR
RNA was extracted using TRI Reagent (MRC; Cincinnati, OH). RNA concentrations were measured using a Bio-Rad Smartspec Plus spectrometer by reading the absorbance at 260 nm and 280 nm. RT-PCR was completed as a two-step process using High Capacity cDNA Reverse Transcription (Applied Biosystems; Foster City, CA) followed by SYBR Green PCR on a 7300 Real Time PCR machine. The real-time PCR conditions were 95 °C for 10 min (1 cycle) and 95 °C for 15 s, 60 °C for 1 min (40 cycles). Data are presented as the level relative to Tata box-binding protein (TBP), which serves as a housekeeping gene. The following equation was used in our data analysis: 2−(CT target gene − CT TATA-binding protein).
Measurement of Apoptosis by Fluorescence Imaging
H123 cells stably expressing Orai1-CFP or Orai1 E106A-CFP were electroporated with either pEYFP (Clontech; Mountain View, CA) or YFP-STIM1 plasmid DNA vectors. 24 h after transfection, viable cells were recovered after Ficoll (Invitrogen) gradient centrifugation. Cells were left untreated or treated with IFN-α (1000 units/ml) for 48 h followed by staining with Annexin V-Alexa Fluor 594 (Molecular Probes; Eugene, OR) for 10 min in Annexin V binding buffer (10 mm Hepes, 140 mm NaCl, 2.5 mm CaCl2, pH 7.4) at 37 °C in the dark. Cells were viewed under a Leica DMI 6000B fluorescence microscope controlled by Slidebook Software. Each group of cells was counted under the microscope with a minimum number of 200 cells. Each independent experiment was performed at least three times.
Measurement of Apoptosis by Flow Cytometry
CEM391 stably expressing empty vector (pIRES) or Orai1 E106A-CFP were left untreated or treated with IFN-α (3000 units/ml) for 72 h followed by staining with FITC Annexin V (Biolegend, San Diego, CA) for 10 min in Annexin V binding buffer (10 mm Hepes, 140 mm NaCl, 2.5 mm CaCl2, pH 7.4) at 37 °C in the dark, and then analyzed by BD FACS Calibur Flow Cytometer. Each experiment was performed at least three times.
Cell Proliferation
Cells were plated on a 96-well plate in quadruplicate at 5 × 103 cells per well. Cells were left untreated or treated with IFN-α and incubated at 37 °C for 72 h. Cell proliferation was assessed by using CellTiter 96-Aqueous One Solution Reagent (Promega, Madison, WI). Absorbance was measured at 490 nm using a VictorX5 multilabel plate reader (Perkin Elmer, Waltham MA). Background values were first subtracted from each well before proceeding with determination of percent of cell growth inhibition.
Luciferase Reporter Assay
Cells were electroporated with 10 μg of 3×-NFAT-luciferase reporter, 5×-ISRE or 5×-NF-κB luciferase reporter plasmids. To normalize for luciferase activity and control for transfection efficiency, 2 μg of pRL-TK (Promega) was also included in the transfection. After overnight incubation at 37 °C, NFAT driven transcriptional activity was measured in cells treated with or without ionomycin (2 μm) plus PMA (10 ng/ml) for 6 h. To determine ISRE driven transcriptional activity, cells were treated with or without IFN-α (1000 units/ml) for 6 h in the absence or presence of 2 μm ionomycin. NF-κB transcriptional activity was measured in cells stimulated with ionomycin plus PMA for 6 h. To determine a role for NF-κB in the ISRE response, cells were pre-incubated with cell permeable NF-κB inhibitor (50 μm) for 1 h prior to treatment with IFN-α in the presence or absence of ionomycin. Cell lysates were prepared and luciferase activity was measured with a Victor X5 multilabel plate reader using the dual-luciferase reporter system according to the manufacturer (Promega).
Statistical Analysis
Standard deviation was calculated using GraphPad Prism 5 software (La Jolla, CA). Statistical differences between groups were determined using unpaired Student's t test. p values of <0.05 between were considered statistically significant. Error bars and shaded regions of calcium traces were shown to represent standard error (S.E.) of the mean.
RESULTS
Impaired SOCE in Somatic Jurkat Variant H123
Chemical mutagenesis of the human T-cell line Jurkat with the ICR-191 frameshift mutagen has led to the identification of a number of critical signaling molecules needed in T cell activation (9, 22). We initially reported that the somatic Jurkat variant H123 harboring a defect in Ca2+ signaling was susceptible to the apoptotic effects of type I IFNs (IFN-α/β) while parental Jurkat was growth inhibited in the absence of apoptosis (8). What remained unclear was whether this gain of apoptotic function induced by IFN-α/β in H123 cells was intimately linked to a defect in the Ca2+ response pathway. First, to confirm that H123 cells had a defect in Ca2+ signaling, parental and H123 Jurkat cells were transiently transfected with a NFAT-responsive luciferase reporter plasmid (Fig. 1A) While robust NFAT transactivation was observed in parental Jurkat T cells after treatment with ionomycin and PMA, no NFAT activity was observed in similarly treated H123 Jurkat cells. Next, we compared the relative abilities of thapsigargin (Tg, Fig. 1B) or ionomycin (Iono, Fig. 1C) to stimulate SOCE in parental and H123 Jurkat cells. Significant endoplasmic reticulum (ER) Ca2+ release was observed in both cell lines after exposure to Tg or Iono (first peak, Fig. 1, B and C); although the total amount of Ca2+ released was moderately less in H123 cells. In contrast, a drastic deficiency of Ca2+ re-entry after store depletion was observed in H123 cells (second peak, Fig. 1, B and C) in comparison to parental Jurkat cells. Of note, IFN-α treatment of either parental or H123 Jurkat cells did not influence Ca2+ mobilization (data not shown). Thus our results show that unlike previously reported somatic Jurkat mutants, H123 Jurkat cells have a deficiency in SOCE, which has been recognized as the major route of Ca2+ entry for T cells.
FIGURE 1.

Jurkat variant H123 cells have impaired SOCE. A, parental and H123 Jurkat cells were transfected with an NFAT-luciferase reporter and left untreated or treated with Iono (2 μm) plus PMA (10 ng/ml) for 6 h. Cells were harvested for determination of luciferase activity. Values were normalized against Renilla luciferase activity. Results from a representative experiment are shown as mean ± S.E. of triplicate samples. B, parental Jurkat (solid line) and the Jurkat variant H123 (dashed line) cells were loaded with 2.5 μm Fura-2/AM. Thapsigargin (Tg, 2 μm) or Ca2+ (1 mm) was added or removed as indicated. C, same as in B except parental Jurkat (solid line) and H123 (dashed line) cells were treated with ionomycin (Iono, 2 μm) before removal and addition of Ca2+ (1 mm).
H123 Cells Lack Orai1 Protein
STIM1, STIM2, and Orai1 are now well recognized as critical components of SOCE (15, 23). Therefore, we assessed their relative levels of expression using quantitative RT-PCR and Western blot analysis. Compared with parental Jurkat cells, H123 cells demonstrated a ∼30% reduction in STIM1 at both the RNA (Fig. 2A) and protein (Fig. 2B, top panel) levels. No obvious changes in the expression of STIM2 were found between parental and H123 Jurkat cells (Fig. 2, A and B). In contrast, although no striking differences in the amount of Orai1 RNA were found; a dramatic loss of Orai1 protein was detected in H123 cells (Fig. 2B). Sequence analysis of Orai1 mRNA revealed a deletion of a single nucleotide mapped to the intracellular portion of the protein that resulted in a frameshift mutation and early termination of translation (supplemental Fig. S1). Thus it is likely that a deficiency in SOCE in H123 cells is due to the combination of loss of Orai1 and reduction in STIM1 protein content.
FIGURE 2.
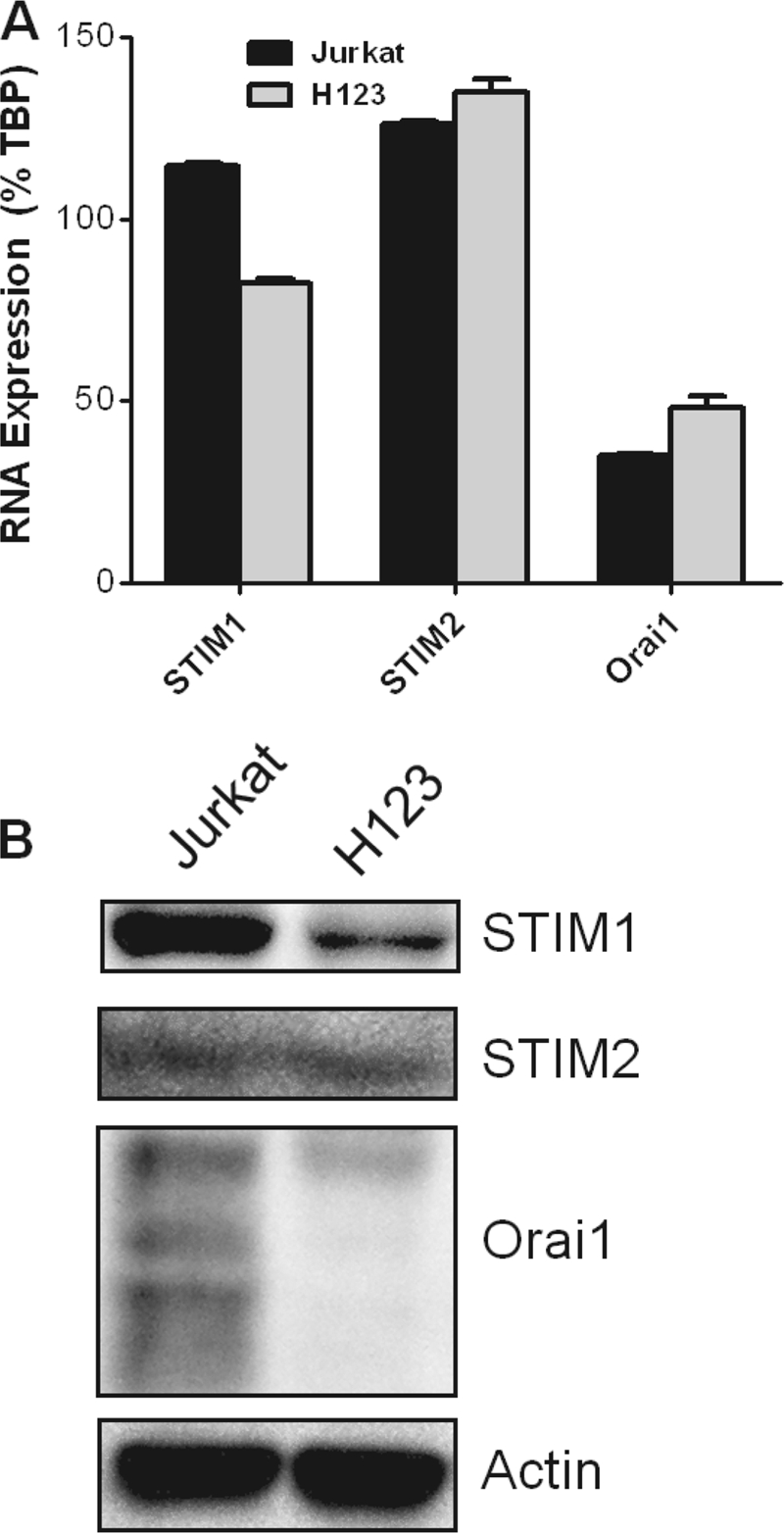
Orai1 and STIM protein expression is impaired in H123 cells. A, STIM1, STIM2, and Orai1 mRNAs were measured in parental and H123 Jurkat cells by quantitative RT-PCR. The level of mRNA transcripts was normalized to TATA box-binding protein (TBP). B, cells extracts were analyzed for detection of the indicated proteins by Western blot analysis. Actin was used to monitor equal loading of proteins.
SOCE Is Restored in H123 Cells by Re-expression of Orai1 and STIM
To determine if the loss of expression of STIM1 and Orai1 was the cause of loss of SOCE in H123 cells, we generated both H123 (Fig. 3A) and parental Jurkat (Fig. 3B) cell lines stably expressing empty vector or Orai1. Cells were then transiently transfected with either YFP or YFP-STIM1 to compare SOCE levels. Consistent with prior studies (16), SOCE was restored only after re-expression of both STIM1 and Orai1 (Fig. 3A); only modest changes in SOCE were observed when expressed individually. In parental Jurkat cells no changes in SOCE were observed in response to either STIM1 expression or STIM1+Orai1 expression (Fig. 3B); consistent with prior studies, expressing Orai1 alone inhibited SOCE (16). We were somewhat surprised by the fact that co-expression of STIM1 and Orai1 failed to further increase SOCE above control levels in parental Jurkat cells. In an effort to determine if this reflected Fura-2 saturation, experiments were also performed using 300 μm extracellular Ca2+ (supplemental Fig. S2A), with similar results. However, examination of YFP intensity revealed ∼50% less STIM1 expression in Jurkat cells stably expressing Orai1 compared with parental Jurkat cells (supplemental Fig S2B). This may indicate that elevated Ca2+ levels due to co-expression of STIM1 and Orai1 may be too high to be tolerated in this cell type. Irrespective, these observations reveal that re-expression of STIM1 and Orai1 in H123 cells restores SOCE.
FIGURE 3.

Re-expression of STIM1 together with Orai1 rescues SOCE in H123 cells. Intracellular Ca2+ concentration was measured in fura-2 loaded (A) H123 Jurkat and (B) parental Jurkat cells. Cells stably expressing either mock vector or Orai1-CFP were transiently transfected with YFP or YFP-STIM1. Thapsigargin (Tg, 2 μm) or Ca2+ (1 mm) was added or removed as indicated.
Orai1 and STIM1 Overexpression Reverses the Apoptotic Effects of IFN-α in H123 Cells
To determine if defective expression of Orai1 and/or STIM1 proteins was associated with H123 cells susceptibility to IFN-α/β-induced apoptosis, H123 cells reconstituted with Orai1 and/or STIM1 were examined for the presence of apoptotic cells following treatment with IFN-α for 48 h. Images of Annexin V-stained cells expressing CFP-Orai1 and/or YFP-STIM1 before and after IFN-α treatment are depicted in Fig. 4A. As predicted, given the absence of any difference in SOCE, expressing pEYFP (empty vector), CFP-Orai1 or YFP-STIM1 had no effect-on IFN-α-induced apoptosis in H123 cells (Fig. 4B). In contrast, H123 cells co-expressing CFP-Orai1 and YFP-STIM1 were fully protected against IFN-α-induced apoptosis (Figs. 4, A, bottom panel and B). Hence, H123 cells exhibiting defective SOCE were vulnerable to the lethal effects of type I IFNs. Given the role of SOCE in the maintenance of endoplasmic reticulum (ER) Ca2+ concentration and the critical link between ER Ca2+ concentration and ER stress (24, 25), we examined the processing of XBP1, an early indicator of ER stress (26). However, we found no evidence that type I IFNs induced ER stress response in either parental or H123 Jurkat cells (supplemental Fig. S3), indicating that ER stress likely has little or no role in protection from IFN-induced apoptosis by Orai1/STIM1 overexpression.
FIGURE 4.

STIM1 and Orai1 inhibit IFN-α-mediated apoptosis of H123 cells. A, H123 cells stably expressing empty vector or Orai1-CFP cells were transiently transfected with either YFP or YFP-STIM1. 24 h after transfection, cells were left untreated or treated with 1000 units/ml IFN-α for 48 h and then stained with Hoechst (blue) and annexin-V (red). Yellow fluorescence indicates cells expressing YFP-STIM1. Apoptotic cells are shown as red. B, percent apoptosis was calculated by counting the number of annexin-V-PE-positive cells divided by the total number of cells (control) or those expressing YFP-STIM1 and/or Orai1-CFP when expressed. A minimum of 200 cells were counted in each individual experiment and data collected from three individual experiments are shown. Results are shown as mean ± S.E.
SOCE Inactivation Causes IFN-α-induced Apoptosis in T Cells
Reciprocal experiments in parental Jurkat cells were performed to confirm if SOCE impairment could switch T cells to now undergo IFN-α-induced apoptosis. To this effect, parental Jurkat cells were transfected with a dominant negative form of CFP-Orai1 (Orai1E106A); known to dramatically inhibit SOCE (see Fig. 5A). As reported before and shown in Fig. 5, B and C, IFN-α stimulation did not induce apoptosis in parental Jurkat cells (Fig. 5C). However, Jurkat cells became sensitive to IFN-α when Orai1E106A was expressed, with apoptosis observed in ∼12–15% of the cells after 48 h of treatment, as detected by Annexin-V staining (Fig. 5, B and C). An increase in the level of IFN-α-induced apoptosis (∼35%) was observed after 72 h of treatment (data not shown). Additional experiments corroborated our finding in that a reduction in Ca2+ entry via incubation of Jurkat cells with the Orai channel blocker BTP2 (Fig. 5D) or the Ca2+ chelator EGTA (Fig. 5E) at doses that minimally affected cell viability for 72 h promoted apoptotic sensitivity to IFN-α to a degree that was similar to that observed in cells expressing dominant-negative Orai1E106A. Interestingly, EGTA (but not BTP2)-induced apoptosis was further enhanced by increasing the IFN-α concentration to 3000 units/ml (compare Fig. 5, D versus E).
FIGURE 5.

Inhibition of SOCE sensitizes parental Jurkat cells to IFN-α-induced apoptosis. A, intracellular Ca2+ concentration was measured in parental Jurkat cells stably expressing empty vector or Orai1 E106A-CFP. Cells were loaded with 2.5 μm fura-2/AM. Thapsigargin (Tg, 2 μm) or Ca2+ (1 mm) was added or removed as indicated. B, parental Jurkat cells stably expressing empty vector or Orai1 E106A-CFP were left untreated or treated with 1000 units/ml IFN-α for 48 h, and then stained with Hoechst (blue) and annexin-V (red). Apoptotic cells are shown as red. C, percent apoptosis was calculated by counting the number of annexin V-positive cells divided by the total number Orai1-CFP-positive cells, from three individual experiments. D, parental Jurkat cells were left untreated, treated with IFN-α alone or pretreated with 3 μm BTP2 for 1 h, followed by treatment with or without IFN-α for 72 h. Quantification of apoptotic cells was measured by flow cytometric analysis of dually stained cells with annexin-V-FITC and propidium iodide. E, same as in D except cells were pretreated with 0.6 mm EGTA. Results from three individual experiments are shown as mean ± S.E.
To confirm that this effect was not restricted to Jurkat T cells, we stably expressed empty vector or CFP-Orai1E106A in the human CD4+ T cell line CEM391. Overexpression of Orai1E106A did not change the growth inhibitory effects of IFN-α when compared with CEM391 expressing empty vector alone (Fig. 6A). However, overexpression of Orai1E106A caused a remarkable apoptotic effect in CEM391 cells as 45% of these cells were in the early stages of apoptosis (Annexin V+/propidium iodide−) 48 h after IFN-α stimulation (Fig. 6, B and C). Similar findings were also observed in the human CD4+ T MOLT-4 cell line stably expressing Orai1E106A; albeit at reduced levels due to low expression of CFP-Orai1E106A (data not shown). Thus our results indicate an unexpected and previously unidentified negative regulatory role for SOCE in the induction of apoptosis by type I IFNs in T cells.
FIGURE 6.

Expression of Orai1E106A sensitizes CEM391 cells to IFN-α-induced apoptosis. CEM391 cells that were untransfected (UnTfx), carrying mock vector (EV) or stably expressing Orai1 E106A-CFP were treated with or without IFN-α (3000 units/ml) for 72 h. A, cell growth inhibition responses were measured by MTS assay. B, apoptosis was shown by Hoechst (blue) and annexin-V (red) stain and expressing of Orai1 E106A-CFP was shown in cyan. C, quantification of apoptosis was measured in cells dually stained with annexin-V-FITC and propidium iodide and analyzed by flow cytometry. Results from three individual experiments are shown as mean ± S.E.
Transcriptional Responses to IFN-α/β Are Negatively Regulated by Ca2+ Signals
Type I IFNs inhibit the proliferation of T cells via utilization of the JAK/STAT pathway (1, 7). Hence, our findings that impaired SOCE changes the IFN-α/β response from growth inhibitory to apoptotic may reveal an inhibitory effect of Ca2+ signaling pathways on IFN-α/β transcriptional responses. To test this possibility, parental and H123 Jurkat cells were transiently transfected with an IFN-responsive ISRE-luciferase reporter plasmid. No ISRE reporter activity was detected in cells treated with ionomycin alone. While IFN-α induced the transactivation of the ISRE reporter to 12-fold in both cell lines (Fig. 7A, second bars in both groups), the reporter activity was inhibited by nearly 50% in parental Jurkat cells co-stimulated with ionomycin. As expected, because of defective SOCE, the inhibitory effect of ionomycin was lost in H123 cells (Fig. 7A, fourth bars in both groups). Furthermore, a reciprocal experiment assessing the ability of ionomycin to affect ISRE reporter activity in parental Jurkat cells stably over-expressing Orai1E106A revealed substantially the same result; loss of ionomycin-mediated inhibition (Fig. 7B). Thus our results reveal SOCE-mediated negative regulation of type I IFN-induced gene transcription.
FIGURE 7.
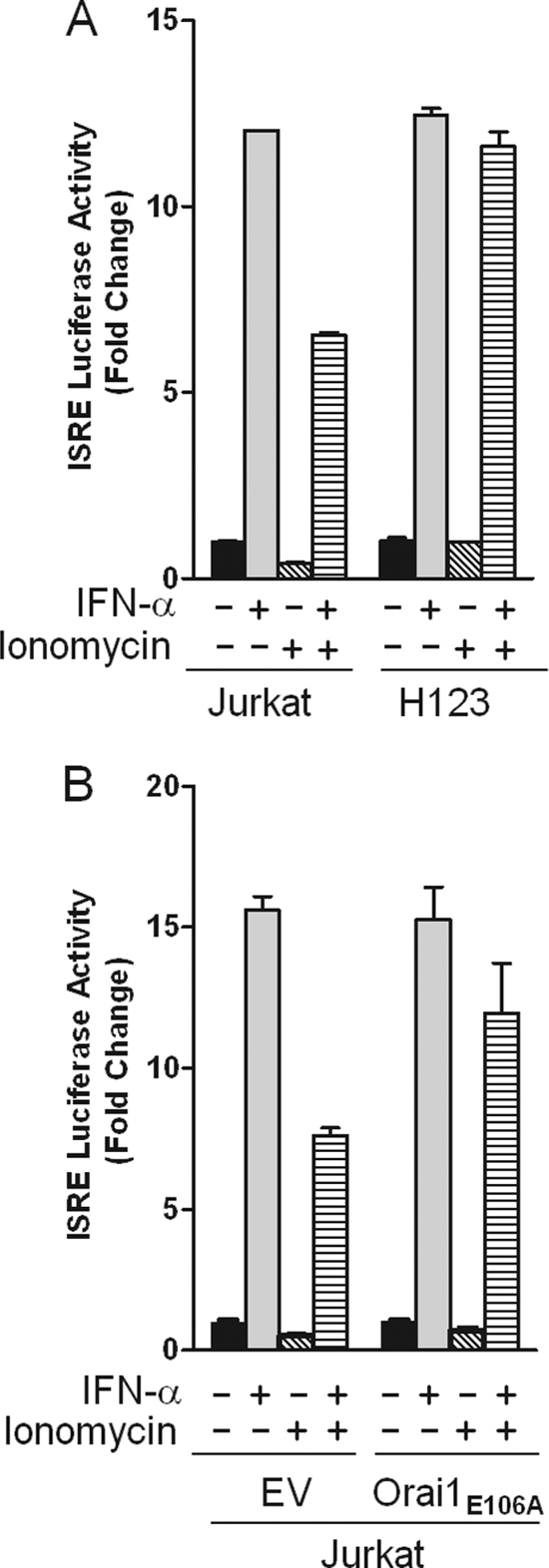
IFN-α stimulated ISRE driven transcriptional activity is not abrogated by ionomycin in H123 cells. A, parental and H123 Jurkat cells were transiently transfected with a 5×-ISRE-luciferase reporter construct. Cells were left untreated or treated with IFN-α (1000 units/ml) for 6 h in the presence or absence of ionomycin (2 μm) and harvested for determination of luciferase activity. B, parental Jurkat cells were transiently co-transfected with either CFP or Orai1 E106A-CFP and a 5×-ISRE luciferase reporter and treated as in A. Values are normalized against Renilla luciferase activity. Results from a representative experiment are shown as mean ± S.E. of triplicate samples.
NF-κB Mediates the Inhibitory Effects of Ca2+ Signals on IFN-α/β-induced Gene Transcription
The transcription factor NF-κB activates cell survival pathways and protects against apoptosis (27). Because Ca2+ signals activate NF-κB (28–31) and NF-κB has been shown to regulate the expression of a subset of IFN-induced ISGs (32), the possibility that NF-κB could be implicated in mediating Ca2+-dependent inhibition of ISG transcription was explored. A selective NF-κB inhibitor that blocks the interaction between NEMO and the IκB kinase complex (33) was used to evaluate in parental Jurkat cells alterations in IFN-responsive ISRE-luciferase activity. As expected, IFN-α transactivated the ISRE reporter to 7-fold (Fig. 8A, second bar), which was inhibited by ionomycin by nearly 50%. Pre-treatment with NF-κB inhibitor followed by co-stimulation with IFN-α plus ionomycin diminished the inhibitory effects of ionomycin by almost 50% (Fig. 8A, fourth bar). Intriguingly, this almost precisely matches the percentage that ionomycin/PMA-induced NF-κB activity was inhibited (Fig. 8B). Additional studies showed that over-expression of Orai1E106A in Jurkat and CEM391 cells impaired dramatically NF-κB activation (supplemental Fig. S4). Both observations reveal a regulatory mechanism by which Ca2+ modulated NF-κB activity suppresses transcriptional responses to type I IFNs.
FIGURE 8.
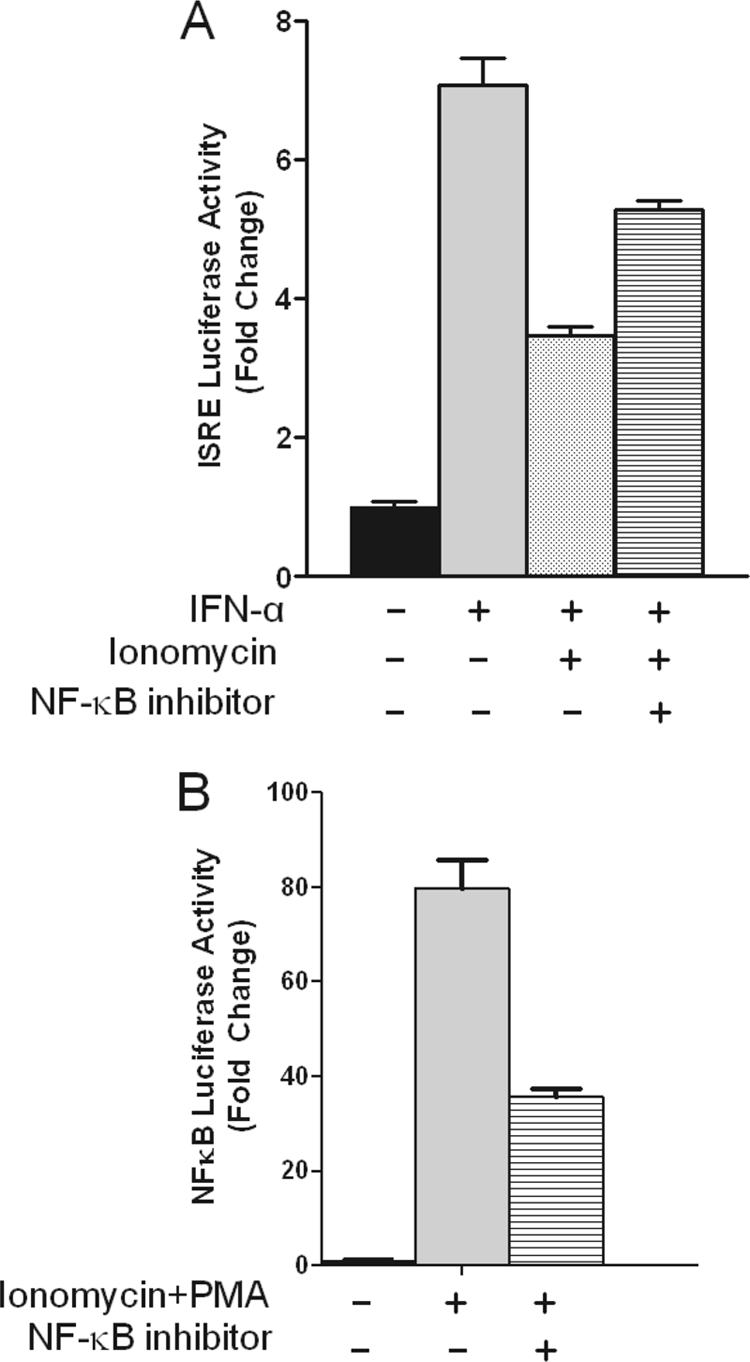
NF-κB mediates Ca2+-induced suppression of IFN-α stimulated ISRE driven transcriptional activity. A, parental Jurkat cells were transiently transfected with a 5X-ISRE-luciferase reporter plasmid. Cells were pre-incubated with or without NF-κB peptide inhibitor (50 μm) for 1 h followed by treatment with IFN-α (1000 units/ml) in the presence or absence of ionomycin (2 μm) and PMA (10 ng/ml) as indicated. B, similar to A except a 5×-NF-κB-luciferase reporter plasmid was used for transfection, and cells were only stimulated with or without ionomycin plus PMA. Values are normalized against Renilla luciferase activity. Results from a representative experiment are shown as mean ± S.E. of triplicate samples.
DISCUSSION
Ca2+ signals are critical secondary messengers that orchestrate T cell activation, differentiation, and transcriptional regulation (34–36). Type I IFNs are essential pleiotropic cytokines known for their role among others in host defense against infection and the development of adaptive immunity via modulation of T cell responses (37, 38). Our earlier work characterized a Ca2+ signaling-defective somatic mutant Jurkat (H123) to be sensitive to the apoptotic effects of type I IFNs (8). What was not established then was whether the lethal effects of type I IFNs were linked to deficiencies in Ca2+ signaling. In the current study, we have characterized the nature of the defect in Ca2+ signaling impairment in H123. Most notably, while H123 cells display normal ER Ca2+ content, they exhibit a marked deficiency in Ca2+ entry due to a lack of Orai1 and reduced expression of STIM1 protein. More remarkably, our findings reveal that SOCE influences the anti-proliferative and apoptotic effects of IFN-α/β.
The increase in intracellular Ca2+ concentration via engagement of the TCR in T cells is divided in two phases. The first phase is dictated by the release of Ca2+ from ER stores that when depleted activates the second phase characterized by prolonged Ca2+ entry via the CRAC channel now recognized as Orai (14, 35). Whereas four previously characterized somatic mutants derived from Jurkat cells exhibit defects in the first phase of TCR-induced Ca2+ mobilization because of loss of Lck, Zap70, CD45, or InsP3R1 (22, 39) H123 cells exhibit defects in the second phase of the TCR Ca2+ response. It is important to note that while T cells devoid of Lck, ZAP70, and CD45 are resistant to the antiproliferative effects of IFN-α/β (7), InsP3R1-null T cells, are highly resistant to apoptotic stimuli, suggesting that calcium release from the ER is an apoptotic trigger in T cells (39–41).
In our study, we found that defective SOCE impacted on T cell survival. Inactivation of SOCE in T cells changed the antiproliferative effects of type I IFNs from cell growth inhibition to apoptosis. While IFN-α slowed down the proliferation of T cells with intact SOCE in the absence of apoptosis (7), overexpression of dominant negative Orai1E106A to inhibit SOCE sensitized Jurkat, CEM391 and MOLT-4 T cells to the apoptotic effects of IFN-α, albeit to varying degrees. Furthermore, we provide additional evidence supporting the importance of Ca2+ entry in regulating the apoptotic effects of type I IFNs. The use of the CRAC channel inhibitor BTP2 or Ca2+ chelator EGTA at doses that minimally affect cell viability (42) were found to also sensitize cells to IFN-α-induced apoptosis similarly to over-expression of Orai1E106A. Although overexpression of Orai1E106A in parental Jurkat cells did not produce the same level of apoptosis detected in H123 cells, further characterization of H123 cells has revealed that additional proteins other than Orai1 are missing (data now shown). This is highly consistent with our observation that IFN-induced apoptosis could be enhanced more effectively by Ca2+ chelation than by BTP2 or Orai1E106A; an indication that some of these additional proteins may provide alternate routes of Ca2+ entry.
Despite differences in the degree of apoptosis observed in different cell lines and/or in response to different inhibitors of Ca2+ entry, we consistently find that IFN-induced apoptosis is stimulated by loss of Ca2+ entry in general and SOCE specifically. Hence, SOCE may have a bona-fide role in the modulation of IFN responses. If so, these findings may provide insight into the reduction of the anti-apoptotic effect of IFN-α in HIV-infected CD4+ T cells (6), since CD4+ T cells incubated with HIV envelope protein gp160 exhibit reduced Ca2+ entry (43).
The biological effects of type I IFNs are mediated by the actions of IFN-stimulated genes (ISGs) (44). While the expression of these genes is primarily driven by STAT transcription factors, there is a subset of genes that is negatively regulated by the actions of the transcription factor NF-κΒ (32). Paradoxically, type I IFNs have been reported to activate the NF-κΒ pathway via PI3K and Akt as a survival mechanism to protect cells against apoptosis (45). Impairment of the NF-κΒ pathway by expression of the IκΒα suppressor sensitizes tumor cells to type I IFN-induced death (46). Further, prior studies have firmly established that Ca2+ signals stimulate NF-κΒ (28–31), supporting the possibility that NF-κB activation may serve a mediatory role in Ca2+-dependent suppression of ISG. Indeed, we find that pharmacological inhibition of NF-κB restores transcriptional responses to IFN indicating that NF-κB has a negative regulatory role in obstructing the apoptotic response to type IFNs. This finding is also supported by the observation that expression of dominant negative Orai1E106A suppresses the induction of NF-κB activity while promoting IFN-induced apoptosis. These observations clearly establish a previously unidentified mechanism whereby, in addition to its well-defined role in T cell activation, SOCE also serves to negatively regulate IFN responses. Given the critical role IFNs play in the control of immune function, these novel findings have wide reaching and highly significant implications.
Supplementary Material
Acknowledgments
We thank Dr. Alexander Tsygankov (Temple University) for generously providing CEM391 cells, Dr. Michael Autieri (Temple University) for generously providing MOLT-4 cells, and Marc Daryl Tan Estioko (undergraduate student at Temple University) for technical assistance. We also thank Dr. Michael May (University of Pennsylvania) for providing a generous amount of NF-κB peptide inhibitor.
This work was supported, in whole or in part, by Grants R01CA140499 and K22CA095326 (to A. M. G.) from the NCI, National Institutes of Health, the American Heart Association (to J. S.), and the Pennsylvania Dept. of Health (to J. S. and A. M. G.).

This article contains supplemental Figs. S1–S4.
- IFN
- interferon
- ISRE
- interferon-stimulated response element
- Tg
- thapsigargin
- Iono
- ionomycin
- SOCE
- store-operated Ca2+ entry
- CRAC
- Ca2+ release-activated Ca2+.
REFERENCES
- 1. Gimeno R., Lee C. K., Schindler C., Levy D. E. (2005) Stat1 and Stat2 but not Stat3 arbitrate contradictory growth signals elicited by αβ interferon in T lymphocytes. Mol. Cell Biol. 25, 5456–5465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Erickson S., Sangfelt O., Castro J., Heyman M., Einhorn S., Grandér D. (1999) Interferon-α inhibits proliferation in human T lymphocytes by abrogation of interleukin 2-induced changes in cell cycle-regulatory proteins. Cell Growth Differ. 10, 575–582 [PubMed] [Google Scholar]
- 3. Dondi E., Rogge L., Lutfalla G., Uzé G., Pellegrini S. (2003) Down-modulation of responses to type I IFN upon T cell activation. J. Immunol. 170, 749–756 [DOI] [PubMed] [Google Scholar]
- 4. Curtsinger J. M., Valenzuela J. O., Agarwal P., Lins D., Mescher M. F. (2005) Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469 [DOI] [PubMed] [Google Scholar]
- 5. Dondi E., Roué G., Yuste V. J., Susin S. A., Pellegrini S. (2004) A dual role of IFN-α in the balance between proliferation and death of human CD4+ T lymphocytes during primary response. J. Immunol. 173, 3740–3747 [DOI] [PubMed] [Google Scholar]
- 6. Rodriguez B., Lederman M. M., Jiang W., Bazdar D. A., Gàrate K., Harding C. V., Sieg S. F. (2006) Interferon-α differentially rescues CD4 and CD8 T cells from apoptosis in HIV infection. AIDS 20, 1379–1389 [DOI] [PubMed] [Google Scholar]
- 7. Petricoin E. F., 3rd, Ito S., Williams B. L., Audet S., Stancato L. F., Gamero A., Clouse K., Grimley P., Weiss A., Beeler J., Finbloom D. S., Shores E. W., Abraham R., Larner A. C. (1997) Antiproliferative action of interferon-α requires components of T-cell-receptor signaling. Nature 390, 629–632 [DOI] [PubMed] [Google Scholar]
- 8. Gamero A. M., Potla R., Sakamoto S., Baker D. P., Abraham R., Larner A. C. (2006) Type I interferons activate apoptosis in a Jurkat cell variant by caspase-dependent and independent mechanisms. Cell. Signal. 18, 1299–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williams B. L., Schreiber K. L., Zhang W., Wange R. L., Samelson L. E., Leibson P. J., Abraham R. T. (1998) Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell Biol. 18, 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hewavitharana T., Deng X., Soboloff J., Gill D. L. (2007) Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium 42, 173–182 [DOI] [PubMed] [Google Scholar]
- 11. Deng X., Wang Y., Zhou Y., Soboloff J., Gill D. L. (2009) STIM and Orai: dynamic intermembrane coupling to control cellular calcium signals. J. Biol. Chem. 284, 22501–22505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr., Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stathopulos P. B., Zheng L., Li G. Y., Plevin M. J., Ikura M. (2008) Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 135, 110–122 [DOI] [PubMed] [Google Scholar]
- 14. Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 [DOI] [PubMed] [Google Scholar]
- 15. Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233 [DOI] [PubMed] [Google Scholar]
- 16. Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., Gill D. L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665 [DOI] [PubMed] [Google Scholar]
- 17. Vig M., Beck A., Billingsley J. M., Lis A., Parvez S., Peinelt C., Koomoa D. L., Soboloff J., Gill D. L., Fleig A., Kinet J. P., Penner R. (2006) CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16, 2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yeromin A. V., Zhang S. L., Jiang W., Yu Y., Safrina O., Cahalan M. D. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hewavitharana T., Deng X., Wang Y., Ritchie M. F., Girish G. V., Soboloff J., Gill D. L. (2008) Location and function of STIM1 in the activation of Ca2+ entry signals. J. Biol. Chem. 283, 26252–26262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou Y., Mancarella S., Wang Y., Yue C., Ritchie M., Gill D. L., Soboloff J. (2009) The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J. Biol. Chem. 284, 19164–19168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abraham R. T. (2000) Mutant T cell lines as model systems for the dissection of T cell antigen receptor signaling pathways. Immunol. Res. 22, 95–117 [DOI] [PubMed] [Google Scholar]
- 23. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang S. H., Zhang W., McGrath B. C., Zhang P., Cavener D. R. (2006) PERK (eIF2α kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem. J. 393, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soboloff J., Berger S. A. (2002) Sustained ER Ca2+ depletion suppresses protein synthesis and induces activation-enhanced cell death in mast cells. J. Biol. Chem. 277, 13812–13820 [DOI] [PubMed] [Google Scholar]
- 26. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 27. Wang C. Y., Mayo M. W., Baldwin A. S., Jr. (1996) TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science 274, 784–787 [DOI] [PubMed] [Google Scholar]
- 28. Chang J. W., Kim C. S., Kim S. B., Park S. K., Park J. S., Lee S. K. (2006) Proinflammatory cytokine-induced NF-κB activation in human mesangial cells is mediated through intracellular calcium but not ROS: effects of silymarin. Nephron Exp. Nephrol 103, e156–165 [DOI] [PubMed] [Google Scholar]
- 29. Li S. Y., Sun W. G., Jia Y. H., Wu G. S., An G. S., Ni J. H., Jia H. T. (2010) Calcium signal-initiated early activation of NF-κB in neurons is a neuroprotective event in response to kainic acid-induced excitotoxicity. Biochemistry 75, 101–109 [DOI] [PubMed] [Google Scholar]
- 30. Palkowitsch L., Marienfeld U., Brunner C., Eitelhuber A., Krappmann D., Marienfeld R. B. (2011) The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-κB activation. J. Biol. Chem. 286, 7522–7534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valdés J. A., Hidalgo J., Galaz J. L., Puentes N., Silva M., Jaimovich E., Carrasco M. A. (2007) NF-κB activation by depolarization of skeletal muscle cells depends on ryanodine and IP3 receptor-mediated calcium signals. Am. J. Physiol. Cell Physiol. 292, C1960–C1970 [DOI] [PubMed] [Google Scholar]
- 32. Wei L., Sandbulte M. R., Thomas P. G., Webby R. J., Homayouni R., Pfeffer L. M. (2006) NFκB negatively regulates interferon-induced gene expression and anti-influenza activity. J. Biol. Chem. 281, 11678–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. May M. J., D'Acquisto F., Madge L. A., Glöckner J., Pober J. S., Ghosh S. (2000) Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science 289, 1550–1554 [DOI] [PubMed] [Google Scholar]
- 34. Feske S. (2007) Calcium signaling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702 [DOI] [PubMed] [Google Scholar]
- 35. Lewis R. S. (2001) Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19, 497–521 [DOI] [PubMed] [Google Scholar]
- 36. Hogan P. G., Chen L., Nardone J., Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes. Dev. 17, 2205–2232 [DOI] [PubMed] [Google Scholar]
- 37. Le Bon A., Tough D. F. (2008) Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Reviews 19, 33–40 [DOI] [PubMed] [Google Scholar]
- 38. Hervas-Stubbs S., Perez-Gracia J. L., Rouzaut A., Sanmamed M. F., Le Bon A., Melero I. (2011) Direct effects of type I interferons on cells of the immune system. Clin. Canc. Res. 17, 2619–2627 [DOI] [PubMed] [Google Scholar]
- 39. Jayaraman T., Marks A. R. (1997) T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol. Cell Biol. 17, 3005–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roifman C. M., Dadi H., Somech R., Nahum A., Sharfe N. (2010) Characterization of zeta-associated protein, 70 kd (ZAP70)-deficient human lymphocytes. J. Allergy Clin. Immunol. 126, 1226–1233.e1221 [DOI] [PubMed] [Google Scholar]
- 41. Belka C., Gruber C., Jendrossek V., Wesselborg S., Budach W. (2003) The tyrosine kinase Lck is involved in regulation of mitochondrial apoptosis pathways. Oncogene 22, 176–185 [DOI] [PubMed] [Google Scholar]
- 42. Zitt C., Strauss B., Schwarz E. C., Spaeth N., Rast G., Hatzelmann A., Hoth M. (2004) Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 279, 12427–12437 [DOI] [PubMed] [Google Scholar]
- 43. Dellis O., Gangloff S. C., Paulais M., Tondelier D., Rona J. P., Brouillard F., Bouteau F., Guenounou M., Teulon J. (2002) Inhibition of the calcium release-activated calcium (CRAC) current in Jurkat T cells by the HIV-1 envelope protein gp160. J. Biol. Chem. 277, 6044–6050 [DOI] [PubMed] [Google Scholar]
- 44. Borden E. C., Williams B. R. (2011) Interferon-stimulated genes and their protein products: what and how? J. Interferon Cytokine Res. 31, 1–4 [DOI] [PubMed] [Google Scholar]
- 45. Yang C. H., Murti A., Pfeffer S. R., Kim J. G., Donner D. B., Pfeffer L. M. (2001) Interferon αβ promotes cell survival by activating nuclear factor κB through phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 276, 13756–13761 [DOI] [PubMed] [Google Scholar]
- 46. Yang C. H., Murti A., Pfeffer S. R., Basu L., Kim J. G., Pfeffer L. M. (2000) IFNαβ promotes cell survival by activating NF-κB. Proc. Natl. Acad. Sci. U.S.A. 97, 13631–13636 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.