

Abstract
The first total synthesis of the dihydrooxepine-containing epidithiodiketopiperazine (ETP) (−)-acetylaranotin (1) is reported. The key steps of the synthesis include an enantioselective azomethine ylide (1, 3)-dipolar cycloaddition reaction to set the absolute and relative stereochemistry, a rhodium-catalyzed cycloisomerization/chloride elimination sequence to generate the dihydrooxepine moiety, and a stereoretentive diketopiperazine sulfenylation to install the epidisulfide. This synthesis provides access to (−)-1 in 18 steps from inexpensive, commercially available starting materials. We anticipate that the approach described herein will serve as a general strategy for the synthesis of additional members of the dihydrooxepine ETP family.
The epidithiodiketopiperazine (ETP) natural products are a large and structurally diverse family of biologically active fungal metabolites that beautifully exemplify the interplay between molecular structure and function in nature (Figure 1).1 One structural subgroup of the ETPs is characterized by the presence of a seven-membered dihydrooxepine ring, and includes acetylaranotin (1),2 MPC1001B (2),3 and emethallicin A (3).4 Compounds 1–3 exhibit an array of biological activities, ranging from the inhibition of viral RNA polymerase2,5 to antiproliferative3 and apoptotic6 activity against various human cancer cell lines. The labile ETP core, in combination with the complex peripheral structures, renders these molecules challenging candidates for chemical synthesis. Indeed, ETPs containing the dihydrooxepine ring at their periphery have remained elusive as synthetic targets,7 despite the fact that 1 was first isolated over 40 years ago, and that the first synthesis of the biosynthetically related compound, gliotoxin (4), was reported in 1976.8 As part of a research program aimed at advancing the chemistry and biology of ETP natural products, we sought to develop a general strategy for the synthesis of the dihydrooxepine ETPs. In this communication we report the results of our synthetic efforts, which have culminated in the first enantioselective total synthesis of (−)-acetylaranotin (1).
Figure 1.

Epidithiodiketopiperazine (ETP) natural products.
Our synthetic planning began with the retrosynthetic simplification of (−)-acetylaranotin (1) to a C2-symmetric diketopiperazine intermediate, 8 (Scheme 1). In the forward sense, this disconnection reserves installation of the redox-active epidisulfide for the final stage of the synthesis, and highlights the first tactical consideration: identification of conditions to oxidize the diketopiperazine C-H bonds in the presence of the dihydrooxepine moiety. The recent syntheses of pyrroloindoline ETPs (such as 5, 6, and 7, Figure 1) have utilized strategies involving diketopiperazine oxidation and thiol trapping of presumed acyliminium intermediates under acidic conditions to achieve late-stage C-S bond formation.9 Due to the sensitivity of dihydrooxepines to both oxidative and acidic conditions, this strategy was not expected to be feasible for the preparation of acetylaranotin (1). Since the dihydrooxepines of 8 were anticipated to be stable under basic conditions, we instead envisioned utilizing a modification of Schmidt’s protocol for diketopiperazine enolization and trapping with S8.10
Scheme 1.
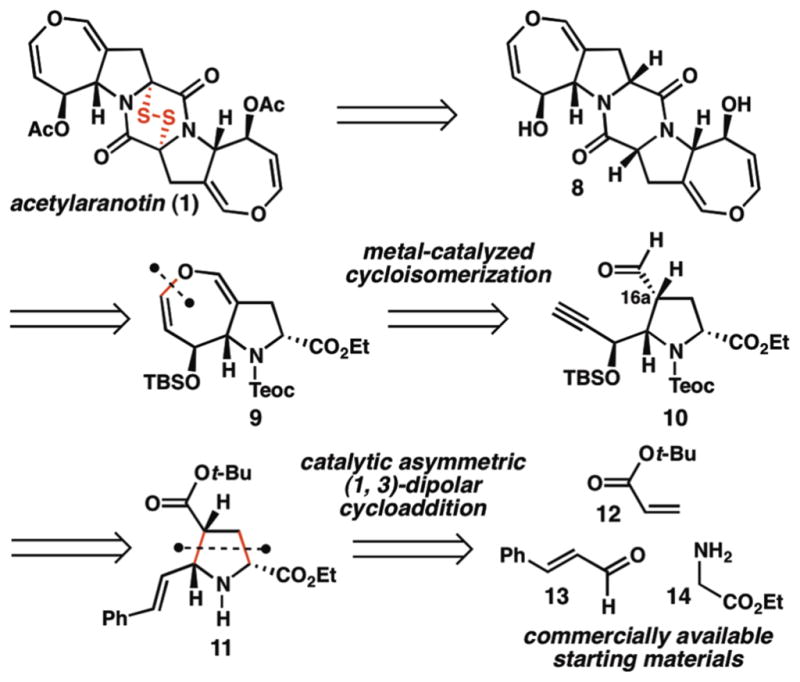
Retrosynthetic analysis for acetylaranotin (1).
Diketopiperazine 8 was envisioned to arise from the dimerization of two equivalents of protected amino ester 9 through a standard peptide coupling sequence. Preparation of 9 raises the second key tactical consideration: construction of the dihydrooxepine moiety. Relatively few general methods have been disclosed for dihydrooxepine formation, and these methods are typically constrained to substitution patterns that are undesirable in the context of preparing 1.11,12 Inspired by recent examples of transition metal-catalyzed hetero-cycloisomerization reactions of alkynes,13 we envisioned preparing dihydrooxepine 9 from alkynal 10 (or from an aldehyde surrogate) through a metal vinylidene-mediated 7-endo cycloisomerization.14,15 Alkynal 10 was expected to arise from pyrrolidine 11, the product of a catalytic asymmetric (1, 3)-dipolar cycloaddition reaction between tert-butyl acrylate (12) and the azomethine ylide derived from ethyl glycinate (14) and cinnamaldehyde (13).
In the forward sense, exposure of cinnamaldimine 15, pre-generated from ethyl glycinate (14) and cinnamaldehyde (13), to tert-butyl acrylate (12) in the presence of catalytic copper iodide and brucin-OL16 as the chiral ligand provided the corresponding endo-pyrrolidine in 50% yield and 96% ee (Scheme 2). Subsequent cleavage of the tert-butyl group using trifluoroacetic acid (TFA) furnished TFA salt 16. Notably, during the trituration process utilized to isolate 16, the enantiomeric excess was enriched to >98%. Although the yield of the (1, 3)-dipolar cycloaddition reaction was modest, the inexpensive starting materials and catalyst system employed in this transformation meant that it could be routinely conducted on multigram scale to furnish ample quantities of 16. Protection of the amine as the trimethylsilylethyl carbamate and ozonolytic cleavage of the alkene delivered hydroxylactone 17 in 77% yield over two steps.
Scheme 2.

Enantioselective synthesis of pyrrolidine 21.
To incorporate the required alkyne for the cycloisomerization reaction, hydroxylactone 17 was treated with excess ethynylmagnesium bromide; a standard acidic workup resulted in spontaneous lactonization. Unfortunately, the major lactone diastereomer (not shown) possessed the undesired stereochemistry at C13 (acetylaranotin numbering) for elaboration to the natural product.17 Whereas efforts to override the observed diastereoselectivity by varying the reaction parameters failed to produce synthetically useful quantities of 18, a procedure involving in situ Mitsunobu lactonization18 of the transiently formed hydroxy acid was more fruitful, delivering 18 in 76% isolated yield. Lactone 18 was then reduced to diol 19 with NaBH4 in EtOH, and bis-silylation with TBSOTf followed by selective cleavage of the primary silyl ether furnished alcohol 20. Finally, oxidation of the primary alcohol with Dess-Martin periodinane19 afforded aldehyde 10 (see Scheme 1) in excellent yield.20
With access to aldehyde 10, we were poised to study the key cycloisomerization reaction. Unfortunately, dihydrooxepine formation was not observed under any of the conditions screened; in all cases, the substrate was either recovered as a mixture with its C16a-epimer or underwent complete decomposition.21 We therefore set out to design an aldehyde surrogate that would demonstrate the desired reactivity, but that would also incorporate the correct oxidation state for conversion to the dihydrooxepine. Given that alkynols have been shown to undergo vinylidene-mediated cycloisomerization under a variety of conditions,14 we turned our attention to chlorohydrin 21 as a potential substrate. Treatment of aldehyde 10 with NCS and pyrrolidine•TFA gave the α-chloroaldehyde as a single diastereomer, which was reduced in situ with NaBH4 to deliver alkynol 21 in excellent yield (Scheme 2). After screening several catalysts and solvents, we were pleased to find that exposure of a solution of the substrate (21) in DMF to catalytic [Rh(cod)Cl]2 and tris-(4-fluorophenyl)-phosphine at 85 °C provided the corresponding chloro-tetrahydrooxepine (22) in 88% yield (Scheme 3).22 After considerable experimentation, elimination of the chloride was achieved using LiCl and Li2CO3 at 100 °C in DMF to yield the desired dihydrooxepine 9.
Scheme 3.

Completion of the synthesis of acetylaranotin (1).
What remained in the synthesis of 1 was diketopiperazine formation, acetylation, and installation of the epidisulfide. Our original plan called for conversion of 9 to the corresponding amino acid, and dimerization of two identical monomers. To this end, chemoselective cleavage of the Teoc group in the presence of the TBS ether was necessary. Unfortunately, exposure of 9 to a variety of conditions provided mixtures of mono- and bis-desilylated products. In contrast, treatment of chloro-tetrahydrooxepine 22 with TBAF at 0 °C cleanly provided the free amine (Scheme 3). Subjection of the amine to the previously optimized chloride elimination conditions delivered dihydrooxepine 23 in 65% yield. Hydrolysis of the ethyl ester using lithium hydroxide in methanol gave the corresponding amino acid, however attempts to form the diketopiperazine by direct dimerization were unfruitful.
Instead, a stepwise approach was pursued in which amine 23 was coupled with carboxylic acid 24 using standard peptide coupling conditions to give 25 (Scheme 3). After a survey of fluoride sources, we were pleased to find that treatment of dipeptide 25 with TBAF•(t-BuOH)423 in acetonitrile at 70 °C effected global desilylation and cyclization to deliver a C2-symmetric compound as the major product (isolated in 27% yield). Interestingly, initial characterization of this compound using standard NMR techniques and high-resolution mass spectrometry suggested that it was a syn-diol, the result of a double C-H oxidation process. Based on the hypothesis that the oxidant was oxygen in the ambient atmosphere, the reaction was repeated under a nitrogen atmosphere using rigorously degassed solvent, which provided diketopiperazine 26 as a single diastereomer in 76% yield. The structure of 26 was confirmed by single crystal X-ray diffraction. Notably, the (S, S)-stereochemistry of the central diketopiperazine is the result of epimerization at both of the diketopiperazine methine positions under the cyclization conditions. At this time, it is uncertain whether epimerization occurs prior to cyclization of the dipeptide – cyclization of the (S, S)-configured dipeptide could potentially be more facile than that of the starting (R, R)-diastereomer – or subsequent to diketopiperazine formation to give a thermodynamically favored product. Isolation and resubjection of 26 to TBAF•(t-BuOH)4 in deuterated acetonitrile at 70 °C under air provided the same oxidation product observed previously, which was confirmed to be syn-diol 27 by X-ray diffraction analysis. Whether this double C-H oxidation proceeds through a radical or anionic mechanism is currently unclear, and understanding this process is the subject of ongoing research in our laboratory. Regardless, the high diastereoselectivity observed in the formation of diketopiperazine 26 is impressive considering the seemingly flat nature of the pentacyclic ring system. Moreover, the high apparent diastereoselectivity of the dihydroxylation suggested that the analogous dithiolation might also proceed stereoselectively.24
With diketopiperazine 26 in hand, attention turned to the epidisulfide formation. In the event, a THF solution of 26 was treated with sodium hexamethyldisilazide (NaHMDS) and the resulting solution was added to a mixture of NaHMDS and S8, after which additional NaHMDS was added (Scheme 3).25 1H NMR analysis of the crude reaction mixture indicated that the major product was a C2-symmetric compound. Upon isolation of this compound, single crystal X-ray diffraction determined it to be tetrasulfide 28, in which C-S bond formation had occurred to give the relative stereochemistry found in 1. Tetrasulfide 28 was unstable to most standard reductants; for example, exposure to sodium borohydride produced a complex mixture of decomposition products. Instead, bis-acetylation of 28 using acetyl chloride furnished the diacetate, and the tetrasulfide was reduced under mild conditions using propanedithiol and triethylamine in acetonitrile. Aerobic oxidation of the resulting dithiol delivered the natural product, 1. The spectroscopic data for synthetic (−)-acetylaranotin are identical to the original isolation data.
In conclusion, we have achieved the enantioselective total synthesis of (−)-acetylaranotin (1) – the first total chemical synthesis of any dihydrooxepine-containing ETP natural product – in 18 steps from inexpensive commercially available materials. Essential to the development of this route was the successful execution of a rhodium-catalyzed cycloisomerization/chloride elimination sequence to furnish the dihydrooxepine ring and complete the monomer subunit (9). This strategy allowed us to exploit the power of an azomethine ylide (1, 3)-dipolar cycloaddition reaction in order to enantio- and diastereoselectively construct the densely functionalized pyrrolidine scaffold of requisite alkynyl alcohol substrate 21. Furthermore, we determined that upon global deprotection, dipeptide 25 could be readily cyclized with concomitant epimerization to afford diketopiperazine 26. Notably, direct sulfenylation of diketopiperazine 26 occurs with complete retention of stereochemistry to provide epitetrathiodiketopiperazine 28. Investigations towards the implementation of these strategies and methods for the synthesis of related dihydrooxepine-containing ETP natural products are ongoing.
Supplementary Material
Acknowledgments
We thank Dr. Michael Day and Mr. Larry Henling for X-ray crystallographic structure determination, as well as Dr. David Van-derVelde for assistance with NMR structure determination. We thank Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. The Bruker KAPPA APEXII X-ray diffractometer was purchased through an award to the California Institute of Technology by the National Science Foundation (NSF) CRIF program (CHE-0639094). NMR spectra were obtained on a spectrometer funded by the National Institutes of Health (NIH) (RR027690). J.A.C. was supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship Program, and by the NSF Graduate Research Fellowship Program (Grant No. DGE-0703267). Financial support from the California Institute of Technology and the NIH (NIGMS RGM097582A) is gratefully acknowledged.
Footnotes
Supporting Information: experimental procedures and characterization and spectral data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Jiang CS, Guo YW. Mini-Rev Med Chem. 2011;11:728–745. doi: 10.2174/138955711796355276. [DOI] [PubMed] [Google Scholar]; (b) Gardiner DM, Waring P, Holwett B. Microbiol. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 2.(a) Neuss N, Boeck LD, Brannon DR, Cline JC, DeLong DC, Gorman M, Huckstep LL, Lively DH, Mabe J, Marsh MM, Molloy BB, Nagarajan R, Nelson JD, Stark WM. Antimicrob Agents Ch. 1968:213–219. [PubMed] [Google Scholar]; (b) Trown PW, Lindh KP, Milstrey KP, Gallo VM, Mayberry BR, Lindsay HL, Miller PA. Antimicrob Agents Ch. 1968:225–228. [PubMed] [Google Scholar]; (c) Nagaraja R, Huckstep LL, Lively DH, Delong DC, Marsh MM, Neuss N. J Am Chem Soc. 1968;90:2980–2982. [Google Scholar]; (d) Nagaraja R, Neuss N, Marsh MM. J Am Chem Soc. 1968;90:6518–6520. doi: 10.1021/ja01025a053. [DOI] [PubMed] [Google Scholar]; (e) Cosulich DB, Nelson NR, Van den Hende JH. J Am Chem Soc. 1968;90:6519–6521. [Google Scholar]
- 3.(a) Onodera H, Hasegawa A, Tsumagari N, Nakai R, Ogawa T, Kanda Y. Org Lett. 2004;6:4101–4104. doi: 10.1021/ol048202d. [DOI] [PubMed] [Google Scholar]; (b) Sumagari N, Nakai R, Onodera H, Hasegawa A, Rahayu ES, Ando K, Yamashita Y. J Antibiotics. 2004;57:532–534. doi: 10.7164/antibiotics.57.532. [DOI] [PubMed] [Google Scholar]
- 4.Kawahara N, Nakajima S, Yamazaki M, Kawai K. Chem Pharm Bull. 1989;37:2592–2595. doi: 10.1248/cpb.37.2592. [DOI] [PubMed] [Google Scholar]
- 5.Murdock KC. J Med Chem. 1974;17:827–835. doi: 10.1021/jm00254a010. [DOI] [PubMed] [Google Scholar]
- 6.Choi EJ, Park J-S, Kim Y-J, Jung J-H, Lee JK, Kwon HC, Yang HO. J Appl Microbiol. 2011;110:304–313. doi: 10.1111/j.1365-2672.2010.04885.x. [DOI] [PubMed] [Google Scholar]
- 7.Synthetic studies toward dihydrooxepine ETPs: Gross U, Nieger M, Bräse S. Chem Eur J. 2010;16:11624–11631. doi: 10.1002/chem.201001169.Peng JB, Clive DLJ. J Org Chem. 2009;74:513–519. doi: 10.1021/jo802344t.Peng JB, Clive DLJ. Org Lett. 2007;9:2938–2941.Goodman RM, Kishi Y. J Am Chem Soc. 1998;120:9392–9393.Goodman RM. PhD Thesis. Harvard University; May, 1998.
- 8.(a) Fukuyama T, Nakatsuka S, Kishi Y. Tetrahedron. 1981;37:2045–2078. [Google Scholar]; (b) Fukuyama T, Kishi Y. J Am Chem Soc. 1976;98:6723–6724. doi: 10.1021/ja00437a063. [DOI] [PubMed] [Google Scholar]
- 9.(a) DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549–6552. doi: 10.1021/ja201789v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J, Movassaghi M. J Am Chem Soc. 2010;132:14376–14378. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]; (d) Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Ohler E, Schmidt U, Tataruch F, Poisel H. Chem Ber. 1972;105:635. doi: 10.1002/cber.19721050229. [DOI] [PubMed] [Google Scholar]; (b) Ohler E, Schmidt U, Tataruch F. Chem Ber. 1972;105:3658. doi: 10.1002/cber.19721051119. [DOI] [PubMed] [Google Scholar]
- 11.Chou WN, White JB, Smith WB. J Am Chem Soc. 1992;114:4658–4667. [Google Scholar]
- 12.Leyhane AJ, Snapper ML. Org Lett. 2006;8:5183–5186. doi: 10.1021/ol061682j. [DOI] [PubMed] [Google Scholar]
- 13.Trost BM, McClory A. Chem As J. 2008;3:164–194. doi: 10.1002/asia.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metal-catalyzed 7-endo cyclization of alcohols and carboxylic acids onto terminal alkynes: Varela-Fernández A, García-Yebra C, Varela JA, Esteruelas MA, Saá C. Angew Chem Int Ed. 2010;49:4278–4281. doi: 10.1002/anie.201000455.Liu PN, Su FH, Wen TB, Sung HHY, Williams ID, Jia G. Chem Eur J. 2010;16:7889–7897. doi: 10.1002/chem.200903441.Koo B, McDonald FE. Org Lett. 2007;9:1737–1740. doi: 10.1021/ol070435s.Alcázar E, Pletcher JM, McDonald FE. Org Lett. 2004;6:3877–3880. doi: 10.1021/ol0483495.Jiménez-Tenorio M, Puerta MC, Valerga P, Moreno-Dorado FJ, Guerra FM, Massanet GM. Chem Commun. 2001:2324–2325. doi: 10.1039/b106647c.
- 15.Metal-catalyzed 6-endo cyclization of aldehydes onto terminal alkynes in the presence of secondary nucleophiles: Yao X, Li CJ. Org Lett. 2006;8:1953–1955. doi: 10.1021/ol060645p.Patil NT, Yamamoto Y. J Org Chem. 2004;69:5139–5142. doi: 10.1021/jo049416b.Asao N, Nogami T, Takahashi K, Yamamoto Y. J Am Chem Soc. 2002;124:764–765. doi: 10.1021/ja017366b.
- 16.Kim HY, Shih H-J, Knabe WE, Oh K. Angew Chem, Int Ed. 2009;48:7420–7423. doi: 10.1002/anie.200903479. For the structure of brucin-OL, see the Supporting Infromation. [DOI] [PubMed] [Google Scholar]
- 17.See Supporting Information for details.
- 18.Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]
- 19.Dess DB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 20.Lactone 18 could be selectively reduced to the corresponding lactol; however, we were unable to advance this compound to the required aldehyde 10.
- 21.Several transition metals catalysts in the presence and absence of ligands and additives were evaluated under a variety of conditions, including [Rh(cod)Cl]2, [Rh(cod)(MeCN)2]BF4, CpRuCl[(4-FC6H4)3P]2, (CO)5W=C(OMe)Me, AuCl, Pd(OAc)2, CuI, and Ag-OTf.
- 22.Conditions were adapted from Trost’s conditions for the preparation of indoles and benzofurans: Trost BM, McClory A. Angew Chem, Int Ed. 2007;46:2074–2077. doi: 10.1002/anie.200604183.
- 23.Kim D-W, Jeong H-J, Lim S-T, Sohn M-H. Angew Chem, Int Ed. 2008;47:8404–8406. doi: 10.1002/anie.200803150. [DOI] [PubMed] [Google Scholar]
- 24.Efforts to convert diol 27 to the corresponding epi-disulfide through standard thiol exchange conditions were unsuccessful; decomposition of the starting material was observed under both Lewis and Brønsted acid-mediated conditions.
- 25.These conditions were adapted from Nicolaou’s recent modification of Schmidt’s sulfenylation procedure: Nicolaou KC, Totokotsopoulos S, Giguére D, Sun Y-P, Sarlah D. J Am Chem Soc. 2011;133:8150–8153. doi: 10.1021/ja2032635.See also reference 9.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.