

Background: Hydrogenases are complex metalloenzymes catalyzing the evolution of hydrogen gas but lacking an efficient system to produce recombinant forms.
Results: An NADP(H)-dependent hydrogenase was overproduced by almost an order of magnitude in a hyperthermophilic microorganism.
Conclusion: Homologous overproduction of an affinity-tagged hydrogenase was achieved.
Significance: Native and mutant forms of hydrogenase can now be generated for in vitro biochemical analyses and bioenergy systems.
Keywords: Bioenergy, Biotechnology, Hydrogenase, Metalloenzymes, Recombinant protein expression, Anaerobic, Fermentation, Hydrogen
Abstract
The cytoplasmic hydrogenase (SHI) of the hyperthermophilic archaeon Pyrococcus furiosus is an NADP(H)-dependent heterotetrameric enzyme that contains a nickel-iron catalytic site, flavin, and six iron-sulfur clusters. It has potential utility in a range of bioenergy systems in vitro, but a major obstacle in its use is generating sufficient amounts. We have engineered P. furiosus to overproduce SHI utilizing a recently developed genetic system. In the overexpression (OE-SHI) strain, transcription of the four-gene SHI operon was under the control of a strong constitutive promoter, and a Strep-tag II was added to the N terminus of one subunit. OE-SHI and wild-type P. furiosus strains had similar rates of growth and H2 production on maltose. Strain OE-SHI had a 20-fold higher transcription of the polycistronic hydrogenase mRNA encoding SHI, and the specific activity of the cytoplasmic hydrogenase was ∼10-fold higher when compared with the wild-type strain, although the expression levels of genes encoding processing and maturation of SHI were the same in both strains. Overexpressed SHI was purified by a single affinity chromatography step using the Strep-tag II, and it and the native form had comparable activities and physical properties. Based on protein yield per gram of cells (wet weight), the OE-SHI strain yields a 100-fold higher amount of hydrogenase when compared with the highest homologous [NiFe]-hydrogenase system previously reported (from Synechocystis). This new P. furiosus system will allow further engineering of SHI and provide hydrogenase for efficient in vitro biohydrogen production.
Introduction
Hydrogenases catalyze the reversible reduction of protons to hydrogen gas (H2) (1–3). Their physiological roles in microorganisms include the reduction of protons to evolve H2 to remove excess reductant generated by oxidative metabolism or, in the reverse reaction, the oxidation of H2 and its use as a source of reductant and energy. Structural and biochemical analyses have revealed that most hydrogenases contain either nickel and iron or only iron at their catalytic sites, and these are referred to as the [NiFe] and [FeFe] enzymes, respectively (4, 5). [NiFe]-hydrogenases have been extensively studied from mesophilic organisms, particularly from species of Desulfovibrio (6–11). With the increasing demand for energy and limiting supply of fossil fuels, carbon-neutral renewable energy sources are receiving increased attention. Biological H2 production is a potentially viable alternative to establish a renewable and low carbon-emitting hydrogen economy (12, 13). One impetus for this is the replacement of expensive palladium- and platinum-based catalysts used in the current chemical generation of hydrogen gas (14). Hence, any future cost-efficient hydrogen production method is likely to have biological or bio-inspired components (15).
Efforts to overproduce hydrogenases in various heterologous systems to decipher their structural and biochemical properties have met with limited success (16). A major limitation is the complex oxygen-sensitive post-translational processing pathway that is required to give a functional [NiFe] catalytic subunit (17–19). For example, assembly of the Hyd3 hydrogenase of E. coli, a membrane-bound [NiFe]-hydrogenase, requires the participation of at least eight processing proteins (17). Consequently, the majority of successful heterologous recombinant expression systems for hydrogenase have been achieved in closely related hosts. For example, the hydrogenase from Desulfovibrio gigas was heterologously expressed in Desulfovibrio fructosovorans (20), and a functional, NAD-dependent [NiFe]-hydrogenase from the Gram-positive organism, Rhodococcus opacus, was produced in the Gram-negative organism, Ralstonia eutropha (21). The membrane-bound hydrogenase of R. eutropha was produced in Pseudomonas stutzeri using a broad host range plasmid containing all the accessory genes required for maturation of the [NiFe] active site (22). The one example of heterologous production of a [NiFe]-hydrogenase in a distantly related organism was the production of the cytoplasmic hydrogenase I (SHI)4 from the hyperthermophilic archaeon P. furiosus, which grows at 100 °C, in the mesophilic bacterium Escherichia coli (23). Interestingly, assembly and maturation of P. furiosus SHI was accomplished by the processing proteins of E. coli, with the exception of the proteolytic C-terminal cleavage to give the functional catalytic subunit, which required the P. furiosus protease (FrxA) specific for SHI.
Unfortunately, however, none of the heterologous systems for hydrogenase have achieved significant overproduction of the enzyme relative to the amount produced in the native host organism (16, 23). An alternative approach is to homologously overproduce hydrogenase, but this obviously requires a genetic system for the host organism. To date, the only successful homologous overexpression of a [NiFe]-hydrogenase was reported with the enzyme from the mesophilic cyanobacterium Synechocystis sp. PCC6803 (24). This enzyme consists of five different subunits and utilizes NAD(P) as an electron carrier. To overexpress the hydrogenase operon and incorporate an affinity Strep-tag II, expression was controlled by a light-induced promoter psbAII. Simultaneously, five hyp accessory genes from the closely related organism Nostoc sp PCC7120 were expressed using the same promoter (24). This resulted in increased expression of the hydrogenase operon by 5-fold, but simultaneous overexpression of the maturation genes was necessary to process the increased amounts of the enzyme, resulting in a 2–3-fold increase in the amount of active hydrogenase.
The goal of the current study was to develop a homologous expression system for P. furiosus SHI. This is a heterotetrameric enzyme that contains flavin and six iron-sulfur clusters, in addition to the [NiFe] catalytic site, and utilizes NADP(H) as an electron carrier (1–3, 23, 25, 26). A diagrammatic representation of the enzyme is shown in Fig. 1, which is based on sequence analyses of the four subunits and the measured cofactor content of the purified enzyme. SHI has been shown to be very efficient in in vitro systems to produce H2 from starch and cellulose in synthetic enzyme pathways (26). These approaches are limited, however, as they utilize SHI purified from P. furiosus biomass. Our goal is, therefore, to take advantage of the genetic system recently reported with P. furiosus (27) to overproduce the holoenzyme and various mutant forms lacking one or more subunit (28). Herein we describe the development of a one-step marked knock-in method using linear DNA fragments to construct a strain that overproduces SHI by at least an order of magnitude more than the wild-type strain. The recombinant hydrogenase has a Strep-tag II affinity tag to facilitate purification and has properties that are comparable, although not identical, with those of SHI purified from wild-type P. furiosus (25). Surprisingly, although an order of magnitude of more fully processed SHI was produced in the recombinant strain, expression of the maturation genes was at the same level as in the parent strain.
FIGURE 1.

Model of affinity-tagged SHI showing subunit and cofactor content. The Strep-tag II is located at the N terminus of PF0891. Adapted from Ref. 25.
EXPERIMENTAL PROCEDURES
Growth of P. furiosus
The strains used in this work are shown in Table 1. Cells were grown in defined medium for all the genetic manipulation work (27). Large scale growth was carried out using a 20-liter fermentor using maltose as the carbon source (29). Cells were grown at 90 °C with constant flushing of N2/CO2 at 90 °C for 14 h. Cells were harvested by centrifugation, flash-frozen in liquid N2, and stored at −80 °C until used for protein purification.
TABLE 1.
Properties of P. furiosus strains used in this study
COM1 is the parent strain that was engineered to generate the OE-SHI strain to overproduce the SHI enzyme.
| Strain designation | Genotype | Deleted or inserted ORF/elements | Source |
|---|---|---|---|
| COM1 | ΔpyrF | PF1114 | Ref. 27 |
| OE-SHI | PslpStrep-tag II-shIβγδα | PslpStrep-tag II | This study |
Construction of Knock-in Cassette by Overlapping PCR
The knock-in cassette was created using overlapping PCR (30) where the primers used contained ∼20-bp overhangs (supplemental Fig. S1A). The selectable marker and flanking regions were amplified from pGLW021 (27) and P. furiosus genomic DNA, respectively. The cassette also had a codon-optimized 8-amino acid long Strep-tag II with an extra 2-amino acid linker sequence (31). PCR products were purified using a commercial extraction protocol (Stratagene, Santa Clara, CA) and were used in overlapping PCR reactions using the Pfx supermix (Invitrogen). Final overlapping PCR products were gel-purified and used for COM1 (the parent strain of P. furiosus) transformation as described previously (27). Standard molecular biology techniques were performed as described (32).
P. furiosus Transformation and Construction of OE-SHI Strain
Transformations were carried out using freshly grown COM1 strain (uracil auxotroph), a competent strain of P. furiosus (27). For transformation, 200 ng of DNA (knock-in cassette, supplemental Fig. S1A) was mixed with 100 μl of an overnight culture of COM1 cells and grown on defined medium. After incubation at 90 °C for 72 h, plates were examined for colonies on defined medium plates for gain of the pyrF marker as transformed cells are able to grow without uracil. Three colonies were picked from defined medium plates and grown overnight in 5 ml of defined medium, and 1.5-ml samples were used to isolate genomic DNA. PCR was used to confirm the correct insertion using the primers listed in supplemental Table S1, which were designed to bind outside of the homologous flanking regions and amplified using the Prime Star HS polymerase mix (Clontech). PCR-positive colonies were further purified by three separate consecutive transfers in the defined medium lacking uracil to confirm the culture phenotype. PCR screening was done after each round to ensure that proper incorporation of the knock-in cassette was maintained. The PCR product amplified from genomic DNA of one positive clone was confirmed by sequencing (Macrogen Sequencing Facility, Rockville, MD).
RNA Isolation and qPCR
Total RNA was extracted from 10 ml of mid-log phase cultures grown in rich maltose medium using the Absolute RNA prep kit (Stratagene) and quantified by Thermo Scientific NanoDrop spectrophotometer. Before quantitative PCR (qPCR) analyses, the RNA was treated with Turbo DNase (Ambion; Applied Biosystems, Bedford, CA) for 30 min at 37 °C and further purified using the DNase inactivation reagent (Ambion; Applied Biosystems, Bedford, CA). cDNA was prepared using the Affinity Script qPCR cDNA synthesis kit (Agilent Technologies, Santa Clara, CA). All quantitative reverse transcription-PCR (RT-qPCR) experiments were carried out with an Mx3000P instrument (Stratagene) with the Brilliant SYBR Green qPCR master mix (Agilent Technologies). The genes encoding the housekeeping enzymes pyruvate ferredoxin oxidoreductase (γ subunit, PF0971) and DNA polymerase sliding clamp (PF0983) were used as internal controls to normalize the amounts of cDNA that were used for qPCR (supplemental Fig. S2).
Growth Studies and Purification of OE-SHI
Growth of and H2 production by the OE-SHI (for overexpressed SHI) strain were carried out in 250-ml cultures at 90 °C in sealed bottles. At various times, the medium was sampled (1 ml) for protein estimation by the Bradford method (33), and the headspace was analyzed for H2 by transferring samples (1 ml) into 10-ml vials containing 1 ml of 0.5 m NaOH. After equilibration for 16 h to remove any residual H2S (which poisons the chromatography column), H2 was estimated by using a 6850 network gas chromatograph (Agilent Technologies, Santa Clara, CA) (23, 27). Samples of the media were also removed throughout the growth phase, and the concentration of maltose was determined. OE-SHI was purified under strictly reducing and anaerobic conditions. Frozen cells (25 g) were thawed and lysed osmotically in 75 ml of Buffer A (50 mm Tris/HCL, pH 8.0, containing 2 mm sodium dithionite) and 50 μg/ml deoxyribonuclease I (Sigma) with stirring for 1 h at 23 °C. The supernatant (cytoplasmic extract (S100), 40 ml, 11.7 mg of protein/ml) was obtained after removal of cell debris by ultracentrifugation at 100,000 × g for 1 h. Avidin (1.0 mg, Sigma) was added to the S100, and it was directly loaded using an ÄKTA purifier system (GE Healthcare) onto three 5-ml StrepTactin Sepharose high performance/StrepTrap HP columns (GE Healthcare) joined in series. The columns were pre-equilibrated and washed, and OE-SHI was eluted using the binding and elution buffers described in the manufacturer's protocol, except that all buffers contained 2 mm sodium dithionite. In addition, to optimize the overall recovery, the flow-through was reloaded onto the columns prior to washing and elution.
Other Methods
Maltose concentrations were measured spectrophotometrically using an assay kit (BioVision). Samples (50 μl) of the medium were diluted 500-fold with distilled water prior to analysis. To measure protein stability using fluorescence spectroscopy, the hydrogenase (0.1 mg/ml in 100 mm EPPS, pH 8.4) was incubated at 90 °C. Samples (50 μl) were periodically removed, and the tryptophan emission spectra were recorded using an RF-5301PC spectrofluorometer (Shimadzu, Columbia, MD). The excitation wavelength was 280 nm using a bandwidth of 5 nm.
Hydrogenase activity was routinely measured by H2 production from methyl viologen (MV, 1 mm) reduced by sodium dithionite (10 mm) at 80 °C in 100 mm EPPS buffer, pH 8.4, using gas chromatography (23, 25). One unit of hydrogenase activity is defined as 1 μmol of H2 evolved min−1. Assays were also carried out using NADPH (1 mm) as the electron donor in place of reduced methyl viologen (23, 25, 34). H2 oxidation activity was measured by the H2-dependent reduction of NADP as described previously (35). Stability assays were performed by exposing hydrogenase samples to air at 23 °C, and thermal stability assays were carried out at 90 °C under argon. Samples of purified hydrogenase, OE-SHI, and native SHI control (0.1 mg/ml) were incubated in 100 mm EPPS buffer, pH 8.0, containing 2 mm sodium dithionite at 90 °C. Western blots were prepared and analyzed using a chemiluminescent dye with the GenScript one-step Western kit (GenScript USA Inc.; Piscataway, NJ) using antibodies for the catalytic subunit of SHI (PF0894) and antibodies to P. furiosus superoxide reductase (PF1281) as the internal control. Nickel and iron were measured using a quadrupole-based ICP-MS (7500cc Agilent Technologies, Tokyo, Japan), equipped with a MicroMist nebulizer as described (36).
RESULTS
One-step Marked Insertion of Pslp with Strep-tag II in P. furiosus Genome
The COM1 mutant P. furiosus strain was utilized to manipulate the native SHI operon. This strain has a deletion in its pyrF gene and cannot grow in a minimal medium lacking uracil (27). It was previously used for markerless gene deletion by selection for uracil prototrophy and counter-selection using resistance to 5-fluoroorotic acid, an inhibitor of uracil biosynthesis. Herein we have developed a variation of that method for inserting a genetic element at any locus using linear DNA and a single double crossover event involving selection for uracil prototrophy. The promoter (Pslp), which in the parent strain drives expression of the gene encoding the S-Layer protein (PF1399), was inserted in front of the four-gene operon (PF0891–PF0894) that encodes SHI. In addition, an affinity Strep-tag II was inserted in-frame with the N terminus of the first gene (PF0891, Fig. 2). The genotypes of the COM1 parent and of the engineered strain, termed OE-SHI, are shown in Table 1. In wild-type cells, the gene encoding the S-layer protein is expressed by an order of magnitude higher than that of the SHI operon according to published DNA microarray data (37). The Strep-tag II was chosen for affinity purification as its function is not affected by the chemical reductant, sodium dithionite, which is used to maintain anaerobic conditions during hydrogenase purification. In addition, this tag was assumed not to interfere with nickel incorporation into SHI, a potential problem with a polyhistidine tag.
FIGURE 2.

Marked knock-in strategy to modify operon (PF0891–0894) encoding SHI. A schematic representation of the knock-in cassette is presented. The abbreviations are: UFR, upstream flanking region; Pgdh-pyrF, marker driven by the promoter for the glutamate dehydrogenase gene; Pslp-strep-tagII, S-Layer promoter with codon-optimized 8-amino acid Strep-tag II sequence; DFR, downstream flanking region.
Constructs were generated by overlapping PCR (supplemental Fig. S1, B and C), transformed into the COM1 parental strain, and transformants were selected on plates containing the minimal medium lacking uracil. Selected colonies were screened by PCR using primers specific for regions outside of the flanking region (supplemental Table S1). A PCR product was expected for both the COM1 and the OE-SHI strains, with the product for OE-SHI being ∼1 kb larger than COM1, indicating correct insertion at the specified locus (supplemental Fig. S1D). One colony was selected and designated as OE-SHI. Correct incorporation without mutation of the Pslp-Strep-tag II upstream of the SHI operon was confirmed by DNA sequencing.
Cytoplasmic Fraction of OE-SHI Strain Shows Increased Hydrogenase Activity
Cytoplasmic extracts (S100) were prepared from fermentor-grown cells of the OE-SHI strain and of the parental strain, and SHI activity was measured using the MV-linked hydrogenase assay (28). Approximately 20 μg of protein was used in the assays for both enzymes. The specific activity at 80 °C of the OE-SHI strain extract was 7.96 ± 3.3 units/mg when compared with 1.23 ± 0.3 units/mg for the COM1 strain (Fig. 3). Hence, the OE-SHI strain had an ∼7-fold higher level of SHI when compared with the parental strain, assuming comparable activities for the native and recombinant enzymes (see below). Immunoanalyses using a polyclonal antibody specific for the catalytic subunit of SHI (PF0894 (23)) also confirmed an increased amount of the catalytic subunit in the OE-SHI strain when compared with the parent (Fig. 3). Quantitative PCR showed that the transcript for PF0894 (the fourth gene in the SHI operon) was 20.2 ± 6.2-fold higher in the OE-SHI strain when compared with the parental strain (Fig. 4), in which transcription of the SHI operon is controlled by the Pslp system and by the native promoter, respectively.
FIGURE 3.

Increased catalytic activity and amount of catalytic subunit of SHI in OE-SHI strain. The bar graph compares the MV-linked hydrogenase activity in cytoplasmic extracts (S100) of the parent COM1 and OE-SHI strains. The error bars represent standard deviations obtained from three independent experiments. The corresponding immunoanalysis of the extracts is shown below using anti-PF0894 (catalytic subunit, see Fig. 1) with anti-PF1281 (superoxide reductase) as the internal loading control.
FIGURE 4.

Relative mRNA abundance in OE-SHI and COM1 strains. The relative levels were determined by qPCR of the mRNA encoding PF0894 (the catalytic subunit of SHI), PF0975 (frxA), and PF0559 (hypF). The error bars represent standard deviations obtained using triplicate independent samples.
Accessory Genes Encoding Hydrogenase Maturation Proteins Are Not Up-regulated in OE-SHI Strain
Because the OE-SHI strain contained 7-fold higher hydrogenase activity in its cytoplasm than the cytoplasm of COM1 cells, it was important to determine whether genes encoding the accessory proteins required for assembly of the [NiFe] active site in the catalytic subunit of SHI (PF0894) were also up-regulated by some type of feedback mechanism. We focused on the hydrogenase protease frxA (PF0975), which specifically cleaves the C terminus of the catalytic subunit (PF0894), and hypF (PF0559), which is involved is the assembly of the diatomic cyanide and carbon monoxide ligands on the iron atom of the [NiFe] catalytic site (17, 19, 23). Linkage between production of hydrogenase and the maturation process is shown by the fact that expression of the genes encoding these two proteins (PF0975 and PF0559) and those encoding SHI are all dramatically down-regulated when P. furiosus is grown on elemental sulfur (38). However, in the OE-SHI strain, the expression of both frxA or hypF was unaffected (Fig. 4) despite an almost order of magnitude increase in the amount of their “substrate,” namely, unprocessed inactive SHI. The activities of these two maturation enzymes at their wild-type levels in the parental strain are apparently high enough to keep up with processing the increased amounts of the SHI protein. This includes the formation of iron-sulfur clusters as well as assembly and insertion of the [NiFe] site and proteolysis of the catalytic subunit (Fig. 1).
OE-SHI Strain Has Similar Growth Properties as Parental Strain
P. furiosus grown on maltose produces H2 as an end product of carbohydrate fermentation, and SHI has been proposed to recycle the H2 for biosynthetic purposes (3). However, the growth of the two strains using maltose as the carbon source was comparable (Fig. 5). Moreover, the amounts of H2 produced by the two strains and the amounts of maltose consumed throughout the growth phase were also similar (Fig. 5). The dramatically increased amount of SHI in the OE-SHI strain might be expected to lead to an increased uptake of H2 and perhaps an increase in cell yield. However, it is clear from these data that the growth of P. furiosus on the maltose-based medium is not limited by the ability of the organism to recycle H2.
FIGURE 5.

Comparison of growth and H2 production and maltose consumption by OE-SHI and COM1 strains. A, growth of the two strains using maltose as the carbon source and consumption of maltose during growth in closed bottles at 95 °C. B, corresponding production of H2 during growth. The error bars represent standard deviation obtained from three independent samples.
Affinity Purification and Characterization of OE-SHI Hydrogenase
The recombinant enzyme containing the Strep-tag II (attached to the PF0891 subunit, Fig. 1), was purified from the OE-SHI strain using a StrepTactin column. The cytoplasmic fraction from the OE-SHI cells was applied directly to the column without any initial purification, and to optimize recovery of the hydrogenase, the material that flowed through the column was reapplied to the column prior to washing the column with buffer. The hydrogenase was eluted with desthiobiotin as determined by its activity. The OE-SHI enzyme was purified 24-fold by the single affinity step with a 21% recovery of activity (Table 2). Note that this is an underestimate because the S100 fraction also contains hydrogenase II (SHII), which represents about 15% of the total cytoplasmic hydrogenase activity of wild-type cells (39). Approximately 4.2 mg of the OE-SHI enzyme was obtained with a specific activity of 120 units/mg from 25 g (wet weight) of cells of the OE-SHI strain by this one-step purification (supplemental Fig. S3). A highly homogeneous preparation of the OE-SHI enzyme (Fig. 6) exhibiting a specific activity of 272 units/mg was obtained by including two additional steps of conventional chromatography (supplemental Table S2), but this yielded much less protein due to the relatively inefficient binding to the hydrophobic interaction column (supplemental Table S2). The overall yield of recombinant OE-SHI enzyme after the single affinity step (4.2 mg/25 g of cells, wet weight) is significantly higher than that reported with SHI after four chromatography steps (0.6 mg/25 g of cells, wet weight (25)).
TABLE 2.
One-step purification of the overproduced affinity-tagged OE-SHI enzyme
The cytoplasmic extract from cells of the OE-SHI strain was purified using a StrepTactin affinity column. Hydrogenase activity was measured by H2 production from reduced methyl viologen, where “unit” is one unit of activity.
| Step | Units | Protein | Specific activity | Yield | -Fold purification |
|---|---|---|---|---|---|
| mg | unit · mg−1 | % | |||
| S100 | 2536 | 468 | 5.4 | 100 | 1 |
| StrepTactin | 530 | 4.2 | 126 | 21 | 24 |
FIGURE 6.
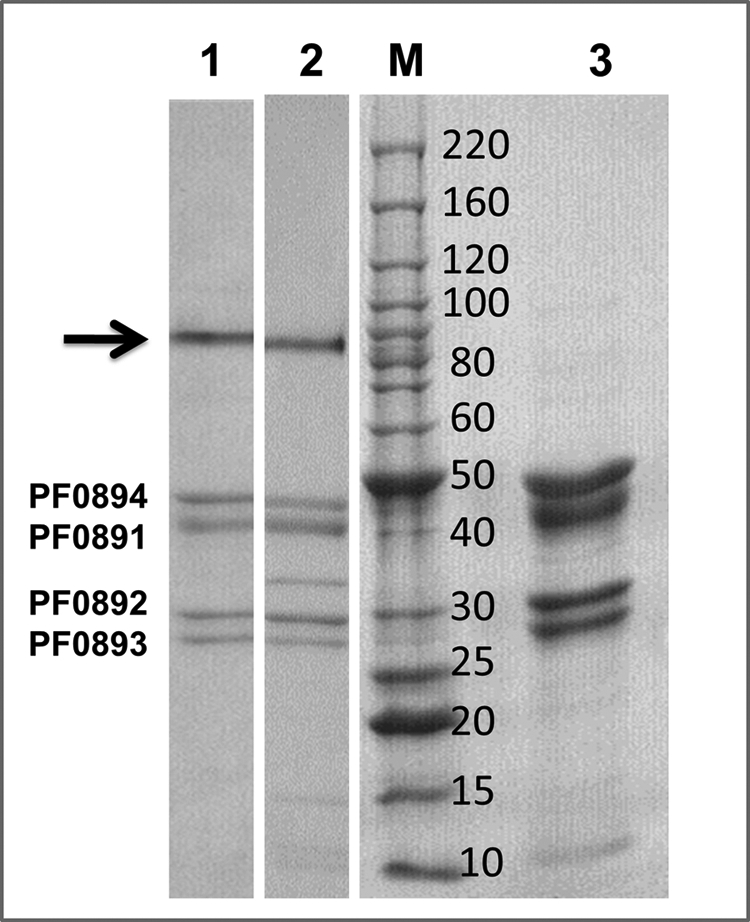
Electrophoretic analysis of OE-SHI hydrogenase. The purified enzyme was analyzed by conventional SDS-PAGE except that the protein was incubated with the SDS-loading buffer for 10 min (lane 1) or for 60 min (lane 3) prior to electrophoresis. Native SHI (treated for 10 min) is shown in lane 2. The arrow indicates the high molecular weight catalytically active protein band seen in lanes 1 and 2 (see Ref. 25). The center lane (M) contains the protein molecular weight ladder (Invitrogen) with corresponding masses as indicated in kDa.
The properties of the hydrogenase purified from the OE-SHI strain were determined and compared with those of the native enzyme purified from the biomass of wild-type cells (25, 39). The results are summarized in Table 3. The specific activity of the recombinant enzyme was about 50% higher than that of the native enzyme in the standard MV-linked H2 production assay measured at 80 °C. This may be related to the finding that the OE-SHI enzyme was not quite as thermostable as the native form, with a half-time for inactivation of 6 rather than 14 h at 90 °C (Table 3). The thermal stability of OE-SHI was also assessed using the H2-dependent reduction of NADP, which involves electron transfer by flavin and iron-sulfur centers and may be a more accurate reflection of protein stability. The results were in accord with those obtained using the H2 evolution assay, with half-life values for OE-SHI and SHI of 5 and 10 h, respectively, at 90 °C (Table 3, supplemental Fig. S6). The OE-SHI enzyme may therefore be more flexible and dynamic at 90 °C when compared with the native enzyme and hence more catalytically active at higher temperatures (supplemental Fig. S4). The lower thermal stability of OE-SHI at the growth temperature of the organism may also explain why there is no significant increase in H2 uptake by the OE-SHI strain in comparison with the parental strain (Fig. 5B). Nevertheless, the high stability of the recombinant enzyme was shown by the presence of the SDS-resistant and catalytically active, high molecular weight band (Fig. 6) that is evident after SDS electrophoresis of the native hydrogenase and represents undenatured holoenzyme (25, 40). That the OE-SHI protein was fully folded and contained the full complements of flavin and iron-sulfur clusters was shown by its ability to use NADPH as the electron donor for H2 production and H2 oxidation (Table 3, supplemental Fig. S6). Furthermore, the tryptophan emission spectrum of both proteins showed the same emission maxima (λmax of 355 nm) with almost the same fluorescence yield (supplemental Fig. S7), which suggests that both have a similar three-dimensional structure. However, both proteins showed little change in their emission spectra even after 16 h at 90 °C, suggesting that the difference in their residual activities after prolonged is not due to gross structural changes.
TABLE 3.
Properties of affinity-tagged SHI purified from the OE-SHI strain and SHI purified from native biomass
| Property | OE-SHI | SHI |
|---|---|---|
| MV-linked specific activity (unit · mg−1) | 272 | 190 |
| NADPH linked specific activity (unit · mg−1) | 2.0 | 1.5 |
| Half-life (t½/h) at 90 °C under argon (H2 evolution) | 6.0 | 14 |
| Half-life (t½/h) at 25 °C under air (H2 evolution) | 25 | 21 |
| Half-life (t½/h) at 90 °C under argon (H2 oxidation) | 5.0 | 10 |
| Fe:Ni ratio | 27:1 | 21:1 |
SHI is predicted to contain 23 iron atoms/heterotetramer together with a single nickel atom (Fig. 1), and the measured ratio for the OE-SHI hydrogenase (27 ± 2.84:1) is slightly higher than that of the native (21 ± 3.23:1, Table 3). However, given the high specific activity of the recombinant enzyme, it would seem unlikely that it contains a significant amount of enzyme lacking a [NiFe] catalytic site. The sensitivity of the OE-SHI enzyme to inactivation by oxygen (air) was also similar to that of the native enzyme with a half-life of about 1 day (Table 3), indicating that the catalytic sites of the two forms of the enzyme are in virtually identical environments within the protein.
DISCUSSION
The development of a genetic system for P. furiosus has important implications for this organism as we now have a tool to manipulate any gene within its genome. In the present study, we focused on SHI, one of three hydrogenases of P. furiosus (3). Overexpression of the four genes encoding this enzyme, which are arranged in a single operon, led to several unexpected results. First, more than a 7-fold increase in specific hydrogenase activity was observed in the OE-SHI strain, but this had little effect on the growth of the organism in closed (non-sparged) cultures, conditions under which H2 accumulates. In fact, the recombinant strain if anything grew marginally better than the COM1 parent (Fig. 5 and supplemental Fig. S5). The physiological effect of an increased amount of SHI is presumably an increase in the amount of NADPH produced, but this is clearly not significant under the growth conditions studied.
We successfully increased the amount of SHI produced by P. furiosus generating an enzyme that can be obtained in a highly purified form by a single affinity step. Remarkably, in comparison with other homologous expression systems for [NiFe]-hydrogenases, that of P. furiosus represents a 100-fold higher yield of the hydrogenase. For the homologous expression system of Synechocystis sp. PCC 6803, a total of 25 g of cells yielded only 0.04 mg of hydrogenase protein, which compares with 4.2 mg from the OE-SHI strain (24). One-step affinity purification based on the Strep-tag II tag was used to purify both types of recombinant enzyme from their cytoplasmic extracts, and in both cases, the recovery of activity was ∼20%. However, there were some important differences between the strategies employed with these two overexpression systems. With Synechocystis sp. PCC 6803, a 5-fold increase in the production of the hydrogenase was obtained, but this also required overexpression of the five hyp genes encoding the maturation and accessory proteins. In contrast, no attempt was made to overexpress the genes encoding the maturation proteins for P. furiosus SHI. In fact, the wild-type expression levels of the genes encoding the protease (frxA) that processes the C terminus of the SHI catalytic subunit and the key processing protein, hypF (17, 19), were unaffected, although expression of the SHI operon increased 20-fold. How the hydrogenase maturation process is regulated is not understood, but clearly, P. furiosus can respond to the increased production of SHI to give the fully functional protein with a full complement of cofactors, including the [NiFe] catalytic site, flavin, and multiple [FeS] clusters. However, the slightly lower Fe:Ni ratio in the OE-SHI enzyme when compared with the native form (Table 3) suggests that the limit may have been reached in processing this amount of the catalytic subunit of the OE-SHI enzyme. Consequently, even more OE-SHI might be produced if frxA and/or hypF, and/or one or more the other six processing proteins in P. furiosus, are also overproduced, and such studies are planned.
The properties of the recombinant affinity-tagged hydrogenase of Synechocystis sp. PCC 6803 were directly compared with that of the unmodified native enzyme because the latter has not been characterized (24). The properties of the purified OE-SHI enzyme from P. furiosus were very similar to those of the native enzyme, although the recombinant form was slightly more active and also slightly less thermostable, suggesting that the OE-SHI hydrogenase was folded differently, although the oxygen sensitivities of the two enzyme forms were the same. These differences in stability and activity of the OE-SHI enzyme are presumably the result of the 8- amino acid Strep-tag II and linker, which interferes with the folding process. The difference in thermal stability is also very evident in its H2 oxidation activity (supplemental Fig. S6). This has been previously reported using the same affinity tag with the d-arabitol dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima. In this case, the enzyme was heterologously produced in E. coli, and the tagless form retained 90% of its original activity after 90 min at 85 °C, whereas the Strep-tagged version had a half-life of only 5 min (41).
The availability a P. furiosus strain that over produces the SHI hydrogenase affords many opportunities for those interested in research on such enzymes. For example, it will now be possible to generate site-directed mutants and active forms lacking one or more subunit of SHI (28). In addition, it provides a means to investigate the roles of the accessory genes hypF and frxA in processing SHI as these are poorly understood. There is also only limited insight into the functions of hypC, hypD, and hypE and SlyD in the maturation mechanism (5, 19, 42). In the engineered P. furiosus OE-SHI strain, the production of the hydrogenase is under control at the transcriptional level by a strong, constitutive promoter (that for the gene encoding the S-Layer protein), which is not known to be down-regulated by conditions that regulate the hydrogenase activity in this organism, such as elemental sulfur (So) (37). Hence, the addition of So to maltose-grown cells would dramatically down-regulate the expression of hypF (PF0559) and frxA (PF0975) significantly (37), but production of OE-SHI would not be affected. It should therefore be possible to trap processing intermediates in SHI maturation by simply adding So to the OE-SHI strain, with and without constitutive expression of one or more of the processing genes that would normally be down-regulated by So. Such an approach might allow the isolation of incompletely processed OE-SHI containing its C-terminal peptide by a single affinity purification step and permit a study of its [NiFe] site and the nature of the other three subunits of the enzyme and their cofactor contents.
In conclusion, we have successfully overexpressed the heterotetrameric metalloenzyme SHI complex by using a marked knock-in method to insert a stronger promoter and an affinity tag for purification purposes. Despite the complex maturation process involved with the [NiFe] active site, the production of SHI was increased by almost an order of magnitude. This is the first example of the overproduction of a thermostable [NiFe]-hydrogenase and provides a method to obtain high amounts of the enzyme necessary for further development of in vitro H2 production systems (15, 43).
Supplementary Material
Acknowledgment
We thank the Bioexpression and Fermentation Facility at the University of Georgia for growing P. furiosus.
This work was supported by Grants DE-FG05-95ER20175 and DE-FG02-05ER15710 from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, Office of Science, United States Department of Energy (to M. W. W. A.).

This article contains supplemental Tables S1 and S2 and Figs. S1–S7.
- SHI
- soluble hydrogenase I
- OE-SHI
- engineered strain of P. furiosus
- MV
- methyl viologen
- qPCR
- quantitative PCR
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid.
REFERENCES
- 1. Vignais P. M., Billoud B. (2007) Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 [DOI] [PubMed] [Google Scholar]
- 2. Vignais P. M., Colbeau A. (2004) Molecular biology of microbial hydrogenases. Curr. Issues Mol. Biol. 6, 159–188 [PubMed] [Google Scholar]
- 3. Jenney F. E., Jr., Adams M. W. (2008) Hydrogenases of the model hyperthermophiles. Ann. N.Y. Acad. Sci. 1125, 252–266 [DOI] [PubMed] [Google Scholar]
- 4. Fontecilla-Camps J. C., Volbeda A., Cavazza C., Nicolet Y. (2007) Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem. Rev. 107, 4273–4303 [DOI] [PubMed] [Google Scholar]
- 5. Pandelia M. E., Ogata H., Lubitz W. (2010) Intermediates in the catalytic cycle of [NiFe]-hydrogenase: functional spectroscopy of the active site. Chemphyschem. 11, 1127–1140 [DOI] [PubMed] [Google Scholar]
- 6. Volbeda A., Charon M. H., Piras C., Hatchikian E. C., Frey M., Fontecilla-Camps J. C. (1995) Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 373, 580–587 [DOI] [PubMed] [Google Scholar]
- 7. Volbeda A., Martin L., Cavazza C., Matho M., Faber B. W., Roseboom W., Albracht S. P., Garcin E., Rousset M., Fontecilla-Camps J. C. (2005) Structural differences between the ready and unready oxidized states of [NiFe]-hydrogenases. J. Biol. Inorg. Chem. 10, 239–249 [DOI] [PubMed] [Google Scholar]
- 8. Ogata H., Hirota S., Nakahara A., Komori H., Shibata N., Kato T., Kano K., Higuchi Y. (2005) Activation process of [NiFe]-hydrogenase elucidated by high resolution X-ray analyses: conversion of the ready to the unready state. Structure. 13, 1635–1642 [DOI] [PubMed] [Google Scholar]
- 9. Marques M. C., Coelho R., De Lacey A. L., Pereira I. A., Matias P. M. (2010) The three-dimensional structure of [NiFeSe]-hydrogenase from Desulfovibrio vulgaris Hildenborough: a hydrogenase without a bridging ligand in the active site in its oxidized, “as-isolated” state. J. Mol. Biol. 396, 893–907 [DOI] [PubMed] [Google Scholar]
- 10. Ogata H., Kellers P., Lubitz W. (2010) The crystal structure of the [NiFe]-hydrogenase from the photosynthetic bacterium Allochromatium vinosum: characterization of the oxidized enzyme (Ni-A state). J. Mol. Biol. 402, 428–444 [DOI] [PubMed] [Google Scholar]
- 11. Lubitz W., Reijerse E., van Gastel M. (2007) [NiFe] and [FeFe]-hydrogenases studied by advanced magnetic resonance techniques. Chem. Rev. 107, 4331–4365 [DOI] [PubMed] [Google Scholar]
- 12. Cammack R., Frey M., Robson R. (2001) Hydrogen as a Fuel: Learning from Nature, pp. 201–230, Taylor & Francis, London [Google Scholar]
- 13. Gupta R. B. (2009) Hydrogen Fuel: Production, Transport, and Storage, pp. 1–31, CRC Press, Boca Raton, FL [Google Scholar]
- 14. Lee H. S., Vermaas W. F., Rittmann B. E. (2010) Biological hydrogen production: prospects and challenges. Trends. Biotechnol. 28, 262–271 [DOI] [PubMed] [Google Scholar]
- 15. Krassen H., Schwarze A., Friedrich B., Ataka K., Lenz O., Heberle J. (2009) Photosynthetic hydrogen production by a hybrid complex of photosystem I and [NiFe]-hydrogenase. ACS Nano. 3, 4055–4061 [DOI] [PubMed] [Google Scholar]
- 16. English C. M., Eckert C., Brown K., Seibert M., King P. W. (2009) Recombinant and in vitro expression systems for hydrogenases: new frontiers in basic and applied studies for biological and synthetic H2 production. Dalton Trans. 9970–9978 [DOI] [PubMed] [Google Scholar]
- 17. Blokesch M., Böck A. (2002) Maturation of [NiFe]-hydrogenases in Escherichia coli: the HypC cycle. J. Mol. Biol. 324, 287–296 [DOI] [PubMed] [Google Scholar]
- 18. Blokesch M., Albracht S. P., Matzanke B. F., Drapal N. M., Jacobi A., Böck A. (2004) The complex between hydrogenase-maturation proteins HypC and HypD is an intermediate in the supply of cyanide to the active site iron of [NiFe]-hydrogenases. J. Mol. Biol. 344, 155–167 [DOI] [PubMed] [Google Scholar]
- 19. Böck A., King P. W., Blokesch M., Posewitz M. C. (2006) Maturation of hydrogenases. Adv. Microb. Physiol. 51, 1–71 [DOI] [PubMed] [Google Scholar]
- 20. Rousset M., Magro V., Forget N., Guigliarelli B., Belaich J. P., Hatchikian E. C. (1998) Heterologous expression of the Desulfovibrio gigas [NiFe]-hydrogenase in Desulfovibrio fructosovorans MR400. J. Bacteriol. 180, 4982–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Porthun A., Bernhard M., Friedrich B. (2002) Expression of a functional NAD-reducing [NiFe]-hydrogenase from the Gram-positive Rhodococcus opacus in the Gram-negative Ralstonia eutropha. Arch. Microbiol. 177, 159–166 [DOI] [PubMed] [Google Scholar]
- 22. Lenz O., Gleiche A., Strack A., Friedrich B. (2005) Requirements for heterologous production of a complex metalloenzyme: the membrane-bound [NiFe]-hydrogenase. J. Bacteriol. 187, 6590–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun J., Hopkins R. C., Jenney F. E., McTernan P. M., Adams M. W. (2010) Heterologous expression and maturation of an NADP-dependent [NiFe]-hydrogenase: a key enzyme in biofuel production. PLoS One. 5, e10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Germer F., Zebger I., Saggu M., Lendzian F., Schulz R., Appel J. (2009) Overexpression, isolation, and spectroscopic characterization of the bidirectional [NiFe]-hydrogenase from Synechocystis sp. PCC 6803. J. Biol. Chem. 284, 36462–36472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bryant F. O., Adams M. W. (1989) Characterization of hydrogenase from the hyperthermophilic archaebacterium, Pyrococcus furiosus. J. Biol. Chem. 264, 5070–5079 [PubMed] [Google Scholar]
- 26. Zhang Y. H., Evans B. R., Mielenz J. R., Hopkins R. C., Adams M. W. (2007) High yield hydrogen production from starch and water by a synthetic enzymatic pathway. PLoS One. 2, e456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lipscomb G. L., Stirrett K., Schut G. J., Yang F., Jenney F. E., Jr., Scott R. A., Adams M. W., Westpheling J. (2011) Natural competence in the hyperthermophilic archaeon Pyrococcus furiosus facilitates genetic manipulation: construction of markerless deletions of genes encoding the two cytoplasmic hydrogenases. Appl. Environ. Microbiol. 77, 2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hopkins R. C., Sun J., Jenney F. E., Jr., Chandrayan S. K., McTernan P. M., Adams M. W. (2011) Homologous expression of a subcomplex of Pyrococcus furiosus hydrogenase that interacts with pyruvate ferredoxin oxidoreductase. PLOS One 6, e26569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verhagen M. F., Menon A. L., Schut G. J., Adams M. W. (2001) Pyrococcus furiosus: large scale cultivation and enzyme purification. Methods Enzymol. 330, 25–30 [DOI] [PubMed] [Google Scholar]
- 30. Horton R. M., Hunt H. D., Ho S. N., Pullen J. K., Pease L. R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68 [DOI] [PubMed] [Google Scholar]
- 31. Maier T., Drapal N., Thanbichler M., Böck A. (1998) Strep-tag II affinity purification: an approach to study intermediates of metalloenzyme biosynthesis. Anal. Biochem. 259, 68–73 [DOI] [PubMed] [Google Scholar]
- 32. Sambrook J., Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., pp. 1.31–1.138, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 33. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 34. Silva P. J., van den Ban E. C., Wassink H., Haaker H., de Castro B., Robb F. T., Hagen W. R. (2000) Enzymes of hydrogen metabolism in Pyrococcus furiosus. Eur. J. Biochem. 267, 6541–6551 [DOI] [PubMed] [Google Scholar]
- 35. Ma K., Zhao Z. H., Adams M. W. (1994) Hydrogen production from pyruvate by enzymes purified from the hyperthermophilic archaeon, Pyrococcus furiosus: a key role for NADPH. FEMS Microbiol. Lett. 122, 245–250 [Google Scholar]
- 36. Cvetkovic A., Menon A. L., Thorgersen M. P., Scott J. W., Poole F. L., 2nd, Jenney F. E., Jr., Lancaster W. A., Praissman J. L., Shanmukh S., Vaccaro B. J., Trauger S. A., Kalisiak E., Apon J. V., Siuzdak G., Yannone S. M., Tainer J. A., Adams M. W. (2010) Microbial metalloproteomes are largely uncharacterized. Nature. 466, 779–782 [DOI] [PubMed] [Google Scholar]
- 37. Lee H. S., Shockley K. R., Schut G. J., Conners S. B., Montero C. I., Johnson M. R., Chou C. J., Bridger S. L., Wigner N., Brehm S. D., Jenney F. E., Jr., Comfort D. A., Kelly R. M., Adams M. W. (2006) Transcriptional and biochemical analysis of starch metabolism in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 188, 2115–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schut G. J., Bridger S. L., Adams M. W. (2007) Insights into the metabolism of elemental sulfur by the hyperthermophilic archaeon Pyrococcus furiosus: characterization of a coenzyme A-dependent NAD(P)H sulfur oxidoreductase. J. Bacteriol. 189, 4431–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma K., Weiss R., Adams M. W. (2000) Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J. Bacteriol. 182, 1864–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukherjee S., Sharma S., Kumar S., Guptasarma P. (2005) Slow irreversible unfolding of Pyrococcus furiosus triosephosphate isomerase: separation and quantitation of conformers through a novel electrophoretic approach. Anal. Biochem. 347, 49–59 [DOI] [PubMed] [Google Scholar]
- 41. Kallnik V., Schulz C., Schweiger P., Deppenmeier U. (2011) Properties of recombinant Strep-tagged and untagged hyperthermophilic D-arabitol dehydrogenase from Thermotoga maritima. Appl. Microbiol. Biotechnol. 90, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 42. Forzi L., Sawers R. G. (2007) Maturation of [NiFe]-hydrogenases in Escherichia coli. Biometals. 20, 565–578 [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y. H., Sun J., Zhong J. J. (2010) Biofuel production by in vitro synthetic enzymatic pathway biotransformation. Curr. Opin. Biotechnol. 21, 663–669 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.