

Abstract
Quantum mechanical calculations have been used to study the intramolecular additions of hydroxylamines to alkenes and alkynes (“reverse Cope eliminations”). In intermolecular reverse Cope eliminations, alkynes are more reactive than alkenes. However, competition experiments have shown that tethering the hydroxylamine to the alkene or alkyne can reverse the reactivity order from that normally observed. The exact outcome depends on the length of the tether. In agreement with experiment, a range of density functional theory methods and CBS-QB3 calculations predict that the activation energies for intramolecular reverse Cope eliminations follow the order 6-exo-dig < 5-exo-trig < 5-exo-dig ≈ 7-exo-dig. The order of the barriers for the 5-, 6-, and 7-exo-dig reactions of alkynes arises mainly from differences in tether strain in the transition states, but is also influenced by the transition-state interaction between the hydroxylamine and alkyne. Cyclization onto an alkene in the 5-exo-trig fashion incurs slightly less tether strain than a 6-exo-dig alkyne cyclization, but its activation energy is higher because the hydroxylamine fragment must distort more before the TS is reached. If the alkene terminus is substituted with two methyl groups, the barrier becomes so much higher that it is also disfavored compared to the 5- and 7-exo-dig cyclizations.
INTRODUCTION
The addition of a hydroxylamine to an alkene, leading to a tertiary amine oxide or hydroxylamine (Scheme 1), is known as the “reverse Cope elimination”. This reaction was first reported by House1 and Black,2 and has been given several other names3 that highlight its considerable utility as a method for C-N bond formation. Alkynes are also suitable substrates.4–6 Early investigations of the reaction concentrated mainly on intramolecular cases, but very recently Beauchemin et al. have demonstrated that intermolecular reverse Cope elimination reactions can be performed with a broad range of substrates, including unstrained alkenes.7 One of the key advantages of intramolecular alkene reverse Cope elimination reactions, on the other hand, is the controlled formation of a stereocenter α to nitrogen. These reactions have been employed in numerous natural product syntheses.3,5-8
Scheme 1.
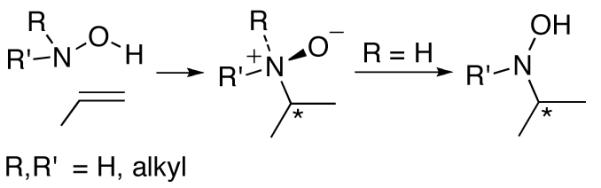
Reverse Cope Elimination Reactions of Alkenes
We report here the factors controlling the reactivities of alkenes and alkynes in intramolecular reverse Cope elimination reactions. Previously, Beauchemin et al.7 showed that alkynes undergo intermolecular reverse Cope elimination reactions more easily than alkenes. However, competition experiments by the Holmes group6 (described in the next section) demonstrated that this reactivity order can be reversed, if the hydroxylamine and alkene/alkyne are connected by a tether. The outcome depends on the tether length.
The entropic advantage of intramolecularity is widely recognized and utilized by synthetic chemists, for example in reactions directed by removable tethers.9 However, even for 5- to 7-membered rings, which are generally the easiest to form, a tether may cause subtle variations in the alignment of the reacting centres that actually disfavor reaction. Numerous examples of reactions showing tether-dependent selectivities have been theoretically characterized.10 We report here a new theoretical basis for analyzing tether length effects. We introduce an extension of the Distortion/Interaction11 (or “Activation Strain”)12 model of reactivity that is applicable to intramolecular reactions. The model allows quantification of individual contributions from distortions within the reacting fragments, interactions between these fragments, and distortion within a tether, which combine with entropic factors to bring about differences in reactivities.
Background
The scope and mechanism of hydroxylamine–alkene cyclizations were investigated by Oppolzer8 and Ciganek.5,13 These additions were established to be concerted processes, involving a 5-membered transition state (Scheme 3a). The same transition state is involved in the forward Cope elimination.14 There had been previous suggestions by House1 that the reverse Cope elimination reactions of alkenes proceed by a different mechanism, involving an intermediate nitroxyl radical (Scheme 3b). However, the radical mechanism was discounted on the basis of the reaction’s stereospecificity5-8 and insensitivity to radical inhibitors.2 Reactions with alkenes bearing electron-withdrawing groups have also been proposed to occur by the 5-centered concerted pathway.15 So too have the related reactions of oximes with electron-deficient alkenes, which were studied by Grigg.3b,16
Scheme 3.
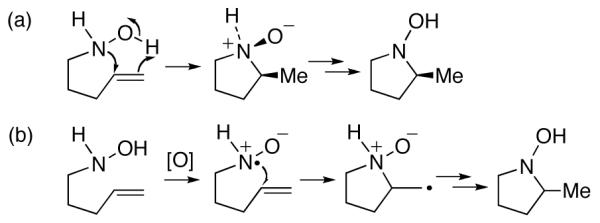
Pericyclic2,5,8 and Radical1 Mechanisms Proposed for Reverse Cope Eliminations involving Alkenes
Alkynes likewise take part in reverse Cope eliminations (Scheme 2). In this case, the immediate product of the cyclization is an enamine N-oxide. When R = H, the N-oxide rearranges to the more stable nitrone. Generation of a nitrone in this manner, and its subsequent capture by a tethered alkene, together constitute a valuable strategy for the construction of polycyclic frameworks.6 We have used the tandem reverse Cope elimination/1,3-dipolar cycloaddition sequence in our syntheses of histrionicotoxin alkaloids.17
Scheme 2.
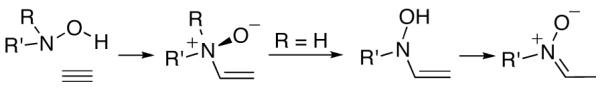
Reverse Cope Elimination Reactions of Alkynes
Hydroxylamine-alkene cyclizations occur most favorably when the products are 5- or 6-membered rings,5 but hydroxylamine–alkyne cyclizations are applicable to 5-, 6-, and 7-membered rings.6 During our studies of tandem reverse Cope elimination/1,3-dipolar cycloaddition reactions, we noted intriguing competition phenomena taking place when a tethered alkene and alkyne were both present in the same molecule.6c,e We have adopted Baldwin’s18 nomenclature for ring closure reactions to describe these cyclization processes. The reaction usually occurs through an exocyclic (exo) transition state, and the ring size is determined by the point of attachment of the nitrogen atom to the alkene (trig) or alkyne (dig). We compared a 5-exo-trig cyclization (onto an alkene) against 5-, 6-, and 7-exo-dig cyclizations (onto alkynes) as shown in Scheme 4. The 5-exo-trig cyclization was favored over the 5- and 7-exo-dig cyclizations (substrates 1 and 3), but not over the 6-exo-dig cyclization (substrate 2). The 5- and 7-exo-dig cyclizations could be forced to prevail over the 5-exo-trig if two methyl groups were included on the terminal carbon of the alkene (4, 5). This type of retardation of hydroxylamine-alkene cyclizations by gem-dimethyl substitution on the alkene terminus had previously been noted by Ciganek13a,5 and by Black.2
Scheme 4.

Competition between Alkenes and Alkynes in Intramolecular Reverse Cope Eliminations6c,e
Beauchemin et al.7 recently reported detailed investigations of intermolecular reverse Cope eliminations, as applied to both alkenes and alkynes. They showed that in intermolecular cases, hydroxylamines react more easily with alkynes than with alkenes. Our competition experiments in Scheme 4 (with substrates 1 and 3) therefore indicate that tether effects can be so substantial as to cause a reversal of the normal reactivity order.
Previous theoretical studies
Transition states for (reverse) Cope eliminations have previously been studied by several groups. Tronchet and Komaromi19 located the 5-membered concerted transition state for the reaction of NH2OH with ethylene, using a range of ab initio and density functional methods. The 5-membered transition state was preferred over an alternative 7-membered transition state, in which a molecule of solvent mediated the proton transfer from O to C.19 Acevedo and Jorgensen20 utilized QM/MM calculations to study the reaction’s solvent dependence, especially the importance of hydrogen-bonding interactions involving the N–O− group. Beauchemin et al.7 reported DFT studies of intermolecular reverse Cope elimination reactions involving both alkenes and alkynes. They found that proton transfer from N to O in the intermediate N-oxide was potentially rate-determining, but could be catalyzed by a hydroxylic molecule. They also noted the higher reactivity of alkynes and accounted for this by means of a Distortion/Interaction11 (“Activation Strain”)12 analysis of the 5-membered transition states. The major factor responsible for the higher reactivity of alkynes was the less advanced elongation of the O-H bond in the TS. The smaller distortion energy of the hydroxylamine resulted in a lower activation barrier overall.
We report calculations to determine the influence of tether length and substituents on intramolecular reverse Cope eliminations. Density functional theory calculations and CBS-QB3 calculations indicate that the parent 6-exo-dig cyclization has a low barrier, but 5- and 7-exo-dig cyclizations suffer from unfavorable tether strain and a weaker interaction between the hydroxylamine and alkyne fragments in the transition state. These factors mean that the 5- and 7-exo-dig cyclizations are unable to compete with the 5-exo-trig reaction in 1 and 3, respectively. However, incorporation of two methyl groups on the alkene terminus raises the 5-exo-trig barrier by 5–8 kcal mol−1, sufficient to reverse the kinetic preference to favor the 5- and 7-exo-dig cyclizations in 4 and 5, respectively.
THEORETICAL CALCULATIONS
Geometry optimizations, conformational searching, and vibrational frequency calculations were performed initially at the B3LYP/6-31G(d) level.21 The nature of each stationary point was determined by vibrational frequency analysis, and transition states were further verified by IRC calculations.22 Enthalpies and free energies (quoted at 298.15 K and 1 atm) were obtained from the unscaled B3LYP frequencies. Single-point energy calculations were subsequently performed on the B3LYP geometries with B3LYP-D3/6-31G(d)23 and M06-2X/6-31G(d)24. D3 provides a better treatment of dispersion interactions than B3LYP, while M06-2X provides better thermodynamics, including on cases where dispersion energy is important. Enthalpies reported at these two levels incorporate the B3LYP zero-point energy and thermal corrections. Where feasible, the DFT data were then validated against benchmark data computed with the high-accuracy CBS-QB3 method.25 Calculations were performed with the Gaussian 0326 and Gaussian 0927 programs. Molecular graphics were produced with the CYLview program.28
RESULTS AND DISCUSSION
Reaction mechanism
Transition states for the concerted additions of MeNHOH to ethylene and acetylene, computed at the B3LYP/6-31G(d) level, are shown in Figure 1. These transition structures are similar to those reported by Tronchet19 and Beauchemin7 for reactions of NH2OH. As found previously,7 the alkene transition state (TSA) shows more advanced cleavage of the O-H bond (1.23 Å) than the alkyne transition state (TSB, 1.08 Å).
Figure 1.
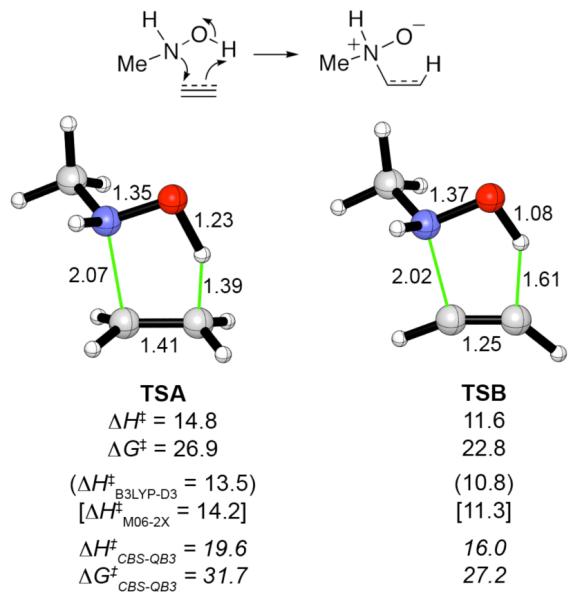
Transition structures for concerted additions of MeNHOH to ethylene and acetylene, calculated at the B3LYP/6-31G(d) level. The B3LYP activation energies (ΔH‡ and ΔG‡) are shown immediately underneath each structure, along with single-point energies from B3LYP-D3 and M06-2X calculations. CBS-QB3 values are shown in italics. Interatomic distances in Å, activation energies in kcal mol−1.
Activation energies were computed with several different density functional methods, and compared with benchmark calculations at the CBS-QB3 level. The results are shown in Figure 1. For these reactions, and others reported below, the geometries obtained by B3LYP with the basis set used for the geometry optimizations in CBS-QB3 [6-311G(2d,d,p)] are very similar to those with the 6-31G(d) basis set. The predicted chemoselectivity at the CBS-QB3 level is 4.5 kcal mol−1 (ΔΔG‡), in favor of addition to the alkyne (TSB). The three density functional methods give lower absolute barriers, but correctly estimate the chemoselectivity to within 1 kcal mol−1 of the benchmark value.29
Although a radical mechanism was discounted for the reverse Cope elimination reactions of alkenes (Scheme 1b),8,13,2 less is known about the mechanism for alkynes. A TMS group was included on the alkynes in 1-5 in the hope of establishing the stereochemistry of the intermediate enamine oxide, but the stereochemical assignments were thwarted by rapid loss of the TMS group under the experimental conditions.6c,e To check the feasibility of a radical pathway, we calculated transition states for additions of the MeNHO* radical to ethylene and acetylene. The transition structures (TSC and TSD) are shown in Figure 2. Their activation energies by CBS-QB3 are 6-10 kcal mol−1 higher than those for the concerted pericyclic reactions (ΔΔG‡). The DFT calculations also disfavor the radical pathway. Thus, even ignoring the initial oxidation step, the radical pathway is not competitive with the concerted mechanism for terminal alkenes and alkynes. Indeed it is easier for the MeNHO* radical to add to these substrates via its oxygen atom (Supporting Information). Moreover, allylic hydrogen abstraction would likely predominate if nitroxyl radicals were involved.30
Figure 2.
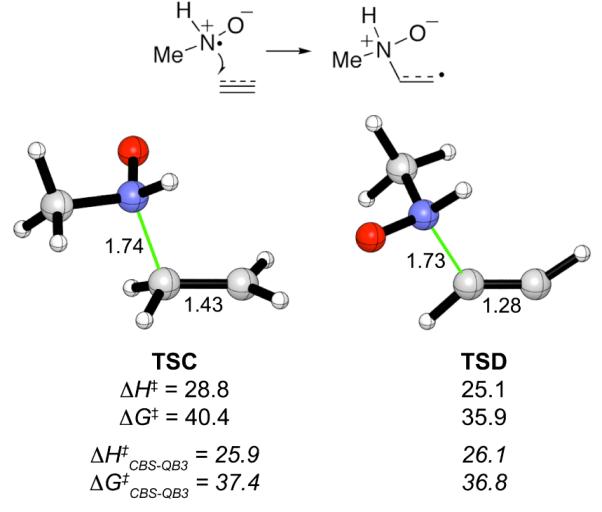
Transition structures for additions of the MeNHO* radical to ethylene and acetylene, calculated at the B3LYP/6-31G(d) level. The B3LYP activation energies (ΔH‡ and ΔG‡) are shown immediately underneath each structure, with CBS-QB3 values in italics. Interatomic distances in Å, activation energies in kcal mol−1.
Tether control
Transition structures for intramolecular reverse Cope eliminations involving an alkene (5-exo-trig) or an alkyne (5-, 6-, and 7-exo-dig) were calculated. The lowest-energy conformer of each transition state is shown in Figure 3. Two views of each TS are given, one showing the geometry of the 5-membered bond-forming array, and another showing the conformation of the tether.
Figure 3.

Transition structures for intramolecular reverse Cope eliminations, calculated at the B3LYP/6-31G(d) level. The B3LYP activation energies (ΔH‡ and ΔG‡) are shown immediately underneath each structure, along with single-point energies from B3LYP-D3 and M06-2X calculations. CBS-QB3 values are shown in italics. Interatomic distances in Å, activation energies in kcal mol−1. Two views of each TS are given, one showing the bond-forming array and the other showing the conformation of the tether.
As with the intermolecular reactions, the DFT activation energies underestimate the CBS-QB3 benchmark values, but all methods used correctly reproduce the reactivity order. The 6-exo-dig transition state (TSG) has the lowest barrier (ΔG‡ = 20.3 kcal mol−1, CBS-QB3), followed by 5-exo-trig (TSE, 23.0 kcal mol−1), and then 5- and 7-exo-dig (TSF, TSH, 24.7 and 25.3 kcal mol−1, respectively). B3LYP underestimates the barriers by 3-4 kcal mol−1. Inclusion of dispersion according to B3LYP-D3 raises the barriers by 1-2 kcal mol−1 compared to B3LYP, while the M06-2X barriers lie −0.6 to +0.9 kcal mol−1 from the B3LYP values.
The 5-exo-trig transition structure (TSE) has an activation enthalpy that is 0.3 kcal mol−1 lower than the corresponding intermolecular TS (TSA), and a free energy of activation that is 8.7 kcal mol−1 lower. TSE shows slightly more advanced transfer of the proton from O to C than TSA, with an O-H distance that is 0.03 Å longer and a C-H distance 0.03 Å shorter. The C-N bond formation in TSE is, however, less advanced (2.15 Å vs. 2.07 Å).
The three exo-dig transition structures (TSF-TSH) also show more advanced proton transfer compared to their corresponding intermolecular TS (TSB). The O-H distances in TSF-TSH are 0.01-0.04 Å longer than in TSB, and the C-H distances are 0-0.11 Å shorter. The C-N distance varies by only 0.02 Å from the intermolecular value. For the intramolecular exo-dig reactions, ΔH‡ is 0.2-5.7 kcal mol−1 higher than for the intermolecular reaction, while ΔG‡ is 1.9–6.9 kcal mol−1 lower.
The bottom structures in Figure 3 illustrate the conformation of the tether in the transition states. The 5-membered tethers in TSE and TSF adopt different envelope-like conformations. The 6-membered tether of TSG has a chair conformation, with the OH group occupying an equatorial position and the alkyne terminus midway between axial and equatorial. The 7-membered tether in TSH adopts a chairlike conformation.
To evaluate the contribution of tether strain to the activation barriers, we calculated distortion energies11 for the transition states in the manner shown in Figure 4. The analysis was performed at two levels of theory: B3LYP/6-31G(d) and CBS-QB3. The transition structures TSE-TSH are reproduced at the top of the figure. Below them, structures E-1-H-1 are models representing the 5-membered bond-forming arrays in TSE-TSH. To obtain these structures, atoms of the tether were removed, and hydrogen atoms were appended, to leave a distorted version of the transition state for addition of MeNHOH to ethylene or acetylene. The energies associated with distorting MeNHOH and ethylene or acetylene to their geometries in these arrays are shown (ΔEdist‡), as are the energies associated with the interaction between the MeNHOH and ethylene or acetylene fragments in the array (ΔEint‡). The bottom structures (E-2-H-2) are models to estimate the tether strain present in the transition states. To obtain these structures, the NHOH group was replaced by NH2 and the alkene/alkyne group was replaced by an H atom, and the bond lengths, angles, and dihedrals of the added H atoms were allowed to relax while the remaining atoms were held fixed. The energies required to distort the amines from reactant to TS geometry are shown (ΔEdist,tether‡).
Figure 4.

Calculated strain-related energies for intramolecular reverse Cope elimination transition states. The TS geometries (TSE-TSH) are reproduced at the top. Structures E-1-H-1 depict the 5-membered bond-forming array after the tether has been removed and replaced by H atoms, leaving a distorted analogue of the TS for addition of MeNHOH to ethylene or acetylene. Structures E-2-H-2 represent the geometry of the tether in the TS, with the NHOH group replaced by NH2 and the alkene/alkyne replaced by H. The bond lengths, angles, and dihedrals of the added H atoms were allowed to relax, while the remaining atoms were held fixed. B3LYP/6-31G(d) potential energies are given, with CBS-QB3 values in parentheses (kcal mol−1).
The distortion energies computed by B3LYP and CBS-QB3 differ by ≤1.8 kcal mol−1. The interaction energies differ more (by up to 7.6 kcal mol−1), and are smaller with CBS-QB3, owing to the larger basis set, better treatment of correlation, and reduction of basis set superposition error. Among the three exo-dig transition states, there is little variation in the energy required to distort the hydroxylamine and alkyne fragments to their TS geometries (F-1–H-1). The values of ΔEdist‡ range from 32 to 35 kcal mol−1 (or 30-34 kcal mol−1 by CBS-QB3). The values of ΔEint‡ vary over a wider range of 5-7 kcal mol−1. The 6-exo-dig arrangement (G-1) has the smallest value of ΔEdist‡ . It also benefits from a relatively strong interaction between the hydroxylamine and alkyne. An even stronger interaction takes place in the 7-exo-dig transition state (H-1), but is negated by the larger ΔEdist‡ . The 6-exo-dig transition state also benefits from a smaller tether distortion penalty. Distortion of the model tether (G-2) is 2-4 kcal mol−1 easier than distortion of the model 5- and 7-exo-dig tethers (F-2 and H-2). The latter two tethers both suffer from unfavorable eclipsing. The eclipsed bond in F-2 is indicated in Figure 4. In H-2, no one bond is so severely eclipsed as in F-2, but numerous small dihedrals (27-45°) are present, introducing destabilizing torsional strain into the tether.
The 5-exo-trig cyclization onto an alkene (TSE) is more difficult than the 6-exo-dig cyclization onto an alkyne (TSG), but easier than the 5- and 7-exo-dig cyclizations. Compared to TSG, TSE benefits from a 2-3 kcal mol−1 smaller tether distortion and a 7-8 kcal mol−1 stronger interaction between the hydroxylamine and alkene, but these are outweighed by the 13-15 kcal mol−1 larger distortion energy for the interacting fragments. The distortion energy of the hydroxylamine alone is 19-21 kcal mol−1 greater. These differences mirror those observed in the fully-optimized transition states (TSA and TSB), reflecting especially the greater O-H elongation in the alkene TSs.7b
The preference for the 5-exo-trig cyclization over the 5- and 7-exo-dig cyclizations arises from a combination of interaction energies and tether strain. The TS interaction energy in the 5-exo-trig cyclization is 4-12 kcal mol−1 more stabilizing than those in the 5- and 7-exo-dig cyclizations, and the tether distortion is 4-7 kcal mol−1 smaller. These two features outweigh the much greater distortions of the hydroxylamine group, and result in a 2-3 kcal mol−1 overall lower activation energy.
Our competition experiments focused on alkene cyclization in the 5-exo-trig mode.6c,e House and Lee found that a 5-exo-trig cyclization occurs more readily than a 6-exo-trig cyclization, as shown in Scheme 5.1b Cyclization of the N-(4-pentenyl)hydroxylamine 11 proceeded readily upon warming (ca. 60 °C), whereas the corresponding N-(5-hexenyl)hydroxylamine 12 had to be heated to 145 °C (refluxing xylene) to bring about cyclization. We computed the transition state for the parent 6-exo-trig cyclization (TSI), and its structure is shown in the Scheme. It favors a boatlike conformation. Its activation energy (ΔG‡) is 3 kcal mol−1 higher than that for the 5-exo-trig cyclization (TSE), consistent with the reactivities reported by House and Lee.
Scheme 5.
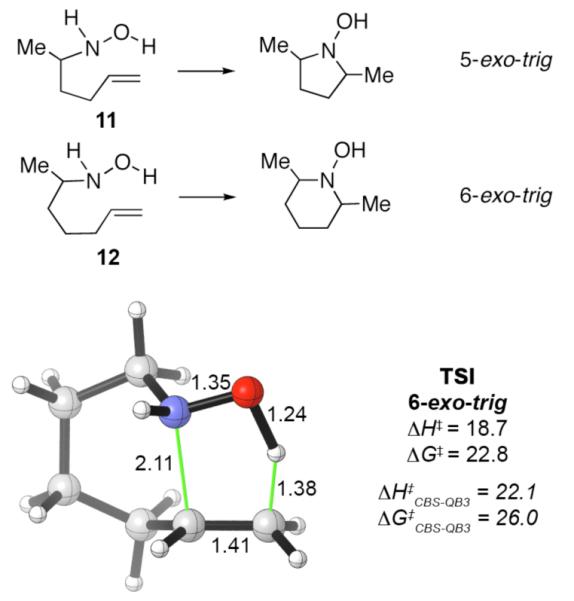
5-exo-trig and 6-exo-trig cyclizations reported by House and Lee,1b and the calculated transition state for the parent 6-exo-trig cyclization
Influence of substituents on activation barriers
The competition between the 5-exo-trig and 5- or 7-exo-dig cyclizations can be reversed to favor the latter if gem-dimethyl groups are included on the alkene terminus.6e To quantify the magnitude of the gem-dimethyl effect, the transition state for addition of MeNHOH to 2-methylpropene was calculated. A transition state was also calculated for the addition of MeNHOH to trimethylsilylacetylene, to measure the effect of the TMS group in the substrates in Scheme 4. The substituted transition structures are shown in Figure 5.
Figure 5.
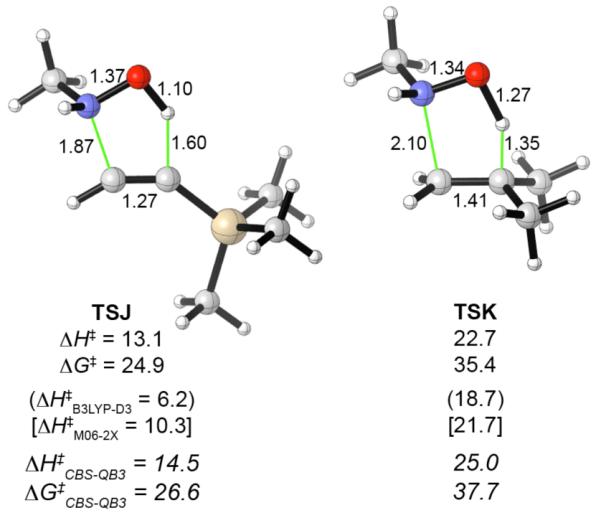
Transition structures for additions of MeNHOH to trimethylsilylacetylene and 2-methylpropene, calculated at the B3LYP/6-31G(d) level. The B3LYP activation energies (ΔH‡ and ΔG‡) are shown immediately underneath each structure, along with single-point energies from B3LYP-D3 and M06-2X calculations. CBS-QB3 values are shown in italics. Interatomic distances in Å, activation energies in kcal mol−1.
A TMS group on the alkyne (TSJ) decreases the activation energy (ΔG‡) by 0.6 kcal mol−1, and the presence of the TMS group in substrates 1-5 is therefore expected not to affect the order of reactivities compared to those calculated for the parent terminal alkynes. The dispersion-corrected functionals B3LYP-D3 and M06-2X likewise suggest that the TMS group should lower the activation barriers. In the case of B3LYP-D3 the TMS effect amounts to 4.6 kcal mol−1 and appears to overestimate the transition state stabilization arising from dispersive interactions. On the other hand, all four levels of theory predict that for an alkene, gem-dimethyl substitution at the terminus (TSK) increases the activation energy by 5-8 kcal mol−1. This effect is sufficient to raise the 5-exo-trig TS well above all three exo-dig TSs. Both the hydroxylamine and the alkene in TSK have higher distortion energies than in TSA. The total distortion energy is 8 kcal mol−1 larger. Approximately 5 kcal mol−1 of this amount is due to the more elongated O-H bond in TSK. The interaction energies, on the other hand, differ by only 0-3 kcal mol−1.
We also examined the effect of N-methyl substitution. Reverse Cope elimination reactions of NMe-substituted hydroxylamines are generally more facile than those of the corresponding NH analogues.1b,2,3a,5,13a Transition states for NMe analogues of the 5-exo-trig and 6-exo-dig cyclizations are shown in Figure 6. The NMe group lowers the barrier for the 5-exo-trig cyclization (TSL) by 2 kcal mol−1 (CBS-QB3), and lowers the barrier for the 6-exo-dig cyclization (TSM) by 1 kcal mol−1. The distortions within the interacting array and the tether are mostly the same or slightly worse in the NMe-substituted transition states, but there is a 2-4 kcal mol−1 stronger interaction between the NMe-substituted hydroxylamine and the alkene or alkyne, which leads to an overall reduction in activation energy.
Figure 6.
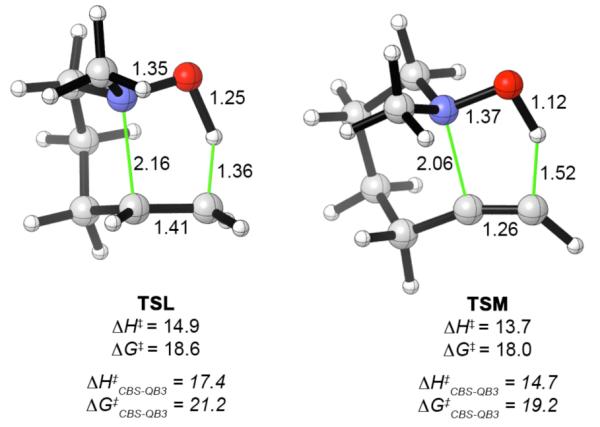
Transition structures for intramolecular reverse Cope eliminations involving N-methyl substituted hydroxylamines, calculated at the B3LYP/6-31G(d) level. The B3LYP activation energies (ΔH‡ and ΔG‡) are shown immediately underneath each structure, with CBS-QB3 values in italics. Interatomic distances in Å, activation energies in kcal mol−1.
CONCLUSIONS
Experimental studies and density functional theory calculations by Beauchemin et al. have previously shown that intermolecular reverse Cope eliminations proceed more easily with alkynes than with alkenes.7b We have found, however, that in intramolecular systems, tether effects can be large enough to reverse this reactivity order. We introduce an extension of the Distortion/Interaction model of reactivity that is applicable to intramolecular reactions, and use it to quantify the role of tether distortion in relation to the individual distortions and interaction involving the reacting fragments as a contribution to the overall barriers. In the absence of further substituents on the alkene or alkyne, the 5-exo-trig reverse Cope elimination is favored over analogous 5- and 7-exo-dig reactions, but not over a 6-exo-dig reaction. The tether effects can be modulated by gem-dimethyl substitution of the alkene, which makes the 5-exo-trig cyclization uncompetitive with the 5-, 6-, and 7-exo-dig reactions. Manipulation of these chemoselectively distinct cyclizations allow access to structurally diverse bicyclic products.
Supplementary Material
ACKNOWLEDGMENT
We thank the US NSF (CHE-0548209 to K.N.H.), the Australian Research Council (DP0985623 to E.H.K.), ARC Centre of Excellence for Free Radical Chemistry and Biotechnology (funding to E.H.K.), the Engineering and Physical Sciences Research Council UK, GlaxoSmithKline, the University of Melbourne and the Commonwealth Scientific and Industrial Research Organisation (Australia) for generous financial support. Computer resources were provided by the NCSA, UCLA ATS (USA) and NCI National Facility (Canberra, Australia, supported by the Australian Commonwealth Government).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Calculated geometries and energies, and complete citations for refs. 26 and 27. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) House HO, Manning DT, Melillo DG, Lee LF, Haynes OR, Wilkes BE. J. Org. Chem. 1976;41:855–863. [Google Scholar]; (b) House HO, Lee LF. J. Org. Chem. 1976;41:863–869. [Google Scholar]
- 2.Black D. St. C., Doyle JE. Aust. J. Chem. 1978;31:2317–2322. [Google Scholar]
- 3.Other names that have been given to this reaction include “reverse Cope cyclization”, “1,3-azaprotio cyclotransfer” and “Cope-type hydroamination”. For a review of the reaction, see: Cooper NJ, Knight DW. Tetrahedron. 2004;60:243–269. Discussion of the various names that have been given to this reaction can be found in Knight’s review, and see also: Grigg R, Markandu J, Perrior T, Surendrakumar S, Warnock WJ. Tetrahedron. 1992;48:6929–6952. For the forward Cope elimination, see: Cope AC, Trumbull ER. Org. React. 1960;11:317–493.
- 4.Winterfeldt E, Krohn W, Stracke HU. Chem. Ber. 1969;102:2346–2361. [Google Scholar]
- 5.Ciganek E, Read JM, Jr., Calabrese JC. J. Org. Chem. 1995;60:5795–5802. [Google Scholar]
- 6.(a) Fox ME, Holmes AB, Forbes IT, Thompson M. Tetrahedron Lett. 1992;33:7421–7424. [Google Scholar]; (b) Fox ME, Holmes AB, Forbes IT, Thompson M, Ziller JW. Tetrahedron Lett. 1992;33:7425–7428. [Google Scholar]; (c) Fox ME, Holmes AB, Forbes IT, Thompson M. J. Chem. Soc. Perkin Trans. 1. 1994:3379–3395. [Google Scholar]; (d) Davison EC, Holmes AB, Forbes IT. Tetrahedron Lett. 1995;36:9047–9050. [Google Scholar]; (e) Davison EC, Forbes IT, Holmes AB, Warner JA. Tetrahedron. 1996;52:11601–11624. [Google Scholar]; (f) Holmes AB, Bourdin B, Collins I, Davison EC, Rudge AJ, Stork TC, Warner JA. Pure & Appl. Chem. 1997;69:531–536. [Google Scholar]
- 7.Beauchemin AM, Moran J, Lebrun M-E, Séguin C, Dimitrijevic E, Zhang L, Gorelsky SI. Angew. Chem. Int. Ed. 2008;47:1410–1413. doi: 10.1002/anie.200703495. Moran J, Gorelsky SI, Dimitrijevic E, Lebrun M-E, Bédard A-C, Séguin C, Beauchemin AM. J. Am. Chem. Soc. 2008;130:17893–17906. doi: 10.1021/ja806300r. Bourgeois J, Dion I, Cebrowski PH, Loiseau F, Bédard A-C, Beauchemin AM. J. Am. Chem. Soc. 2009;131:874–875. doi: 10.1021/ja8077895. For synthetic development and applications, see: Cebrowski PH, Roveda J-G, Moran J, Gorelsky SI, Beauchemin AM. Chem. Commun. 2008:492–493. doi: 10.1039/b715022a. Bourgeois J, Dion I, Cebrowski PH, Loiseau F, Bédard A-C, Beauchemin AM. J. Am. Chem. Soc. 2009;131:874–875. doi: 10.1021/ja8077895. Moran J, Pfeiffer JY, Gorelsky SI, Beauchemin AM. Org. Lett. 2009;11:1895–1898. doi: 10.1021/ol900292r. Lebrun M-E, Pfeiffer JY, Beauchemin AM. Synlett. 2009:1087–1090. Loiseau F, Clavette C, Raymond M, Roveda J-G, Burrell A, Beauchemin AM. Chem. Commun. 2011;47:562–564. doi: 10.1039/c0cc02403a. Dion I, Beauchemin AM. Angew. Chem. Int. Ed. 2011 doi: 10.1002/anie.201102408. DOI: 10.1002/anie/201102408.
- 8.Oppolzer W, Spivey AC, Bochet CG. J. Am. Chem. Soc. 1994;116:3139–3140. For early work, see:Oppolzer W, Siles S, Snowden RL, Bakker BH, Petrzilka M. Tetrahedron Lett. 1979;45:4391–4394. Oppolzer W, Siles S, Snowden RL, Bakker BH, Petrzilka M. Tetrahedron. 1985;41:3497–3509.
- 9.Reviews: Bols M, Skrydstrup T. Chem. Rev. 1995;95:1253–1277. Fensterbank L, Malacria M, Sieburth SM. Synthesis. 1997:813–854. Gauthier DR, Jr., Zandi KS, Shea KJ. Tetrahedron. 1998;54:2289–2338. Knapp S. Chem. Soc. Rev. 1999;28:61–72. Breslow R. In: Templated Organic Synthesis. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 2000. pp. 159–188. Cox LR, Ley SV. In: Templated Organic Synthesis. Diederich F, Stang PJ, editors. Wiley-VCH; Weinheim: 2000. pp. 275–396. Padwa A. Pure. Appl. Chem. 2003;71:47–62. Aron ZD, Overman LE. Chem. Commun. 2004:253–265. doi: 10.1039/b309910e. Sugimura T. Eur. J. Org. Chem. 2004:1185–1192. Thilgen C, Sergeyev S, Diederich F. Top. Curr. Chem. 2005;248:1–61. Hansen EC, Lee D. Acc. Chem. Res. 2006;39:509–519. doi: 10.1021/ar050024g. Bracegirdle S, Anderson EA. Chem. Soc. Rev. 2010;39:4114–4129. doi: 10.1039/c0cs00007h. Evans PA, Cossy J. In: Metathesis in Natural Product Synthesis. Arseniyadis S, Meyer C,, editors. Wiley-VCH; Weinheim: 2010. pp. 225–259.
- 10.(a) Himo F, Demko ZP, Noodleman L. J. Org. Chem. 2003;68:9076–9080. doi: 10.1021/jo030137i. [DOI] [PubMed] [Google Scholar]; (b) Norrby P-O, Mader MM, Vitale M, Prestat G, Poli G. Organometallics. 2003;22:1849–1855. [Google Scholar]; (c) Soriano E, Marco-Contelles J. J. Org. Chem. 2005;70:9345–9353. doi: 10.1021/jo0514265. [DOI] [PubMed] [Google Scholar]; (d) Aubert C, Gandon V, Geny A, Heckrodt TJ, Malacria M, Paredes E, Vollhardt KPC. Chem. Eur. J. 2007;13:7466–7478. doi: 10.1002/chem.200601822. [DOI] [PubMed] [Google Scholar]; (e) Hashmi SK, Pankajakshan S, Rudolph M, Enns E, Bander T, Rominger F, Frey W. Adv. Synth. Catal. 2009;351:2855–2875. [Google Scholar]; (f) Lemière G, Gandon V, Cariou K, Hours A, Fukuyama T, Dhimane A-L, Fensterbank L, Malacria MJ. Am. Chem. Soc. 2009;131:2993–3006. doi: 10.1021/ja808872u. [DOI] [PubMed] [Google Scholar]; (g) Siebert MR, Karaman R. J. Mol. Struct: THEOCHEM. 2010;940:70–75. [Google Scholar]; (h) Osbourn JM, Brummond KM, Tantillo DJ. J. Am. Chem. Soc. 2010;132:11952–11966. doi: 10.1021/ja102848z. [DOI] [PubMed] [Google Scholar]; (i) Fernández I, Solé D, Sierra MA. J. Org. Chem. 2011;76:1592–1598. doi: 10.1021/jo1020954. [DOI] [PubMed] [Google Scholar]
- 11.(a) Ess DH, Houk KN. J. Am. Chem. Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; (b) Ess DH, Houk KN. J. Am. Chem. Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]; (c) Hayden AE, Houk KN. J. Am. Chem. Soc. 2009;131:4084–4089. doi: 10.1021/ja809142x. [DOI] [PubMed] [Google Scholar]; (d) Krenske EH, Houk KN, Holmes AB, Thompson J. Tetrahedron Lett. 2011;52:2181–2184. [Google Scholar]
- 12.For a review: van Zeist W-J, Bickelhaupt FM. Org. Biomol. Chem. 2010;8:3118–3127. doi: 10.1039/b926828f.
- 13.(a) Ciganek E. J. Org. Chem. 1990;55:3007–3009. [Google Scholar]; (b) Ciganek E. J. Org. Chem. 1995;60:5803–5807. [Google Scholar]
- 14.(a) Cram DJ, McCarty JE. J. Am. Chem. Soc. 1954;76:5740–5745. [Google Scholar]; (b) Bach RD, Andrzejewski D, Dusold LR. J. Org. Chem. 1973;38:1742–1743. [Google Scholar]; (c) Chiao W-B, Saunders WH., Jr. J. Am. Chem. Soc. 1978;100:2802–2805. [Google Scholar]; (d) Kwart H, George TJ, Louw R, Ultee W. J. Am. Chem. Soc. 1978;100:3927–3928. [Google Scholar]
- 15.(a) Niu D, Zhao K. J. Am. Chem. Soc. 1999;121:2456–2459. [Google Scholar]; (b) O’Neil IA, Cleator E, Southern JM, Bickley JF, Tapolczay DJ. Tetrahedron Lett. 2001;42:8251–8254. [Google Scholar]; (c) Moglioni AG, Muray E, Castillo JA, Álvarez-Larena Á, Moltrasio GY, Branchadell V, Ortuño RM. J. Org. Chem. 2002;67:2402–2410. doi: 10.1021/jo0159082. [DOI] [PubMed] [Google Scholar]
- 16.(a) Grigg R, Markandu J, Surendrakumar S, Thornton-Pett M, Warnock WJ. Tetrahedron. 1992;48:10399–10422. [Google Scholar]; (b) Grigg R, Perrior TR, Sexton GJ, Surendrakumar S, Suzuki TJ. Chem. Soc., Chem. Commun. 1993:372–374. [Google Scholar]; (c) Dondas HA, Grigg R, Hadjisoteriou M, Markandu J, Thomas WA, Kennewell P. Tetrahedron. 2000;56:10087–10096. [Google Scholar]; (d) Dondas HA, Grigg R, Hadjisoteriou M, Markandu J, Kennewell P, Thornton-Pett M. Tetrahedron. 2001;57:1119–1128. [Google Scholar]; (e) Dondas HA, Fishwick CWG, Grigg R, Thornton-Pett M. Tetrahedron. 2003;59:9997–10007. [Google Scholar]
- 17.(a) Williams GM, Roughley SD, Davies JE, Holmes AB, Adams JP. J. Am. Chem. Soc. 1999;121:4900–4901. [Google Scholar]; (b) Davison EC, Fox ME, Holmes AB, Roughley SD, Smith CJ, Williams GM, Davies JE, Raithby PR, Adams JP, Forbes IT, Press NJ, Thompson MJ. J. Chem. Soc., Perkin Trans. 1. 2002:1494–1514. [Google Scholar]; (c) Horsley HT, Holmes AB, Davies JE, Goodman JM, Silva MA, Pascu SI, Collins I. Org. Biomol. Chem. 2004;2:1258–1265. doi: 10.1039/b402307b. [DOI] [PubMed] [Google Scholar]; (d) Macdonald JM, Horsley HT, Ryan JH, Saubern S, Holmes AB. Org. Lett. 2008;10:4227–4229. doi: 10.1021/ol801604z. [DOI] [PubMed] [Google Scholar]; (e) Brasholz M, Johnson BA, Macdonald JM, Polyzos A, TsanakTSJdis J, Saubern S, Holmes AB, Ryan JH. Tetrahedron. 2010;66:6445–6449. [Google Scholar]; (f) Brasholz M, Macdonald JM, Saubern S, Ryan JH, Holmes AB. Chem. Eur. J. 2010;16:11471–11480. doi: 10.1002/chem.201001435. [DOI] [PubMed] [Google Scholar]
- 18.Baldwin JE. J. Chem. Soc., Chem. Comm. 1976:734–736. For a recent reexamination, see: Alabugin IV, Gilmore K, Manoharan M. J. Am. Chem. Soc. 2011;133:12608–12623. doi: 10.1021/ja203191f.
- 19.Komaromi I, Tronchet JMJ. J. Phys. Chem. A. 1997;101:3554–3560. [Google Scholar]
- 20.Acevedo O, Jorgensen WL. J. Am. Chem. Soc. 2006;128:6141–6146. doi: 10.1021/ja057523x. [DOI] [PubMed] [Google Scholar]
- 21.(a) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (b) Becke AD. J. Chem. Phys. 1993;98:1372–1377. [Google Scholar]; (c) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 22.(a) Gonzalez C, Schlegel HB. J. Chem. Phys. 1989;90:2154–2161. [Google Scholar]; (b) Gonzalez C, Schlegel HB. J. Phys. Chem. 1990;94:5523–5527. [Google Scholar]
- 23.Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132:154104-1–154104-19. doi: 10.1063/1.3382344. Zero-damping was employed. [DOI] [PubMed] [Google Scholar]
- 24.(a) Zhao Y, Truhlar DG. Theor. Chem. Account. 2008;120:215–241. [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 25.(a) Montgomery JA, Jr., Frisch MJ, Ochterski JW, Petersson GA. J. Chem. Phys. 1999;110:2822–2827. [Google Scholar]; (b) Montgomery JA, Jr., Frisch MJ, Ochterski JW, Petersson GA. J. Chem. Phys. 2000;112:6532–6542. [Google Scholar]
- 26.Frisch MJ, et al. Gaussian 03, Revision E.01. Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 27.Frisch MJ, et al. Gaussian 09, Revision A.02. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 28.Legault CY. CYLview, 1.0b. Université de Sherbrooke; 2009. ( http://www.cylview.org) [Google Scholar]
- 29.Basis set effects were also considered. The values of ΔH‡ for TSA and TSB at the M06-2X/6-311+G(d,p) level are 18.2 and 14.5 kcal mol-1, respectively, closer to the CBS-QB3 values.
- 30.Coseri S, Mendenhall GD, Ingold KU. J. Org. Chem. 2005;70:4629–4636. doi: 10.1021/jo0504060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.