

Abstract
The central zone of the supramolecular activation cluster (c-SMAC) is a zone of T cell receptor (TCR) enrichment that forms at a T cell/antigen-presenting cell (APC) junction in response to antigen stimulation. We demonstrate that there is a surprisingly complex relocalization process that brings PKCθ and Bcl10, two intermediates in TCR activation of NF-κB, to the cytoplasmic face of the c-SMAC. TCR activation causes enrichment of PKCθ at the c-SMAC, followed by Bcl10 relocalization to punctate cytoplasmic structures, often at sites distant from the c-SMAC. These Bcl10 structures then undergo further relocalization, becoming enriched at the c-SMAC. TCR activation of NF-κB therefore involves the dynamic relocalization of multiple signaling intermediates, with distinct phases proximal to and distant from the c-SMAC.
Tcell receptor (TCR)-regulated activation of the transcription factor NF-κB is of paramount importance in the T cell response to antigen. Because T cells cannot enter S-phase when TCR-modulated NF-κB activation is blocked (1), the ability of T cells to divide and acquire effector functions crucially depends on delivery of this signal. Recent data from gene-targeted mice have demonstrated that the serine-threonine kinase, PKCθ, the caspase recruitment domain (CARD) proteins, Bcl10 and CARMA1, and the “paracaspase” MALT1, are intermediates in TCR activation of NF-κB (2–8).
A subset of T cell transmembrane and cytoplasmic molecules segregate into discrete zones of enrichment, after interaction with stimulatory antigen-presenting cells (APCs) (9, 10). These regions, called supramolecular activation clusters (SMACs), form a bull's-eye pattern at the interface between the T cell and the APC, which has been termed the immunological synapse (11, 12). The TCR is concentrated in the central zone of the SMAC, called the c-SMAC. PKCθ is also enriched in this zone, associated indirectly with the cytoplasmic face of the TCR. The adhesion molecule LFA-1 is enriched in a peripheral zone called the p-SMAC, which surrounds the c-SMAC (9). The cytoskeletal organizing protein talin is also enriched in the p-SMAC (9) and may be directly associated with the cytoplasmic tail of LFA-1 (13).
Thus, gene inactivation studies have established that PKCθ is required for TCR activation of NF-κB, and microscopy analyses have shown that PKCθ translocates to the c-SMAC in response to antigen stimulation. Because recent studies have suggested that Bcl10 is downstream of PKCθ in TCR activation of NF-κB (14, 15), we decided to examine the spatial and kinetic redistribution of PKCθ and Bcl10 in single cells in response to antigen stimulation.
We find that these two NF-κB mediators undergo a surprisingly complex series of redistribution events. Shortly after contact between a T cell and an antigen-loaded APC, PKCθ translocates to the c-SMAC. Within a period of several minutes, enrichment of PKCθ generally reaches its maximal value and then begins to reverse. At approximately the point of maximal PKCθ enrichment at the c-SMAC, Bcl10 begins to coalesce into punctate structures throughout the cytoplasm, often at points distant from the c-SMAC. These punctate Bcl10 structures then migrate to the c-SMAC, where they continue to become enriched. Blockade of PKC activity prevents coalescence and phosphorylation of Bcl10, and NF-κB activation requires both PKC activity and Bcl10 phosphorylation.
The c-SMAC is thus a site of sequential enrichment of NF-κB signaling intermediates, and this enrichment appears to regulate delivery of essential activating signals. These data strongly suggest that the c-SMAC is of unique importance in TCR-regulated activation of NF-κB.
Materials and Methods
Cell Culture. D10-IL2 (henceforth referred to as D10) is an IL-2-dependent subclone of the CD4+ T cell clone D10 (16), maintained as described (17). Primary T cell blasts were prepared by stimulation of spleen and lymph node cells with 100-μg/ml plate-bound anti-TCRβ for 48 h, with IL-2 added for the final 24 h of stimulation. CH12 H-2k B cells and CHb H-2b B cells (18) were maintained as described (17).
Retroviral Constructions and Infections. Murine PKCθ was fused to a C-terminal linker (GRVELGR) followed by GFP or cyan fluorescent protein (19), modified by mutations that improve folding at 37°C (20). Two I.M.A.G.E. EST clones encoding murine Bcl10 were purchased (2123397 and 3026026; ResGen/Invitrogen). Clone 3026026 was used for PCR amplification of Bcl10, adding the C-terminal linker GR, which was then fused to the previously described modified yellow fluorescent protein (YFP) gene (17). The PKCθ–cyan fluorescent protein and Bcl10-YFP gene fusions were cloned into the retroviral vectors pEneo and pEhyg (21), respectively. Retroviral infection and cell line production were as described (17).
Microscopy. Fluorescent and Nomarski images were acquired on an Olympus IX70 inverted microscope (Olympus, Melville, NY) or a Zeiss Axiovert 200M inverted microscope (Zeiss) by using a ×100 plan-apochromat oil objective. The microscope was controlled by a tillvistrac imaging system, driven by tillvision 4.0 software (TILL Photonics, Planegg, Germany). Excitation light was provided by a Polychrome IV monochromator (TILL Photonics). Dichroic mirrors and excitation filters appropriate for each fluorophore were purchased from Chroma Technology (Brattleboro, VT). For further details, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Western Blotting. T cells were stimulated with 100-μg/ml plate-bound anti-TCRβ. Cell lysates were prepared on ice by using ×1 Laemmli buffer, followed by sonication. For immunoprecipitations, 5 × 106 cells were lysed for 1 h on ice in 360 μl of a buffer consisting of 0.5% Nonidet P-40, 0.5% deoxycholate, 20 mM Tris (pH 7.6), 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 1 mM PMSF, 20 μg/ml aprotinin, 100 μg/ml leupeptin, 1 mM Na3VO4, 10 mM NaF, and 1 mM DTT. Nuclei were removed by centrifugation, and lysates were incubated with polyclonal rabbit anti-Bcl10 antibody (Santa Cruz Biotechnology) for 2–3 h at 4°C and then for 1 h at 4°C with Protein A Sepharose. Lysates and immunoprecipitates were separated by SDS/PAGE and blotted onto nitrocellulose. Proteins were detected by monoclonal antibodies against Bcl10 (Santa Cruz Biotechnology) and PKCθ (BD/Transduction Laboratories, San Diego) and polyclonal antibodies against phospho-IκBα, anti-phospho-PKCθ/Thr-538 (Cell Signaling Technologies) and actin (Santa Cruz Biotechnology). For phosphatase treatment, immunoprecipitates were incubated for 45 min at 30°C with 400 units of λ-phosphatase (New England Biolabs), according to the instructions of the manufacturer.
Results
Redistribution of Endogenous Bcl10 in Response to Specific Antigen Stimulation. We sought to establish whether Bcl10, like PKCθ, might be recruited to the c-SMAC in response to specific antigen-loaded APC. Recent work using antibody capping (22) or antibody-coated beads (4) has shown that Bcl10 can be recruited to the cytoplasmic face of the engaged TCR, but no analysis of Bcl10 localization to SMACs in a T cell/APC interaction model has been reported. We produced T cell blasts from mice transgenic for a TCR specific for an ovalbumin peptide (Ova 323–339) bound to IAb [OTII mice (23)]. OTII T cell blasts were incubated for 45 min with CHb APC that had been loaded with either no antigen, or with the ovalbumin peptide. T cell/APC conjugates were detected by immunofluorescence microscopy, using monoclonal antibodies directed against Bcl10, PKCθ, and CD4 (Fig. 1a). In the absence of specific antigen, no enrichment of PKCθ was seen at the T cell/APC interface (Fig. 1a, top two rows), and T cell Bcl10 was observed throughout the cytoplasm.
Fig. 1.

Antigen-regulated relocalization of Bcl10 to the T cell/APC interface. (a) OTII T cells were incubated with CHb B cells (APC) for 45 min in the absence (CHb + No antigen) or presence of 10 μg/ml OVA 323–339 (CHb + Ovalbumin). Images of T cell/APC conjugates are arranged as follows (left to right): Nomarski, anti-Bcl10 (red), anti-PKCθ (green), anti-CD4 (blue), and an overlay of the three fluorescent images. (b) Quantitative representation of anti-Bcl10 staining data from a. Images are represented by using a pseudocolor intensity scale (red, most intense staining; dark blue, least intense; scale bar indicates range). Images have been scaled such that the maximal intensity is set as the highest value of anti-Bcl10 staining in the T cell (i.e., APC staining was not considered). (c) Quantification of maximal T cell Bcl10 fluorescence intensity data, with error bars representing SEM. Note that the 7-fold increase in maximal Bcl10 enrichment of ovalbumin-stimulated cells relative to unstimulated cells underestimates the average increase, because two of eight ovalbumin-stimulated cells displayed Bcl10 fluorescence above the maximal detection limit (65,534).
In contrast, in T cells stimulated by specific antigen-loaded CHb APC, PKCθ became enriched at the region of contact between the T cell and the APC. Bcl10 was preferentially relocalized to a region of the T cell that was at or near the T cell/APC interface (Fig. 1a, bottom two rows). The amount of Bcl10 in the region of enrichment near the T cell/APC contact in ovalbumin-stimulated cells was much greater than the most intensely staining regions in the no antigen controls. The mean maximal anti-Bcl10 fluorescence intensity of the enriched regions of the antigen-stimulated T cells was 7-fold higher than the maximal T cell anti-Bcl10 fluorescence of the no antigen controls (Fig. 1 b and c). These results thus show that endogenous T cell Bcl10 both relocalizes to and becomes substantially enriched at the T cell/APC interface in response to specific antigen stimulation.
Fluorescent Protein Fusions of Bcl10 Recapitulate the Behavior of Endogenous Bcl10. To study antigen-mediated relocalization of Bcl10 in more detail, we constructed a fluorescent protein fusion between Bcl10 and YFP. For these experiments, we used the CD4+ T cell clone D10, which is specific for a conalbumin peptide bound to IAk, and we infected D10 cells with a Bcl10-YFP retrovirus. In these cells, Bcl10-YFP was diffusely localized throughout the cell, and this distribution was unchanged when the cells were conjugated with CH12 APC in the absence of antigen (Fig. 2a Left). However, a dramatic redistribution of Bcl10-YFP into punctate and filamentous structures was observed when these T cells were conjugated with conalbumin-loaded CH12 APC (Fig. 2a Right). The Bcl10-YFP fusion protein is therefore dramatically relocalized in response to TCR stimulation.
Fig. 2.

Relocalization of Bcl10 in response to TCR stimulation. (a) D10 T cells expressing Bcl10-YFP were incubated with CH12 B cells (APC) in the absence (No Ag) or presence of 500 μg/ml of conalbumin (+Conalbumin). The fluorescent image is overlayed on the Nomarski image. (b) Bcl10-YFP-expressing D10 T cells were cloned by limiting dilution, and a clone with low Bcl10-YFP expression was identified by FACS [green histogram peak; red peak shows fluorescence of the YFP-negative D10 parent (p)]. Western blotting analysis shows that Bcl10-YFP is expressed at levels lower than the endogenous Bcl10. Note that the increased level of endogenous Bcl10 in this clone is proportional to the increased size of cl.24 cells relative to the D10 parent, as measured by FACS (data not shown). Microscopy images of D10-Bcl10-YFP cl.24 cells conjugated to CH12 B cells (preloaded with 250 μg/ml conalbumin protein) are arranged as follows (left to right): Nomarski, anti-Bcl10 (red), anti-GFP (green), and an overlay of the fluorescent images. The anti-GFP monoclonal antibody JL-8 (BD/Clontech) was used to boost the YFP signal in this cell line for detection by fluorescence microscopy. (c) D10 T expressing the CARD mutant Bcl10-G78R-YFP and PKCθ-CFP were incubated with CH12 B cells (APC) in the presence of 250 μg/ml conalbumin protein for 30 min. The images are arranged as follows (left to right): Nomarski, Bcl10-G78R-YFP (red), PKCθ-CFP (green), and an overlay of the fluorescent images.
In some cells, the punctate pattern was very similar to that observed for endogenous Bcl10 (compare T cell at far right of Fig. 2a Right, to antigen-stimulated T cells in Fig. 1a), whereas the Bcl10 filaments observed in a subset of cells (e.g., T cell at far left of Fig. 2a Right) were more similar to the “spontaneous” filaments of Bcl10 that have been reported to occur in a subset of Bcl10-transfected HeLa cells (24). Further characterization of D10 subclones has shown that filamentous structures occur in TCR-stimulated cells with the highest level of Bcl10-YFP expression, suggesting that the punctate structures more accurately represent the structures formed by endogenous Bcl10 in non-transduced antigen-stimulated T cells.
To establish that endogenous Bcl10 and the fluorescent fusion protein undergo equivalent redistribution, we subjected D10 T cells that express Bcl10-YFP to limiting dilution cloning and identified a subclone (cl.24) with low uniform expression of Bcl10-YFP (Fig. 2b). Western blotting showed that the level of Bcl10-YFP expression was 10% lower than the level of endogenous Bcl10 in this subclone, and an antibody-staining experiment demonstrated that endogenous Bcl10 and the fluorescent protein fusion redistribution in an indistinguishable manner in response to antigen stimulation (Fig. 2b). This experiment demonstrated both that the fluorescent protein tag does not affect relocalization of the Bcl10 fusion protein, and that the monoclonal anti-Bcl10 antibody is indeed highly specific for the Bcl10 protein.
To further characterize the mechanism of redistribution of Bcl10, we introduced the G78R mutation into the Bcl10-YFP CARD. This mutation is known to inactivate the Bcl10 CARD, and it has previously been shown to prevent the formation of “spontaneous” Bcl10 filaments in HeLa cells (23). As anticipated, this mutation completely blocked the antigen-driven redistribution of Bcl10-YFP without influencing the antigen-stimulated redistribution of PKCθ (Fig. 2c). Thus, Bcl10 redistribution requires a functional Bcl10 CARD.
The punctate and filamentous structures formed by Bcl10 in antigen-stimulated T cells are quite similar to oligomeric filamentous structures, called death effector filaments, which are formed by specific mediators of apoptosis (25, 26). Interestingly, Bcl10 and its tightly associated binding partner, a caspase-like protein called MALT1 (27), contain protein interaction motifs of the death domain-fold family (28, 29), also found in mediators of cell death. Thus, these punctate and filamentous structures may represent a higher-order signaling complex common to death domain-fold protein activators of caspases and caspase-like proteins. Because these cellular domains are visually quite similar, and because they both appear to perform a similar function (i.e., signal transduction), we have named these structures punctate and oligomeric killing or activating domains transducing signals (POLKADOTS).
Redistribution of Bcl10 Does Not Depend on Cytoskeletal Filaments. Because TCR stimulation causes formation of Bcl10 POLKADOTS, which sometimes adopt a filamentous appearance, we decided to examine the possibility that Bcl10 clustering might involve cytoskeletal filaments. To circumvent the problem that many cytoskeletal inhibitors also impair T cell/APC conjugate formation, we activated D10 T cells with phorbol ester (PMA), a stimulator of PKC. Indeed, PMA stimulation of D10 T cells expressing PKCθ–cyan fluorescent protein (CFP) and Bcl10-YFP led to the translocation of PKCθ-CFP to the cell membrane (data not shown) and the formation of Bcl10-YFP POLKADOTS (Fig. 5a, which is published as supporting information on the PNAS web site).
In PMA-stimulated cells pretreated with nocodazole, microtubules were disrupted, as indicated by the diffuse cytoplasmic distribution of tubulin. Cells pretreated with latrunculin A also did not appear to have reduced levels of Bcl10-YFP POLKADOTS (Fig. 5a). Thus, the formation of Bcl10 POLKADOTS does not depend on the presence of either microtubules or actin microfilaments.
To assess potential associations between POLKADOTS and intermediate filaments, we introduced a CFP-vimentin fusion protein into D10 T cells expressing Bcl10-YFP [lymphocyte intermediate filaments are primarily composed of vimentin (30), and CFP-vimentin fusion proteins have been shown to be functional (31)]. These T cells were incubated with APC either in the absence or presence of specific antigen (Fig. 5b). In unstimulated cells, CFP-vimentin intermediate filaments showed the expected uropod localization (32) (Fig. 5b, top row), and Bcl10-YFP showed the expected diffuse cellular distribution. In antigen-stimulated cells, the CFP-vimentin reoriented toward the site of TCR clustering, as has been previously described (30), and Bcl10-YFP formed POLKADOTS (Fig. 5b, bottom three rows). Because Bcl10-YFP POLKADOTS and CFP-vimentin frequently did not colocalize, the data strongly suggest that POLKADOTS are not associated with intermediate filaments, although association with a minority population of intermediate filaments cannot be ruled out.
Bcl10 POLKADOTS are thus an oligomeric structure distinct from cytoskeletal filaments. It thus seems most likely that POLKADOTS are de novo oligomers formed in T cells as the result of a TCR-dependent posttranslational modification of Bcl10 or a Bcl10-associated protein.
Bcl10 POLKADOTS Relocalize to the c-SMAC After PKCθ Redistribution. Because TCR stimulation causes the relocalization of both PKCθ (33) and Bcl10 (see above), we wished to investigate the potential kinetic relationship between the redistribution of these two molecules. D10 T cells expressing both PKCθ-CFP and Bcl10-YFP were mixed with conalbumin-loaded CH12 B cells, and the redistribution of PKCθ-CFP and Bcl10-YFP was monitored in seven-parameter live cell imaging experiments. In these image series, red represents PKCθ-CFP, green represents Bcl10-YFP, and blue is an infrared/bright-field image. Z-series data were collected once per minute for 40 min, and selected frames are shown in Fig. 3a. (The complete image series is available as Movie 1, which is published as supporting information on the PNAS web site).
Fig. 3.

Live cell imaging analysis of PKCθ-CFP and Bcl10-YFP redistribution in response to antigen stimulation. (a) D10 T cells expressing PKCθ-CFP and Bcl10-YFP were incubated with conalbumin-loaded CH12 B cells (APC). Data are displayed as follows: PKCθ-CFP (red), Bcl10-YFP (green), and bright field (blue). Frames at the indicated times are shown; the complete 39 frame series is in Movie 1. An additional cell, which appears to have been activated and then separated from the activating APC, is seen at the right of the T cell/APC conjugate beginning at 7 min. (b) An additional example of the experiment in a is shown. In this case, the display shows six panels of three frames each. The first two frames are pseudocolor intensity representations of PKCθ-CFP and Bcl10-YFP fluorescence (scale bar represents fluorescence intensity in relative units). The third frame is a red/green/blue (RGB) overlay image of the same data, with color representation as in a. (c) The data in b were quantitatively analyzed. Colored points show relative enrichment of PKCθ-CFP (red) and Bcl10-YFP (green) at the T cell/APC contact over time. The relative enrichment of each fluorescent protein in the whole cell is defined as 1 (dashed line).
Within the first minute after initial contact between the D10 T cell and the CH12 B cell, PKCθ translocated to the T cell/APC interface. Several minutes later, Bcl10-YFP began to assemble into POLKADOTS at many sites in the cytoplasm. Over time, many of the Bcl10-YFP POLKADOTS were rearranged so that they became condensed at the T cell/APC interface, at the area of PKCθ-CFP enrichment. Thus PKCθ-CFP and Bcl10-YFP cluster in an overlapping region at the c-SMAC (see also Movies 2 and 3, which are published as supporting information on the PNAS web site).
A second series of images is shown in Fig. 3b (for the complete series, see Movies 4 and 5, which are published as supporting information on the PNAS web site), but in this case, the data for PKCθ-CFP and Bcl10-YFP are shown as side-by-side pseudocolor intensity panels, followed by a composite red/green/blue (RGB) image. This figure shows an early T cell/APC interaction, in which PKCθ-CFP has already translocated to the interface between the cells, but Bcl10-YFP has not yet formed POLKADOTS. In this experiment, Bcl10-YFP POLKADOTS were initially observed at approximately the 3-min time point, scattered throughout the cytoplasm. As seen in Fig. 3a, the Bcl10-YFP POLKADOTS became redistributed over time to the contact between the T cell and the APC, overlapping with the site of enrichment of PKCθ. Additionally, the intensity of Bcl10-YFP enrichment at the T cell/APC contact increased over time, apparently due both to the continued redistribution of cytoplasmic Bcl10-YFP to existing POLKADOTS and to the recruitment of additional POLKADOTS to this site.
Fig. 3c shows quantification of data from Fig. 3b, displaying the relative enrichment of both PKCθ-CFP and Bcl10-YFP at the T cell/APC contact. During minutes 0–2, the enrichment of PKCθ-CFP at the contact increased to a level ≈3-fold over the mean fluorescence intensity for the entire cell. This period of enrichment was followed by a reversal of this translocation during minutes 2–39. In contrast, Bcl10-YFP enrichment at the contact did not begin until POLKADOTS began to form, and this enrichment continued throughout the time course. Thus, PKCθ translocation to the T cell/APC interface precedes Bcl10 POLKADOTS formation and the enrichment of Bcl10 at the T cell/APC interface.
TCR Stimulation Causes PKC-Dependent Phosphorylation of Bcl10. We next investigated whether Bcl10 redistribution reflects biochemical modification of Bcl10. Interestingly, a recent study of CARMA1 mutant mice has shown that CARMA1 is required for PMA-dependent Bcl10 phosphorylation in B cells (7). However, there has been no demonstration of TCR-dependent Bcl10 phosphorylation in T cells. We stimulated D10 T cells expressing PKCθ-CFP and Bcl10-YFP with anti-TCR, and performed Western blotting analyses of Bcl10-YFP and endogenous Bcl10 (Fig. 4a). These data show that Bcl10-YFP and Bcl10 exist single predominant isoforms (≈60 and 30 kDa, respectively) in unstimulated D10 T cells, whereas much of the 60- and 30-kDa isoforms were converted to two more slowly migrating forms during 60 min of anti-TCR stimulation. Phosphatase treatment of immunoprecipitated Bcl10-YFP and endogenous Bcl10 from anti-TCR-stimulated D10 T cells converted the slowly migrating forms to the most rapidly migrating form (Fig. 4b), demonstrating that the slowly migrating forms are phosphorylated.
Fig. 4.

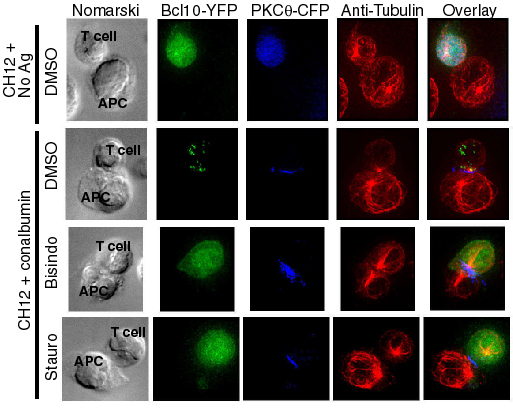
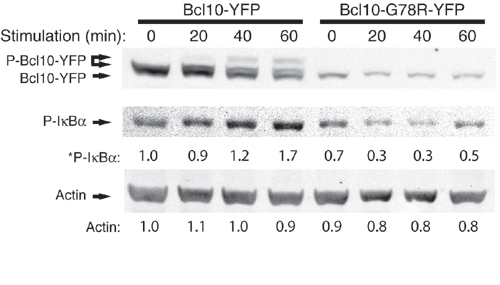
TCR ligation stimulates PKC-dependent Bcl10 phosphorylation and NF-κB activation. (a) D10 T cells expressing PKCθ-CFP and Bcl10-YFP were stimulated for 60 min with no antibody or with anti-TCRβ. Bcl10-YFP, endogenous Bcl10, and phosphorylated forms of these proteins (P-) were detected by Western blotting. (b) Bcl10-YFP and endogenous Bcl10 from TCR-stimulated cells were immunoprecipitated and incubated with no enzyme or with λ-phosphatase and detected by SDS/PAGE and Western blotting. (c) D10 T cells expressing PKCθ-CFP and Bcl10-YFP were pretreated for 30 min with 0.1% DMSO or bisindolylmaleimide (2 μM) and treated for the indicated times with anti-TCRβ. Proteins were detected by Western blotting, and relative amounts of the phosphorylated forms of Bcl10, IκBα, and ERK1 (* indicates values were normalized to actin; actin values are relative to t = 0) are shown below the respective blot for each time point. (d) D10 T cells expressing Bcl10-YFP were pretreated for 30 min with 0.1% DMSO, staurosporine (100 nM), or bisindolylmaleimide (2 μM). CH12 B cells that had been preincubated with no antigen or with 100 μg/ml conalbumin protein were then added. Cells were fixed after 20 min and examined by fluorescence microscopy. For each group, >200 T cells were counted, and percentages with and without POLKADOTS were determined.
To determine whether TCR-dependent phosphorylation of Bcl10 requires PKC activity, we stimulated the D10 T cells expressing PKCθ-CFP and Bcl10-YFP with anti-TCRβ, after pretreatment with either DMSO or 2 μM bisindolylmaleimide (bisindo), a specific protein kinase C inhibitor. As shown in Fig. 4c, bisindo treatment completely blocked formation of phosphorylated Bcl10-YFP. Furthermore, Bcl10-YFP phosphorylation was closely associated with activation of NF-κB (as assessed by monitoring phosphorylation of IκBα), and both signaling events were blocked by treatment with bisindo. Additionally, the kinetics of phosphorylation of Bcl10-YFP matched the kinetics of POLKADOTS formation in response to this immobilized anti-TCR antibody treatment (data not shown). Notably, the activation of the ERK MAP kinases was not blocked but was partially inhibited by bisindo, in agreement with published data (34). The data in Fig. 4 a–c thus demonstrate that TCR-dependent phosphorylation of Bcl10 and concomitant activation of NF-κB are PKC-dependent signaling events.
We next examined whether formation of Bcl10 POLKADOTS is PKC-dependent (Fig. 4d and Fig. 6, which is published as supporting information on the PNAS web site). D10 T cells were incubated with CH12 B cells with no antigen, or with conalbumin-loaded CH12 B cells. Some of the antigen-stimulated samples were also treated with bisindo, and some were treated with the broad-spectrum protein kinase inhibitor staurosporine (stauro). Antigen stimulation resulted in formation of Bcl10-YFP POLKADOTS in >80% of the cells. In contrast, POLKADOTS formation was blocked by treatment with stauro or bisindo. Thus, both TCR-stimulated Bcl10-YFP POLKADOTS formation and Bcl10 phosphorylation require PKC activity.
Finally, to determine the dependence of NF-κB activation on Bcl10 phosphorylation, we examined IκBα phosphorylation in D10 T cells expressing the G78R CARD mutant of Bcl10. As shown in Fig. 7, which is published as supporting information on the PNAS web site, the CARD mutant of Bcl10 does not become phosphorylated in response to anti-TCR stimulation, and it acts as a dominant-negative, blocking TCR-dependent IκBα phosphorylation. Bcl10 phosphorylation is thus a necessary intermediate in TCR activation of NF-κB.
Discussion
In this study, we have used antibody staining and fluorescent protein fusions to examine the distribution of critical TCR-regulated transducers of NF-κB signals. We have shown that Bcl10 forms punctate cytoplasmic structures (POLKADOTS) in response to antigen stimulation. Bcl10 POLKADOTS then undergo further reorganization, coalescing at the c-SMAC. We have provided evidence that Bcl10 redistribution is functionally linked to TCR-mediated NF-κB activation, by showing that Bcl10 phosphorylation, POLKADOTS formation, and NF-κB activation in response to TCR stimulation all depend on PKC activity. Furthermore, we have shown that Bcl10 phosphorylation is required for NF-κB activation. Our data thus demonstrate that Bcl10 reorganization and phosphorylation occurs in response to antigen plus APC signaling, and that these events are of critical importance in the delivery of the NF-κB-activating signal.
Our data suggest two probable mechanisms whereby Bcl10 is phosphorylated. The first is that PKCθ constantly shuttles into and away from the c-SMAC, rapidly diffusing throughout the cell. In this model, the enrichment of PKCθ at the c-SMAC reflects a zone of concentration where individual PKCθ molecules reside for a short time before becoming activated and diffusing throughout the cytoplasm to activate Bcl10. This model is supported to some degree by our observation that the enrichment of PKCθ at the c-SMAC is transient and begins to reverse at about the time POLKADOTS are first evident. The second possibility is that PKCθ activates another kinase, and this second kinase diffuses to distant sites to activate Bcl10, stimulating POLKADOTS formation.
In either case, we find it surprising that PKCθ and Bcl10 relocalization is so complex. Although both proteins eventually concentrate at the c-SMAC, there is an intermediate period during which Bcl10 POLKADOTS form, and these structures are often distant from the region of PKCθ enrichment at the c-SMAC (note particularly 8 min, Fig. 3b, and Movies 4 and 5). These data thus demonstrate that ligand engagement by a transmembrane receptor does not simply result in the clustering of signaling molecules at the cytoplasmic face of the receptor. Rather, other redistribution events distant from the point of receptor engagement are also involved in specific signal transduction processes.
We propose that the existence of two distinct reorganization events that bring two different mediators of TCR activation of NF-κB to the c-SMAC provides evidence that the c-SMAC is of functional importance in the regulation NF-κB. Although SMACs may not be involved in the delivery of certain very early signals from the TCR (35), a consensus view is emerging postulating that SMACs exist for the purpose of providing a stable signaling platform for the lengthy series of biochemical signals originating from antigen stimulation of the TCR (12, 36, 37). We postulate that the c-SMAC regulates activation of a subset of TCR-controlled signaling pathways, which require continued TCR signaling following SMAC organization (38). Based on our observation of sequential localization of NF-κB signal transducers to the c-SMAC, we propose that TCR activation of NF-κB is “validated” at multiple steps, via delivery of signals from the engaged TCR to critical NF-κB signaling intermediates that traffic to the c-SMAC in a spatially organized and temporally ordered manner. Such validation would ensure that the NF-κB signal, which commits a T cell to S-phase, is only delivered to the nucleus on sustained TCR engagement by antigen.
Supplementary Material
Acknowledgments
We thank T. Vass for care of mice, W. Townend for help with microscopy, and S. Sobus and K. Wolcott for cell sorting. B.C.S. was a Special Fellow of the Leukemia and Lymphoma Society. This work was supported by National Institutes of Health Grants AI-17134, AI-18785, AI-52225, AI-22295, and AI-23764 (to P.M. and J.W.K.) and Uniformed Services University of the Health Sciences Grant CO73JN (to B.C.S.).
Abbreviations: CARD, caspase recruitment domain; TCR, T cell receptor; APC, antigen-presenting cell; SMAC, supramolecular activation cluster; c-SMAC, central zone of the SMAC; POLKADOTS, punctate and oligomeric killing or activating domains transducing signals; PMA, phorbol ester; YFP, yellow fluorescent protein; CFP, cyan fluorescent protein.
References
- 1.Boothby, M. R., Mora, A. L., Scherer, D. C., Brockman, J. A. & Ballard, D. W. (1997) J. Exp. Med. 185, 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruland, J., Duncan, G. S., Elia, A., del Barco Barrantes, I., Nguyen, L., Plyte, S., Millar, D. G., Bouchard, D., Wakeham, A., Ohashi, P. S., et al. (2001) Cell 104, 33–42. [DOI] [PubMed] [Google Scholar]
- 3.Sun, Z., Arendt, C. W., Ellmeier, W., Schaeffer, E. M., Sunshine, M. J., Gandhi, L., Annes, J., Petrzilka, D., Kupfer, A., Schwartzberg, P. L., et al. (2000) Nature 404, 402–407. [DOI] [PubMed] [Google Scholar]
- 4.Egawa, T., Albrecht, B., Favier, B., Sunshine, M. J., Mirchandani, K., O'Brien, W., Thome, M. & Littman, D. R. (2003) Curr. Biol. 13, 1252–1258. [DOI] [PubMed] [Google Scholar]
- 5.Newton, K. & Dixit, V. M. (2003) Curr. Biol. 13, 1247–1251. [DOI] [PubMed] [Google Scholar]
- 6.Hara, H., Wada, T., Bakal, C., Kozieradzki, I., Suzuki, S., Suzuki, N., Nghiem, M., Griffiths, E. K., Krawczyk, C., Bauer, B., et al. (2003) Immunity 18, 763–775. [DOI] [PubMed] [Google Scholar]
- 7.Jun, J. E., Wilson, L. E., Vinuesa, C. G., Lesage, S., Blery, M., Miosge, L. A., Cook, M. C., Kucharska, E. M., Hara, H., Penninger, J. M., et al. (2003) Immunity 18, 751–762. [DOI] [PubMed] [Google Scholar]
- 8.Ruefli-Brasse, A. A., French, D. M. & Dixit, V. M. (2003) Science 302, 1581–1584. [DOI] [PubMed] [Google Scholar]
- 9.Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. (1998) Nature 395, 82–86. [DOI] [PubMed] [Google Scholar]
- 10.Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M. & Dustin, M. L. (1999) Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- 11.Dustin, M. L. & Cooper, J. A. (2000) Nat. Immunol. 1, 23–29. [DOI] [PubMed] [Google Scholar]
- 12.Krummel, M. F. & Davis, M. M. (2002) Curr. Opin. Immunol. 14, 66–74. [DOI] [PubMed] [Google Scholar]
- 13.Kupfer, A., Burn, P. & Singer, S. J. (1990) J. Mol. Cell Immunol. 4, 317–325. [PubMed] [Google Scholar]
- 14.Wang, D., You, Y., Case, S. M., McAllister-Lucas, L. M., Wang, L., DiStefano, P. S., Nunez, G., Bertin, J. & Lin, X. (2002) Nat. Immunol. 3, 830–835. [DOI] [PubMed] [Google Scholar]
- 15.McAllister-Lucas, L. M., Inohara, N., Lucas, P. C., Ruland, J., Benito, A., Li, Q., Chen, S., Chen, F. F., Yamaoka, S., Verma, I. M., et al. (2001) J. Biol. Chem. 276, 30589–30597. [DOI] [PubMed] [Google Scholar]
- 16.Kaye, J., Porcelli, S., Tite, J., Jones, B. & Janeway, C. A., Jr. (1983) J. Exp. Med. 158, 836–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer, B. C., Ware, M. F., Marrack, P., Fanger, G. R., Kappler, J. W., Johnson, G. L. & Monks, C. R. (1999) Immunity 11, 411–421. [DOI] [PubMed] [Google Scholar]
- 18.Haughton, G., Arnold, L. W., Bishop, G. A. & Mercolino, T. J. (1986) Immunol. Rev. 93, 35–51. [DOI] [PubMed] [Google Scholar]
- 19.Heim, R. & Tsien, R. Y. (1996) Curr. Biol. 6, 178–182. [DOI] [PubMed] [Google Scholar]
- 20.Crameri, A., Whitehorn, E. A., Tate, E. & Stemmer, W. P. (1996) Nat. Biotechnol. 14, 315–319. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer, B. C., Mitchell, T. C., Kappler, J. W. & Marrack, P. (2001) Anal. Biochem. 297, 86–93. [DOI] [PubMed] [Google Scholar]
- 22.Gaide, O., Favier, B., Legler, D. F., Bonnet, D., Brissoni, B., Valitutti, S., Bron, C., Tschopp, J. & Thome, M. (2002) Nat. Immunol. 3, 836–843. [DOI] [PubMed] [Google Scholar]
- 23.Barnden, M. J., Allison, J., Heath, W. R. & Carbone, F. R. (1998) Immunol. Cell Biol. 76, 34–40. [DOI] [PubMed] [Google Scholar]
- 24.Guiet, C. & Vito, P. (2000) J. Cell Biol. 148, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez, D. & White, E. (1998) J. Cell Biol. 141, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel, R. M., Martin, D. A., Zheng, L., Ng, S. Y., Bertin, J., Cohen, J. & Lenardo, M. J. (1998) J. Cell Biol. 141, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uren, A. G., O'Rourke, K., Aravind, L. A., Pisabarro, M. T., Seshagiri, S., Koonin, E. V. & Dixit, V. M. (2000) Mol. Cell 6, 961–967. [DOI] [PubMed] [Google Scholar]
- 28.Fairbrother, W. J., Gordon, N. C., Humke, E. W., O'Rourke, K. M., Starovasnik, M. A., Yin, J. P. & Dixit, V. M. (2001) Protein Sci. 10, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinon, F., Hofmann, K. & Tschopp, J. (2001) Curr. Biol. 11, R118–R120. [DOI] [PubMed] [Google Scholar]
- 30.Dellagi, K. & Brouet, J. C. (1982) Nature 298, 284–286. [DOI] [PubMed] [Google Scholar]
- 31.Yoon, K. H., Yoon, M., Moir, R. D., Khuon, S., Flitney, F. W. & Goldman, R. D. (2001) J. Cell Biol. 153, 503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown, M. J., Hallam, J. A., Colucci-Guyon, E. & Shaw, S. (2001) J. Immunol. 166, 6640–6646. [DOI] [PubMed] [Google Scholar]
- 33.Monks, C. R., Kupfer, H., Tamir, I., Barlow, A. & Kupfer, A. (1997) Nature 385, 83–86. [DOI] [PubMed] [Google Scholar]
- 34.Puente, L. G., Stone, J. C. & Ostergaard, H. L. (2000) J. Immunol. 165, 6865–6871. [DOI] [PubMed] [Google Scholar]
- 35.Lee, K. H., Holdorf, A. D., Dustin, M. L., Chan, A. C., Allen, P. M. & Shaw, A. S. (2002) Science 295, 1539–1542. [DOI] [PubMed] [Google Scholar]
- 36.Dustin, M. L. & Chan, A. C. (2000) Cell 103, 283–294. [DOI] [PubMed] [Google Scholar]
- 37.Delon, J. & Germain, R. N. (2000) Curr. Biol. 10, R923–R933. [DOI] [PubMed] [Google Scholar]
- 38.Freiberg, B. A., Kupfer, H., Maslanik, W., Delli, J., Kappler, J., Zaller, D. M. & Kupfer, A. (2002) Nat. Immunol. 3, 911–917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}