

Abstract
Previous research demonstrates increased prostate cancer risk for pesticide applicators and pesticide manufacturing workers. Although underlying mechanisms are unknown, human biomonitoring studies indicate increased genetic damage (e.g. chromosomal aberrations) with pesticide exposure. Given that the nucleotide excision repair (NER) pathway repairs a broad range of DNA damage, we evaluated interactions between pesticide exposure and 324 single-nucleotide polymorphisms (SNPs) tagging 27 NER genes among 776 prostate cancer cases and 1444 male controls in a nested case–control study of white Agricultural Health Study pesticide applicators. We determined interaction P values using likelihood ratio tests from logistic regression models and three-level pesticide variables (none/low/high) based on lifetime days of use weighted to an intensity score. We adjusted for multiple comparisons using the false discovery rate (FDR) method. Of the 17 interactions that met FDR <0.2, 3 displayed a monotonic increase in prostate cancer risk with increasing exposure in one genotype group and no significant association in the other group. Men carrying the variant A allele at ERCC1 rs2298881 exhibited increased prostate cancer risk with high versus no fonofos use [odds ratio (OR) 2.98; 95% confidence interval (CI) 1.65–5.39; Pinteract = 3.6 × 10−4; FDR-adjusted P = 0.11]. Men carrying the homozygous wild-type TT genotype at two correlated CDK7 SNPs, rs11744596 and rs2932778 (r2 = 1.0), exhibited increased risk with high versus no carbofuran use (OR 2.01; 95% CI 1.31–3.10 for rs11744596; Pinteract = 7.2 × 10−4; FDR-adjusted P = 0.09). In contrast, we did not observe associations among men with other genotypes at these loci. While requiring replication, our findings suggest a role for NER genetic variation in pesticide-associated prostate cancer risk.
Introduction
Previous research has demonstrated a significantly increased risk of prostate cancer for pesticide applicators and pesticide manufacturing workers compared with the general population (1–3), suggesting a role for pesticides in prostate cancer etiology; however, underlying mechanisms are unknown. Although few pesticides have consistently demonstrated genotoxicity in standard in vitro and animal assays (4), a number of human biomonitoring studies (5,6) have observed increased genetic damage with exposure to mixtures of pesticides based on increased sister chromatid exchanges and micronuclei formation, as well as chromosomal aberrations (7). Other studies of workers exposed to various groups of pesticides (8–12) have observed increased damage based on the alkaline Comet assay (13). These findings suggest that pesticides might influence prostate cancer risk in humans in part by inducing DNA damage.
The nucleotide excision repair (NER) pathway is perhaps the broadest DNA repair pathway based on its role in repairing damage ranging from bulky adducts and cross-links to oxidative damage (14). NER is thought to be important in repairing damage induced by several putative prostate carcinogens, including pesticides, polycyclic aromatic hydrocarbons and heterocyclic amines (14), as well as other exogenous carcinogens such as ultraviolet radiation and aflatoxin B1 (15). NER capacity has also been associated with prostate cancer risk, with one case–control study observing a dose–response relationship between lower NER capacity and increased prostate cancer risk (16). Additionally, some studies have observed altered prostate cancer risk with genetic variation in several NER genes, including XPC, XPD (ERCC2) and XPF (ERCC4) (17). Although genome-wide association studies have not implicated NER gene loci in prostate cancer risk (18,19), these studies have not focused on populations exposed to DNA-damaging agents, in which NER genetic variation may be more important.
Given the growing evidence for a role of DNA damage in pesticide-associated prostate carcinogenesis in humans and the known involvement of the NER pathway in repairing a wide range of damage, we conducted a nested case–control study of white male pesticide applicators within the Agricultural Health Study (AHS) to evaluate interactions between pesticides from a range of chemical and functional classes and genetic variation in 27 NER genes with respect to prostate cancer. We hypothesized that NER gene variants may modify the risk of prostate cancer associated with pesticide exposure.
Materials and methods
Study population
The AHS cohort has been described in detail elsewhere (20). Briefly, 57 310 licensed restricted-use pesticide applicators in Iowa and North Carolina, representing 82% of the target population, were enrolled between December 1993 and December 1997. As part of a follow-up of the cohort (Phase two, from 1999 to 2005), participants were asked to provide a mouthwash rinse sample for extraction of buccal cell DNA, with ∼40% of the cohort returning a sample. In addition, participants with an incident prostate cancer diagnosis that did not return a sample at follow-up and met inclusion criteria for the current study were asked separately to provide a mouthwash rinse sample, with 307 of 561 (55%) returning a sample. Minimal differences were observed with respect to a variety of demographic, lifestyle, disease and occupational characteristics between cohort participants that did and did not provide a sample (21). For this nested case–control study in the AHS, which has been described previously (22), eligible cases were white pesticide applicators who were diagnosed with prostate cancer between 1993 and 2004 after enrollment in the AHS, provided a buccal cell sample and had no previous history of cancer except non-melanoma skin cancer. Eligible controls were white male applicators in the cohort who provided a buccal cell sample, had no previous history of cancer except non-melanoma skin cancer and were alive at the time of case diagnosis. Controls were frequency matched 2:1 to cases by date of birth (±1 year). Based on these inclusion criteria, 841 cases and 1659 controls were identified. As described previously (22), exclusions due to insufficient number of available chips (n = 164), quality control issues (insufficient/poor DNA quality, n = 20, or <90% completion rate for genotyping assays, n = 88) or a genetic background that was inconsistent with European ancestry (<80% European ancestry using STRUCTURE software, n = 3, or significant deviation from the first two components in principal components analysis, n = 5) resulted in a final sample size of 776 cases and 1444 controls. On average, these cases were diagnosed ∼5.3 years after enrollment in the AHS. The majority of the cases and controls (∼83%) were still farming or registered as a commercial applicator/handler of pesticides at the time of the nested case–control study.
Exposure assessment
Information on lifetime use of 50 pesticides was captured in two self-administered questionnaires (http://aghealth.org/questionnaires.html) completed during cohort enrollment (1993–97). All nested case–control study participants completed the first (enrollment) questionnaire, which inquired about ever/never use of the 50 pesticides, as well as duration (years) and frequency (average days/year) of use for a subset of 22 of the pesticides, whereas 1439 of these men (60.4% of cases and 67.2% of controls) completed the second (take home) questionnaire, which inquired about duration and frequency of use for the remaining 28 pesticides. A previous analysis in the AHS demonstrated similar characteristics, except for age, between cohort participants that completed and returned the take-home questionnaire and those that did not (23). For each pesticide, we computed total lifetime days of application (number of years × days/year applied) using midpoints of the indicated categories. We also computed an intensity-weighted metric by multiplying the total lifetime days by an intensity score that was derived from an algorithm based on whether the applicator mixed pesticides, application method used, whether the applicator repaired equipment used for pesticide application and use of personal protective equipment (24) that was recently updated (Coble,J., submitted for publication). For permethrin, we summed exposure variables for applications to crops and animals since these were asked about separately. We categorized lifetime days and intensity-weighted lifetime days of application for each pesticide into a three-level ordinal-valued variable (none/low/high), with low and high categories distinguished by the median among exposed controls. Due to statistical power limitations, we excluded pesticides with <10% prevalence among the cases (trichlorfon, ziram, aluminum phosphide, ethylene dibromide, maneb/mancozeb, chlorothalonil, carbon tetrachloride/carbon disulfide, dieldrin, aldicarb and 2,4,5-trichlorophenoxypropionic acid), leaving 39 available for analysis. All analyses were based on AHS data release version P1REL0712.04.
Genotyping and single-nucleotide polymorphism selection
DNA was extracted from buccal cells using the Autopure protocol (Qiagen, Valencia, CA). Genotyping was performed at the National Cancer Institute’s Core Genotyping Facility (http://cgf.nci.nih.gov/operations/multiplex-genotyping.html) using a custom Infinium® BeadChip assay (iSelect™) from Illumina (San Diego, CA) as part of an array of 26 512 single-nucleotide polymorphisms (SNPs) in 1291 candidate genes. Blinded duplicate samples (2%) were included, and SNP concordance ranged from 96 to 100%. Tag SNPs were chosen to cover candidate DNA repair genes for three ancestry populations (Caucasian, Japanese Tokyo + Chinese Beijing and Yoruba people of Ibadan, Nigeria) in the HapMap Project [Data Release 20/Phase II, National Center for Biotechnology Information Build 36.1 assembly, dbSNPb126 (http://hapmap.ncbi.nlm.nih.gov/index.html.en)]. Tag SNPs were chosen using a modified version of the method described by Carlson et al. (25) as implemented in the Tagzilla module of the GLU software package (http://code.google.com/p/glu-genetics/). For each candidate gene, SNPs within the region spanning 20 kb 5′ of the start of transcription to 10 kb 3′ of the end of the stop codon were grouped using a binning threshold of r2 = 0.80, and one tag SNP per bin was selected. Select SNPs that were considered to be potentially functional (e.g. amino acid change or altered protein expression or function) based on a literature review were also included.
There were 27 NER genes included in the iSelect platform, which were primarily selected based on two reviews (26,27) and supplementary review information (see http://sciencepark.mdanderson.org/labs/wood/DNA_Repair_Genes.html). Of the 646 tag SNPs selected and genotyped for these genes, 553 remained after quality control exclusions (completion rate <90% or Hardy–Weinberg Equilibrium P value < 1 × 10−6). We further restricted SNPs to those with a minor allele frequency (MAF) of at least 10% among controls due to limited power for interaction assessments with rarer variants, which resulted in 324 SNPs.
Statistical analysis
We used unconditional logistic regression models adjusted for age (<60, 60–69, ≥70) and state of residence (Iowa or North Carolina) to estimate main effect odds ratios (ORs) and 95% confidence intervals (CIs) for the 39 pesticides and 324 NER SNPs with prostate cancer risk and to evaluate pesticide × SNP interactions. We adjusted for state because different patterns of pesticide use are observed by state, and this variable demonstrated a borderline significant association with prostate cancer risk in our study population. The addition of family history of prostate cancer and ever/never use of the five pesticides most highly correlated with a given pesticide did not alter the inference, and thus, these variables were not included in the models.
While we examined both intensity-weighted and unweighted pesticide exposure variables, the results were similar, and we present results only for the intensity-weighted variables. For pesticide main effects analysis and interaction analysis, we used the three-level ordinal-valued pesticide variables. For the tests of trend with pesticide exposure, we created new variables from the ordinal pesticide variables by assigning participants the median intensity-weighted (or unweighted) lifetime days value among controls for their respective exposure category (none/low/high). We determined P values for trend by entering the variable as a single term in the model (i.e. one degree of freedom) and testing using the Wald test. For SNP main effects analysis, we used ordinal variables coded as the number of variant alleles, zero, one or two, assuming a log-additive genetic model. To test for interaction, we computed P values from a one degree of freedom likelihood ratio test comparing nested models with and without the interaction term, using the three-level ordinal-valued pesticide variables and assuming the dominant genetic model for SNPs. We used SAS, version 9.1 (SAS Institute, Cary, NC), to estimate ORs for pesticide main effects and stratified effects by genotype, as well as P values for interaction, and PLINK (http://pngu.mgh.harvard.edu/purcell/plink/) (28) to estimate ORs for SNP main effects. We evaluated interactions between pesticides and haplotypes for SNPs in a gene of interest (including all tag SNPs with MAF ≥ 1% among controls) using generalized linear models, assuming the additive genetic model for haplotypes and treating the most common haplotype as the referent group, using the haplostats package in R (http://www.r-project.org) (29). Haplotypes with frequency <1% were collapsed into a single group. We used Haploview software (http://www.broadinstitute.org/scientific-community/science/programs/medical-and-population-genetics/haploview/downloads) (30) to compute r2 values among controls for pairings of SNPs.
We used SAS to calculate false discovery rate (FDR)-adjusted interaction P values with the intensity-weighted pesticide variables (31). We conducted the FDR analysis by gene (number of comparisons = 39 pesticides × number of tag SNPs for gene) to account for the differing numbers of SNPs by gene. Interactions meeting FDR <0.2 were considered robust to adjustment for multiple comparisons.
We presented two sets of results for pesticide × SNP interactions. One set encompassed interactions meeting FDR <0.2. The second set (as shown in Supplementary Table III, available at Carcinogenesis online) encompassed interactions that did not meet FDR <0.2, but that had a P value for interaction <0.01 for both intensity-weighted and unweighted exposure metrics and involved a significantly increased risk of prostate cancer (α = 0.05) following a monotonic pattern with increasing pesticide exposure in one genotype group and no significant association in the other group. We presented the second set of interactions because these interactions fit an expected biological pattern, although the P value for interaction was not low enough to meet FDR <0.2 in our study population.
Results
Nested case–control study participants were representative of prostate cancer cases and prostate cancer-free participants in the cohort with respect to state of residence, applicator type, family history of prostate cancer and disease characteristics for the cases (Table I). Cases and their matched controls were on average older at enrollment than men in the cohort as a whole. At the time of enrollment in the cohort, the average age among the nested case–control study participants was 61 years (range 34–88 years), compared with 46 years for the entire cohort.
Table I.
Characteristics of prostate cancer cases and controls in the AHS nested case–control study compared with the entire AHS cohort
| Characteristic | Nested case–controla |
AHS cohorta |
||
| Cases, n (%), total = 776 | Controls, n (%), total = 1444 | Prostate cancer, n (%), total = 1275 | Non-cancer, n (%), total = 48 286 | |
| Age at enrollment | ||||
| <40 | 3 (0.4) | 5 (0.4) | 9 (0.7) | 17 801 (36.9) |
| 40–49 | 74 (9.5) | 144 (10.0) | 111 (8.7) | 13 592 (28.2) |
| 50–59 | 259 (33.4) | 491 (34.0) | 409 (32.1) | 9515 (19.7) |
| 60–69 | 355 (45.8) | 634 (43.9) | 573 (44.9) | 5657 (11.7) |
| ≥70 | 85 (11.0) | 170 (11.8) | 173 (13.6) | 1721 (3.6) |
| State of residence | ||||
| Iowa | 520 (67.0) | 991 (68.6) | 789 (61.9) | 32 740 (67.8) |
| North Carolina | 256 (33.0) | 453 (31.4) | 486 (38.1) | 15 546 (32.2) |
| Applicator type | ||||
| Private | 741 (95.5) | 1363 (94.4) | 1219 (95.6) | 43 895 (90.9) |
| Commercial | 35 (4.5) | 81 (5.6) | 56 (4.4) | 4391 (9.1) |
| Family history of prostate cancerb | ||||
| No | 575 (74.1) | 1193 (82.6) | 924 (72.5) | 41 365 (85.7) |
| Yes | 130 (16.8) | 145 (10.0) | 212 (16.6) | 3748 (7.8) |
| Missing | 71 (9.1) | 106 (7.3) | 139 (10.9) | 3173 (6.6) |
| Prostate cancer stage | ||||
| Local | 579 (74.3) | — | 945 (74.1) | — |
| Regional | 156 (20.1) | — | 247 (19.4) | — |
| Distant | 12 (1.5) | — | 33 (2.6) | — |
| Not staged | 29 (3.7) | — | 50 (3.9) | — |
| Prostate cancer grade | ||||
| Well differentiated | 38 (4.9) | — | 60 (4.7) | — |
| Moderately differentiated | 547 (70.5) | — | 855 (67.1) | — |
| Poorly differentiated | 168 (21.6) | — | 302 (23.7) | — |
| Undifferentiated | 4 (0.5) | — | 6 (0.5) | — |
| Not graded | 19 (2.4) | — | 52 (4.1) | — |
Excludes females, non-white subjects and subjects with any previous cancer.
Family history of prostate cancer in first-degree relative.
Estimated pesticide main effects on prostate cancer risk in the nested case–control study have been published previously (32) and were largely null, except for six pesticides with inverse trends [carbaryl, chlordane, cyanazine, 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), metolachlor and imazethapyr] (Supplementary Table I, available at Carcinogenesis online). In the SNP main effect analysis, we observed 23 SNPs that were associated with prostate cancer risk with Ptrend < 0.05, including four SNPs with Ptrend < 0.01: rs4150351, located in ERCC5 (Ptrend = 1.7 × 10−3); rs1264308, located in GTF2H4 (Ptrend = 3.1 × 10−3) and rs4151330 and rs4151351, both located in MNAT1 (Ptrend = 4.9 × 10−3 and Ptrend = 6.6 × 10−3, respectively) (Table II). Supplementary Table II (available at Carcinogenesis online) presents main effect estimates for the remaining NER SNPs evaluated.
Table II.
Associations between NER gene SNPs and prostate cancer with Ptrend <0.05 in the AHS
| SNP | Gene | Location | Variant (effect) allele | Chromosome | MAFa | OR (95% CI)b | Ptrendb |
| rs4150351 | ERCC5 | IVS12-1581A>C | C | 13 | 0.20 | 0.77 (0.65–0.91) | 1.7 × 10−3 |
| rs1264308 | GTF2H4 | IVS10+64C>T | T | 6 | 0.14 | 0.75 (0.61–0.91) | 3.1 × 10−3 |
| rs4151330 | MNAT1 | IVS7+24992A>G | G | 14 | 0.34 | 0.83 (0.73–0.94) | 4.9 × 10−3 |
| rs4151351 | MNAT1 | IVS7+36194G>A | A | 14 | 0.21 | 0.80 (0.69–0.94) | 6.6 × 10−3 |
| rs13246995 | RPA3 | IVS2+714T>C | C | 7 | 0.43 | 1.17 (1.04–1.33) | 0.01 |
| rs12702628 | RPA3 | IVS4-142A>G | G | 7 | 0.47 | 1.17 (1.04–1.33) | 0.01 |
| rs11587393 | RPA2 | −19365G>A | A | 1 | 0.14 | 0.79 (0.65–0.95) | 0.01 |
| rs11656931 | RPA1 | −15692G>A | A | 17 | 0.17 | 1.22 (1.04–1.43) | 0.01 |
| rs12953068 | RPA1 | −16112C>A | A | 17 | 0.14 | 1.24 (1.04–1.47) | 0.02 |
| rs7503422 | RPA1 | −17952A>G | G | 17 | 0.18 | 1.21 (1.04–1.42) | 0.02 |
| rs8065937 | RPA1 | −410T>G | G | 17 | 0.19 | 1.20 (1.03–1.40) | 0.02 |
| rs1859604 | RPA3 | IVS4-7020G>T | T | 7 | 0.36 | 1.16 (1.03–1.32) | 0.02 |
| rs2108003 | RPA3 | IVS2-2752G>C | C | 7 | 0.39 | 1.16 (1.02–1.32) | 0.02 |
| rs4150393 | ERCC5 | c404A>G | G | 13 | 0.13 | 0.80 (0.66–0.97) | 0.02 |
| rs2296926 | MNAT1 | c8471T>C | C | 14 | 0.29 | 1.17 (1.02–1.34) | 0.02 |
| rs4151182 | MNAT1 | IVS1-21185C>A | A | 14 | 0.27 | 1.17 (1.02–1.34) | 0.02 |
| rs7503173 | RPA1 | IVS6+1080G>T | T | 17 | 0.13 | 1.22 (1.02–1.45) | 0.03 |
| rs17257294 | RPA2 | −17022T>C | C | 1 | 0.30 | 0.85 (0.74–0.98) | 0.03 |
| rs11867830 | RPA1 | IVS5+3896A>G | G | 17 | 0.13 | 1.21 (1.02–1.44) | 0.03 |
| rs7792009 | RPA3 | IVS4-214C>T | T | 7 | 0.29 | 0.86 (0.75–0.99) | 0.04 |
| rs1557997 | RPA3 | IVS4+21501A>G | G | 7 | 0.31 | 1.15 (1.00–1.31) | 0.04 |
| rs2882684 | MNAT1 | IVS7-18485G>A | A | 14 | 0.28 | 1.15 (1.00–1.32) | 0.05 |
| rs973063 | MNAT1 | IVS5-88G>A | A | 14 | 0.38 | 0.88 (0.78–1.00) | 0.05 |
OR, odds ratio per allele.
Among controls.
Effect of variant allele, assuming a log-additive genetic model and adjusting for age and state.
Indicates located 3' of the stop codon.
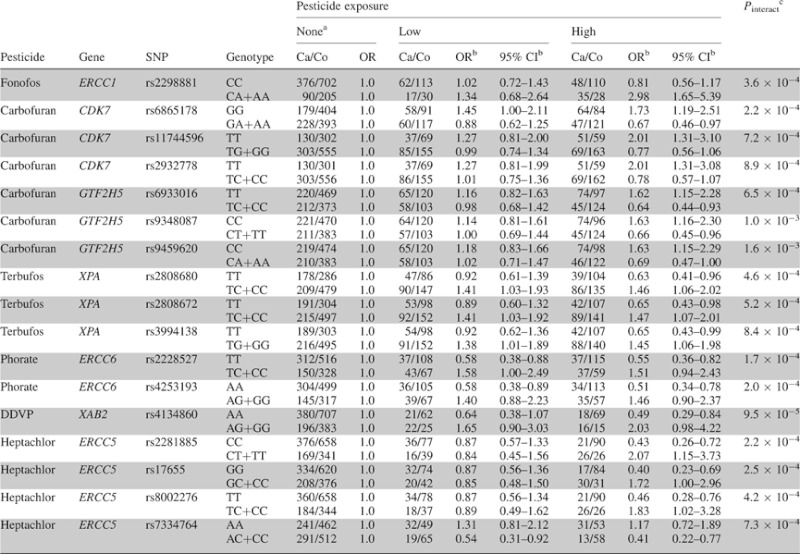
We examined whether genetic variation in the NER genes modified the risk of prostate cancer associated with pesticide exposure and observed 17 significant interactions with SNPs in seven NER genes (CDK7, ERCC1, ERCC5, ERCC6, GTF2H5, XAB2 and XPA) at FDR <0.2 (Table III). Table IV presents pesticide associations with prostate cancer stratified by genotype for these 17 pesticide × SNP interactions. Of these 17 interactions, 14 appeared to be qualitative with an increased risk of prostate cancer with exposure in one genotype group and decreased risk in the other group, including the interaction between rs4134860 in XAB2 and dichlorvos, which was the most significant overall (Pinteract = 9.5 × 10−5; FDR-adjusted P = 0.04) (Table III). The remaining three interactions meeting FDR <0.2 displayed a significant monotonic increase in prostate cancer risk with pesticide exposure in one genotype group and no significant association in the other group: fonofos and ERCC1 rs229881 and carbofuran and two highly correlated SNPs tagging CDK7, rs11744596 and rs2932778 (r2 = 1.0) (Table IV). The interaction between fonofos and ERCC1 rs2298881 was the most significant interaction observed for the ERCC1 gene (Table III). Men carrying the variant A allele at ERCC1 rs2298881 exhibited a monotonic increase in prostate cancer risk with increasing fonofos exposure (OR for low compared with no use: 1.34, 95% CI 0.68–2.64 and OR for high compared with no use: 2.98, 95% CI 1.65–5.39; Pinteract = 3.6 × 10−4; FDR-adjusted P = 0.11) (Table IV). Men who were homozygous for the wild-type T allele at CDK7 rs11744596 or rs2932778 exhibited a monotonic increase in risk with increasing carbofuran exposure (OR for low compared with no use: 1.27, 95% CI 0.81–2.00 and OR for high compared with no use: 2.01, 95% CI 1.31–3.10 for rs11744596; Pinteract = 7.2 × 10−4; FDR-adjusted P = 0.09). In contrast, men with other genotypes at these loci did not exhibit significant associations (Table IV).
Table III.
NER genes evaluated, number of pesticide × SNP interactions meeting FDR <0.2 and the most significant interaction in each gene in the AHS
| Gene | Number of SNPs evaluated | Number of interactions with FDR <0.2 | Pesticide × SNP combination with most significant interaction | Pinteract for most significant interactiona | FDR P valueb |
| CCNH | 6 | 0 | Phorate × rs7702514 | 1.9 × 10−3 | 0.44 |
| CDK7 | 8 | 3 | Carbofuran × rs6865178 | 2.2 × 10−4 | 0.07 |
| CETN2 | 2 | 0 | Chlorpyrifos × rs5925234 | 0.01 | 0.46 |
| DDB1 | 1 | 0 | 2,4-D × rs9651726 | 0.03 | 0.98 |
| DDB2 | 9 | 0 | Parathion × rs4647709 | 9.0 × 10−4 | 0.32 |
| ERCC1 | 8 | 1 | Fonofos × rs2298881 | 3.6 × 10−4 | 0.11 |
| ERCC2 (XPD) | 8 | 0 | Cyanazine × rs238417 | 3.4 × 10−3 | 0.68 |
| ERCC3 (XPB) | 11 | 0 | Dicamba × rs3088374 | 2.7 × 10−3 | 0.87 |
| ERCC4 (XPF) | 20 | 0 | Petroleum oil × rs9646271 | 2.5 × 10−3 | 0.45 |
| ERCC5 (XPG) | 16 | 4 | Heptachlor × rs2281885 | 2.2 × 10−4 | 0.07 |
| ERCC6 (CSB) | 23 | 2 | Phorate × rs2228527 | 1.7 × 10−4 | 0.09 |
| ERCC8 | 11 | 0 | Carbofuran × rs17332824 | 6.8 × 10−3 | 0.45 |
| GTF2H1 | 11 | 0 | Paraquat × rs10741741 | 4.1 × 10−3 | 0.56 |
| GTF2H3 | 12 | 0 | Butylate × rs2048802 | 0.02 | 1.00 |
| GTF2H4 | 11 | 0 | Heptachlor × rs1049623 | 9.2 × 10−4 | 0.39 |
| GTF2H5 (TTDA) | 9 | 3 | Carbofuran × rs6933016 | 6.5 × 10−4 | 0.18 |
| MMS19L (MMS19) | 14 | 0 | Coumaphos × rs10786353 | 0.02 | 0.96 |
| MNAT1 | 12 | 0 | Metribuzin × rs2020892 | 5.4 × 10−3 | 0.57 |
| RAD23A (HR23A) | 5 | 0 | Parathion × rs2967890 | 0.01 | 0.97 |
| RAD23B (HR23B) | 15 | 0 | Metalaxyl × rs10816481 | 5.9 × 10−3 | 0.99 |
| RPA1 | 23 | 0 | Carbaryl × rs17761467 | 5.9 × 10−4 | 0.53 |
| RPA2 | 12 | 0 | Metalaxyl × rs2236074 | 5.8 × 10−4 | 0.26 |
| RPA3 | 28 | 0 | Heptachlor × rs1557997 | 3.9 × 10−4 | 0.36 |
| RPA4 | 2 | 0 | Carbaryl × rs233214 | 0.05 | 0.94 |
| XAB2 (HCNP) | 11 | 1 | DDVP × rs4134860 | 9.5 × 10−5 | 0.04 |
| XPA | 13 | 3 | Terbufos × rs2808680 | 4.6 × 10−4 | 0.13 |
| XPC | 23 | 0 | Carbaryl × rs2733587 | 3.2 × 10−3 | 0.69 |
2,4-D, 2,4-dichlorophenoxyacetic acid; DDVP, dichlorvos; LRT, likelihood ratio test.
P value for interaction from LRT, treating pesticide exposure variables as ordinal variables, assuming the dominant genetic model and adjusting for age and state.
FDR-adjusted P value for the most significant interaction.
Table IV.
Pesticide exposure and prostate cancer risk stratified by NER SNP genotype for interactions meeting FDR <0.2 in the AHS
 |
Ca, cases; Co, controls; DDVP, dichlorvos; LRT, likelihood ratio test.
Referent group for estimated effects of low and high pesticide use.
Adjusted for age and state.
P value for interaction from LRT, treating pesticide exposure variables as ordinal variables, assuming the dominant genetic model and adjusting for age and state.
ERCC1 rs2298881 was weakly to moderately correlated (r2 range: 0.002–0.38) with the other ERCC1 tag SNPs genotyped; however, interactions with the other SNPs did not meet FDR <0.2 (data not shown). The most correlated SNP with rs2298881 was rs928911 (r2 = 0.38), which we did not include in our interaction analysis due to MAF <10% (MAF = 0.05). When we included both interactions for these SNPs with fonofos together in the model, only the interaction for rs2298881 remained statistically significant (Pinteract = 0.03 and Pinteract = 0.21, respectively), although rs928911 demonstrated a similar pattern of interaction with fonofos, such that men carrying the variant allele at rs928911 exhibited significantly increased risk with high compared with no fonofos use (OR 3.26; 95% CI 1.40–7.60), whereas men carrying the homozygous wild-type genotype did not exhibit a significant association (OR 1.04; 95% CI 0.76–1.42) (data not shown).
Interactions that did not meet FDR <0.2, but that had a P value for interaction <0.01 for both intensity-weighted and unweighted exposure metrics and involved a significant monotonic increase in risk with increasing exposure in one genotype group and no significant association in the other group are presented in Supplementary Table III (available at Carcinogenesis online). These included interactions between two CDK7 tag SNPs (rs7706902 and rs17331590) and carbofuran. Rs7706902 was highly correlated with the CDK7 SNPs (rs6865178, rs11744596 and rs2932778) that interacted with carbofuran at the FDR <0.2 level (r2 > 0.70) and demonstrated a similar pattern of interaction, such that men carrying the homozygous wild-type genotype exhibited increased prostate cancer risk with carbofuran exposure. In contrast, for rs17331590, which showed low correlations with the other SNPs that interacted with carbofuran (r2 < 0.09), men carrying the variant allele exhibited increased risk. Findings from including the interactions for rs11744596 and rs17331590 together in the same model, such that the interaction for rs11744596 remained statistically significant (Pinteract = 8.3 × 10−3) and the interaction for rs17331590 was borderline significant (Pinteract = 0.06), as well as analysis of interactions between carbofuran and CDK7 haplotypes (data not shown), further suggested that the interaction for rs17331590 might represent a separate CDK7 signal.
We observed several additional interactions that did not meet FDR <0.2, but that had a P value for interaction <0.01 for both intensity-weighted and unweighted exposure metrics and involved a significant monotonic increase in risk with increasing exposure in one genotype group and no significant association in the other group (Supplementary Table III, available at Carcinogenesis online). These included interactions between parathion and a SNP tagging DDB2, as well as interactions between petroleum oil/petroleum distillate and five SNPs tagging ERCC4, four of which were very highly correlated (r2 > 0.91). The interaction between parathion and DDB2 rs4647709 and that between petroleum oil/petroleum distillate and ERCC4 rs9646271 also represented the most significant interactions for their respective genes (Table III).
Discussion
Our study is the first to evaluate interactions between pesticides and genetic variation in the NER pathway with respect to prostate cancer risk. We observed 17 interactions that were robust to multiple comparison adjustment based on FDR <0.2. Of these 17 interactions, three displayed a significant monotonic increase in prostate cancer risk with increasing pesticide exposure in one genotype group (e.g. TT carriers) and no significant association in the other group: fonofos × ERCC1 rs2298881 and carbofuran × rs11744596 and rs2932778, two highly correlated SNPs tagging CDK7. The remaining 14 interactions that met FDR <0.2 were the result of a positive association in one genotype group and an inverse association in the other (i.e. qualitative interactions), which could arise because of a chance effect of the exposure of interest in one population subgroup when there is no main effect of the exposure. We also presented interactions that displayed the stratified pattern of interest and had a P value for interaction <0.01, although the P value did not meet FDR <0.2 in our study population and thus should be interpreted with caution.
Our finding of a significant interaction for fonofos, an organophosphate insecticide that is no longer registered for use in the USA (since 1998) (33), but was used by ∼25% of the AHS prostate cancer nested case–control study participants, is consistent with previous AHS findings of increased prostate cancer risk with fonofos exposure among participants with a family history of prostate cancer (34,35), which suggested a role of genetic susceptibility to carcinogenic effects of this chemical. Specifically, fonofos interacted with rs2298881, an intronic SNP located before the first coding exon in ERCC1, such that men carrying at least one variant allele exhibited increasing prostate cancer risk with increasing fonofos exposure, whereas men who were homozygous wild-type at this locus did not exhibit a significant association. The ERCC1 protein forms a heterodimer with XPF endonuclease (encoded by ERCC4), which catalyzes the 5′ incision in NER removal of DNA lesions and also plays a role in the repair of interstrand cross-links and double-stranded DNA breaks. Rs2298881 was moderately correlated with rs928911, located in an intronic region of the gene PPP1R13L (also known as IASPP), which is important in p53 inhibition and located <10 kb downstream of ERCC1. However, only the interaction with rs2298881 remained significant when both interaction terms were included in the same model, suggesting that rs2298881 was the SNP driving the risk. It is also possible that a third SNP in linkage disequilibrium with these two SNPs might be driving our findings, and further research is needed to identify the SNP responsible for the observed effect.
There is some plausibility for a role of DNA damage in our finding of an interaction between fonofos and genetic variation in ERCC1. Whereas one report did not find evidence of fonofos genotoxicity based on several in vitro assays (4), another study observed positive results for two assays in bacteria and yeast (36), suggesting that fonofos may induce some DNA damage. To our knowledge, no human biomonitoring studies have specifically evaluated fonofos in relation to DNA damage endpoints (e.g. DNA adducts and chromosomal aberrations). However, our fonofos interaction finding appears consistent with numerous human biomonitoring studies suggesting the genotoxicity of organophosphate insecticides as a group (5,6,10,11). In addition, in a previous analysis in the AHS prostate cancer nested case–control study (32), fonofos demonstrated a significant interaction with a SNP in the promoter region of the base excision repair gene NEIL3. Although the NER pathway is predominantly involved in repairing relatively bulky helix-distorting DNA lesions and the base excision repair pathway in repairing smaller lesions with minimal helix-distorting effect, there is some overlap between the two pathways (14,15).
Carbofuran (a carbamate insecticide) interacted with several highly correlated SNPs tagging CDK7 (rs6865178, rs11744596, rs2932778 and rs7706902), such that men who were homozygous wild-type exhibited increasing prostate cancer risk with increasing carbofuran exposure. Additionally, carbofuran interacted with another CDK7 tag SNP (rs17331590), such that men carrying at least one variant allele exhibited increasing risk with increasing exposure. Taken together with the low correlations between rs17331590 and the other CDK7 SNPs that interacted with carbofuran and the findings of haplotype analysis, our results suggest that there might be two separate carbofuran × CDK7 interaction signals, emphasizing the potential importance of this region in modifying the effects of carbofuran exposure. However, only the carbofuran interactions with the very highly correlated SNPs rs11744596 and rs2932778 met FDR <0.2 and did not appear to be qualitative. Rs11744596 is located in an intronic region of MRPS36, which encodes a 28S subunit of the mitochondrial ribosome and is located ∼17 kb upstream of CDK7, and rs2932778 is located in the promoter region of CDK7. CDK7 encodes a protein that is a member of the cyclin-dependent kinase family and complexes with cyclin H and MAT1 to form a cyclin-dependent kinase-activating kinase. As a component of the transcription factor TFIIH, the cyclin-dependent kinase-activating kinase complex plays a role in DNA repair and DNA transcription. This complex is also important in the regulation of cell cycle progression.
Although one study found no evidence of genotoxicity for carbofuran based on several assays (36), other studies have observed increased mutations, chromosomal aberrations and micronuclei formation with carbofuran exposure in bacterial or animal systems (37–39), suggesting some biologic plausibility for a role of DNA damage in our interaction findings for carbofuran. Another study observed increased mutations associated with the N-nitroso derivative N-nitrosocarbofuran, but not carbofuran itself, in a bacterial system (40). Several human biomonitoring studies have observed increased genetic damage (based on micronuclei formation, chromosomal aberrations and damage detected by the Comet assay) among workers exposed to carbofuran (41–43). However, these populations were often exposed to other chemicals as well, and there was generally limited exposure assessment, which precluded estimation of the specific contribution of carbofuran to the findings.
It is possible that mechanisms other than DNA damage repaired by the NER pathway could have contributed to our interaction findings. For example, CDK7 has other functions beyond DNA repair as described above. In addition, the SNP(s) driving our findings could act in part by affecting the expression or function of nearby genes. For example, ERCC1 overlaps with CD3EAP, which has been proposed to play a role in T-cell activation (44), among other activities. Although fonofos has not been specifically implicated, organophosphate insecticides in general have been associated with immunotoxicity in experimental animal studies and epidemiologic studies (45). There are several genes located nearby CDK7, including CCNB1, which contributes to the regulation of mitosis by encoding a protein that complexes with p34 (cdc2) to form the mitosis-promoting factor. CCNB1 may be relevant to the carcinogenicity of carbofuran as exposure to carbofuran has been associated with mitotic inhibition in mice (37).
Although our findings may be due to chance, we took several steps to help reduce false-positive results in our study. We used the FDR method to adjust interaction P values for multiple comparisons. Additionally, we highlighted interactions with a significant monotonic increase in prostate cancer risk with increasing exposure in one genotype group and no significant association in the other. However, we recognize that by focusing on this subset of interaction findings, we might have missed some true-positive results among our remaining findings.
Our study was limited in power, and we may have missed some interactions by excluding SNPs with MAF <10% due to power concerns. The number of participants often became small when stratifying by genotype, particularly for the homozygous variant group. To help reduce this problem, we chose to use the dominant genetic model for the interaction assessments; however, this choice could have resulted in a loss of power if another genetic model was more appropriate. Additionally, there were insufficient case numbers to evaluate interactions by prostate cancer stage or grade. However, to our knowledge, no other study has greater power to evaluate pesticide–gene interactions for individual pesticides with prostate cancer.
There are also several strengths of our study. We were able to evaluate a number of pesticides from a range of chemical and functional classes. Previous findings in the AHS have suggested that the effects for pesticides within a chemical class are heterogeneous (46). Thus, our examination of effect modification of individual as opposed to grouped pesticides is a further strength. Furthermore, self-reported pesticide information in the AHS has been demonstrated to be reliable and consistent with the dates of introduction to the market (47,48). We focused our analyses on the intensity-weighted exposure metric, which incorporates an intensity score that has shown moderate correlation with biomarkers of pesticide exposure in post-application urine samples (49). Additionally, availability of genotyping data for a large number of tag SNPs across the NER pathway allowed us to comprehensively explore the hypothesis that NER genetic variation might modify pesticide-associated risk of prostate cancer.
In conclusion, although we did not observe highly significant NER SNP main effects, our interaction findings between SNPs tagging ERCC1 and CDK7 and fonofos and carbofuran, respectively, suggest the importance of the NER pathway in prostate cancer risk in the presence of certain pesticide exposures. While requiring replication, our interaction results are consistent with a pesticide mechanism of effect involving DNA damage. Additional studies among pesticide-exposed populations are needed to evaluate interactions between pesticides and genetic variation in DNA repair genes and to assess the genotoxicity of individual pesticides in humans. Investigation of the mechanisms of action, metabolism and bioavailability of the different pesticides may help clarify their relationship with cancer risk.
Supplementary material
Supplementary Tables I–III can be found at http://carcin.oxfordjournals.org/.
Funding
National Institutes of Health (Intramural Research Program of the National Cancer Institute, Division of Cancer Epidemiology and Genetics, Z01CP010119; Intramural Research Program of the National Institute of Environmental Health Sciences, Z01ES049030 and National Cancer Institute grant T32 CA105666 to K.H.B.).
Supplementary Material
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AHS
Agricultural Health Study
- CI
confidence interval
- FDR
false discovery rate
- MAF
minor allele frequency
- NER
nucleotide excision repair
- OR
odds ratio
- SNP
single-nucleotide polymorphism
References
- 1.Koutros S, et al. An update of cancer incidence in the Agricultural Health Study. J. Occup. Environ. Med. 2010;52:1098–1105. doi: 10.1097/JOM.0b013e3181f72b7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Maele-Fabry G, et al. Prostate cancer among pesticide applicators: a meta-analysis. Int. Arch. Occup. Environ. Health. 2004;77:559–570. doi: 10.1007/s00420-004-0548-8. [DOI] [PubMed] [Google Scholar]
- 3.Van Maele-Fabry G, et al. Review and meta-analysis of risk estimates for prostate cancer in pesticide manufacturing workers. Cancer Causes Control. 2006;17:353–373. doi: 10.1007/s10552-005-0443-y. [DOI] [PubMed] [Google Scholar]
- 4.Garrett NE, et al. Evaluation of the genetic activity profiles of 65 pesticides. Mutat. Res. 1986;168:301–325. doi: 10.1016/0165-1110(86)90024-2. [DOI] [PubMed] [Google Scholar]
- 5.Bolognesi C. Genotoxicity of pesticides: a review of human biomonitoring studies. Mutat. Res. 2003;543:251–272. doi: 10.1016/s1383-5742(03)00015-2. [DOI] [PubMed] [Google Scholar]
- 6.Sanborn M, et al. Non-cancer health effects of pesticides: systematic review and implications for family doctors. Can. Fam. Physician. 2007;53:1712–1720. [PMC free article] [PubMed] [Google Scholar]
- 7.Hagmar L, et al. Chromosomal aberrations in lymphocytes predict human cancer: a report from the European Study Group on Cytogenetic Biomarkers and Health (ESCH) Cancer Res. 1998;58:4117–4121. [PubMed] [Google Scholar]
- 8.Bhalli JA, et al. DNA damage in Pakistani agricultural workers exposed to mixture of pesticides. Environ. Mol. Mutagen. 2009;50:37–45. doi: 10.1002/em.20435. [DOI] [PubMed] [Google Scholar]
- 9.Grover P, et al. Evaluation of genetic damage in workers employed in pesticide production utilizing the Comet assay. Mutagenesis. 2003;18:201–205. doi: 10.1093/mutage/18.2.201. [DOI] [PubMed] [Google Scholar]
- 10.Kisby GE, et al. Oxidative stress and DNA damage in agricultural workers. J. Agromedicine. 2009;14:206–214. doi: 10.1080/10599240902824042. [DOI] [PubMed] [Google Scholar]
- 11.Shadnia S, et al. Evaluation of oxidative stress and genotoxicity in organophosphorus insecticide formulators. Hum. Exp. Toxicol. 2005;24:439–445. doi: 10.1191/0960327105ht549oa. [DOI] [PubMed] [Google Scholar]
- 12.Wong RH, et al. Polymorphisms in metabolic GSTP1 and DNA-repair XRCC1 genes with an increased risk of DNA damage in pesticide-exposed fruit growers. Mutat. Res. 2008;654:168–175. doi: 10.1016/j.mrgentox.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Singh NP, et al. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- 14.Lockett KL, et al. Nucleotide-excision repair and prostate cancer risk. Cancer Lett. 2005;220:125–135. doi: 10.1016/j.canlet.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 15.Weinberg R. In The Biology of Cancer. New York, NY: Garland Science, Taylor & Francis Group, LLC.; 2007. Maintenance of genomic integrity and the development of cancer; pp. 463–526. [Google Scholar]
- 16.Hu JJ, et al. Deficient nucleotide excision repair capacity enhances human prostate cancer risk. Cancer Res. 2004;64:1197–1201. doi: 10.1158/0008-5472.can-03-2670. [DOI] [PubMed] [Google Scholar]
- 17.Park JY, et al. Single nucleotide polymorphisms in DNA repair genes and prostate cancer risk. Methods Mol. Biol. 2009;471:361–385. doi: 10.1007/978-1-59745-416-2_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eeles RA, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 19.Thomas G, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 20.Alavanja MC, et al. The Agricultural Health Study. Environ. Health Perspect. 1996;104:362–369. doi: 10.1289/ehp.96104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engel LS, et al. Factors associated with refusal to provide a buccal cell sample in the Agricultural Health Study. Cancer Epidemiol. Biomarkers Prev. 2002;11:493–496. [PubMed] [Google Scholar]
- 22.Koutros S, et al. Pesticide use modifies the association between genetic variants on chromosome 8q24 and prostate cancer. Cancer Res. 2010;70:9224–9233. doi: 10.1158/0008-5472.CAN-10-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarone RE, et al. The Agricultural Health Study: factors affecting completion and return of self-administered questionnaires in a large prospective cohort study of pesticide applicators. Am. J. Ind. Med. 1997;31:233–242. doi: 10.1002/(sici)1097-0274(199702)31:2<233::aid-ajim13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 24.Dosemeci M, et al. A quantitative approach for estimating exposure to pesticides in the Agricultural Health Study. Ann. Occup. Hyg. 2002;46:245–260. doi: 10.1093/annhyg/mef011. [DOI] [PubMed] [Google Scholar]
- 25.Carlson CS, et al. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood RD, et al. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 27.Wood RD, et al. Human DNA repair genes, 2005. Mutat. Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinnwell J, et al. haplo.stats: Statistical Analysis of Haplotypes with Traits and Covariates when Linkage Phase is Ambiguous. 2009. R package version 1.4.4. http://cran.r-project.org/web/packages/haplo.stats/index.html (19 October 2011, date last accessed) [Google Scholar]
- 30.Barrett JC, et al. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 31.Benjamini Y, et al. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 32.Barry KH, et al. Genetic variation in base excision repair pathway genes, pesticide exposure, and prostate cancer risk. Environ. Health Perspect. 2011;119:1726–1732. doi: 10.1289/ehp.1103454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.U.S. Environmental Protection Agency. R.E.D. Facts: O-Ethyl S-Phenyl Ethylphosphonodithiolate (Fonofos) 1999. http://www.epa.gov/oppsrrd1/REDs/factsheets/0105fact.pdf (22 April 2011, date last accessed) [Google Scholar]
- 34.Alavanja MC, et al. Use of agricultural pesticides and prostate cancer risk in the Agricultural Health Study cohort. Am. J. Epidemiol. 2003;157:800–814. doi: 10.1093/aje/kwg040. [DOI] [PubMed] [Google Scholar]
- 35.Mahajan R, et al. Fonofos exposure and cancer incidence in the agricultural health study. Environ. Health Perspect. 2006;114:1838–1842. doi: 10.1289/ehp.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gentile JM, et al. An evaluation of the genotoxic properties of insecticides following plant and animal activation. Mutat. Res. 1982;101:19–29. doi: 10.1016/0165-1218(82)90161-6. [DOI] [PubMed] [Google Scholar]
- 37.Chauhan LK, et al. Induction of chromosome aberrations, micronucleus formation and sperm abnormalities in mouse following carbofuran exposure. Mutat. Res. 2000;465:123–129. doi: 10.1016/s1383-5718(99)00219-3. [DOI] [PubMed] [Google Scholar]
- 38.Moriya M, et al. Further mutagenicity studies on pesticides in bacterial reversion assay systems. Mutat. Res. 1983;116:185–216. doi: 10.1016/0165-1218(83)90059-9. [DOI] [PubMed] [Google Scholar]
- 39.Hour TC, et al. Comparative investigation on the mutagenicities of organophosphate, phthalimide, pyrethroid and carbamate insecticides by the Ames and lactam tests. Mutagenesis. 1998;13:157–166. doi: 10.1093/mutage/13.2.157. [DOI] [PubMed] [Google Scholar]
- 40.Yoon JY, et al. N-nitrosocarbofuran, but not carbofuran, induces apoptosis and cell cycle arrest in CHL cells. Toxicology. 2001;169:153–161. doi: 10.1016/s0300-483x(01)00502-9. [DOI] [PubMed] [Google Scholar]
- 41.Davies HW, et al. Cytogenetic analysis of South Asian berry pickers in British Columbia using the micronucleus assay in peripheral lymphocytes. Mutat. Res. 1998;416:101–113. doi: 10.1016/s1383-5718(98)00071-0. [DOI] [PubMed] [Google Scholar]
- 42.Zeljezic D, et al. Effect of occupational exposure to multiple pesticides on translocation yield and chromosomal aberrations in lymphocytes of plant workers. Environ. Sci. Technol. 2009;43:6370–6377. doi: 10.1021/es900824t. [DOI] [PubMed] [Google Scholar]
- 43.Zeljezic D, et al. Comparative evaluation of acetylcholinesterase status and genome damage in blood cells of industrial workers exposed to carbofuran. Food Chem. Toxicol. 2007;45:2488–2498. doi: 10.1016/j.fct.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 44.Yamazaki T, et al. CAST, a novel CD3epsilon-binding protein transducing activation signal for interleukin-2 production in T cells. J. Biol. Chem. 1999;274:18173–18180. doi: 10.1074/jbc.274.26.18173. [DOI] [PubMed] [Google Scholar]
- 45.Galloway T, et al. Immunotoxicity of organophosphorous pesticides. Ecotoxicology. 2003;12:345–363. doi: 10.1023/a:1022579416322. [DOI] [PubMed] [Google Scholar]
- 46.Weichenthal S, et al. A review of pesticide exposure and cancer incidence in the Agricultural Health Study cohort. Environ. Health Perspect. 2010;118:1117–1125. doi: 10.1289/ehp.0901731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blair A, et al. Reliability of reporting on life-style and agricultural factors by a sample of participants in the Agricultural Health Study from Iowa. Epidemiology. 2002;13:94–99. doi: 10.1097/00001648-200201000-00015. [DOI] [PubMed] [Google Scholar]
- 48.Hoppin JA, et al. Accuracy of self-reported pesticide use duration information from licensed pesticide applicators in the Agricultural Health Study. J. Expo. Anal. Environ. Epidemiol. 2002;12:313–318. doi: 10.1038/sj.jea.7500232. [DOI] [PubMed] [Google Scholar]
- 49.Thomas K, et al. Assessment of a pesticide exposure intensity algorithm in the agricultural health study. J. Expo. Sci. Environ. Epidemiol. 2010;20:559–569. doi: 10.1038/jes.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.