

Abstract
Context:
Gliosarcoma is a rare variant of glioblastoma multiforme containing distinct gliomatous and sarcomatous components. Gliosarcoma comprise 1.8–8% of glioblastoma multiforme and are clinically similar to them, affecting adults in the fourth and sixth decades of life, with a higher proportion found in males. The survival for patients with Gliosarcoma is equally poor as for those with glioblastoma multiforme, and there is a greater propensity for extracranial metastasis in Gliosarcoma. Clinical treatment-related experience reported in the literature is limited, and Gliosarcoma are currently treated in a similar fashion to glioblastoma multiforme, with modalities including tumor resection, postoperative radiation therapy, and chemotherapy. Gliosarcoma can arise secondarily, after conventional adjuvant treatment of high-grade glioma. The current literature on the occurrence of secondary gliosarcoma after glioblastoma multiforme is limited, with only 54 reported cases.
Case Report:
The authors present a 48-year-old Caucasian male who had previously received postoperative combined radiation and temozolomide chemotherapy for glioblastoma multiforme. After a free disease period of 9 months the disease recurs as Gliosarcoma. The patient underwent a Total surgical excision and received chemotherapy with a basis of bevacizumab and irinotecan. The patient died from tumor progression 5 months after gliosarcoma diagnosis.
Conclusion:
The poor survival of patients with secondary gliosarcoma who had previously received combined radiation and temozolomide chemotherapy for glioblastoma multiforme may reflect a unique molecular profile of glioblastoma multiforme that eventually recurs as secondary gliosarcoma. We have to keep in mind the possibility of gliosarcomatous change in the recurrence of malignant glioma. Awareness of this pathological entity will allow more rapid diagnosis and treatment.
Keywords: High-grade glioma, tumor resection, secondary gliosarcoma, glioblastoma multiforme, radiotherapy, chemotherapy
Introduction
Gliosarcoma (GS) is a very rare primary mixed tumor in the central nervous system (CNS), with a biphasic pattern consisting of glial and malignant mesenchymal elements[1]. It was described for the first time in 1895 by Stoebe[2]. In the 2007 World Health Organization (WHO) classification of tumors of CNS, gliosarcoma is considered a subtype of glioblastoma (GB).This conclusion is supported by finding identical genetic alterations in both tumor elements[3]. Most GS are the novo, and are hence termed primary GS, whereas those detected at subsequent surgery for previously resected and irradiated glioblastoma multiforme (GBM) are termed secondary gliosarcoma (SGS) .We report a case of SGS and literature review.
Case Report
A 48-year-old Caucasian male consulted for dizziness, headache and epileptic crisis. On neurological examination there were no deficits in sensory or motor functions. Brain Magnetic resonance imaging (MRI) showed a Right temporal process measuring 32 mm and an accompanying peritumoral Edema (Figure 1). A complete macroscopic resection was performed. Histological examination demonstrated a hypocellular tumor with pleomorphic astrocytes, endothelial proliferation and wide range of necrosis Immunohistochemistry was positive for Glial fibrillary acidic protein (GFAP) and negative for anti cytokeratine. A diagnosis of GBM was made.
Fig. 1.
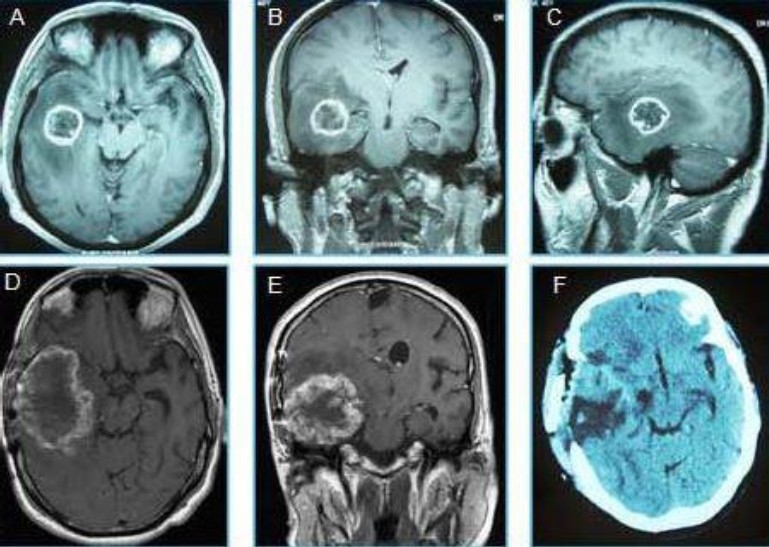
Neuroimages of glioblastoma recur after treatment in the same place as gliosarcoma. A: axial, B: Coronary and D: sagittal MRI showed a Right temporal process corresponded to a glioblastoma multiforme before treatment. D: Axial and E: Coronary MRI image of a secondary gliosarcoma, later found at the same location as the previously treated GBM. F: axial CT, performed after the total resection of the gliosarcoma.
The patient was then referred to our department and underwent a post-operative radiotherapy treatment using an isocentric field technique, with 6MV and 23MV photons of a linear accelerator, receiving a total dose of 59.4 Gy in 33 fractions (1.8 Gy per fraction) and concurrent temozolomide 75mg/m2 per day 1h before radiotherapy and at weekends,. Adjuvant temozolomide therapy, 150mg/m2 per day 5 days per month, was administered in 5 cycles. Sixteen months after the radiotherapy had been administered; the patient developed a severe headache, a decreased visual acuity, memory problems, sixth nerve palsy and left hemiparesis. Brain MRI showed a right temporoparietal process isointense inT1 sequence, hyperintense in flair, measuring 72/56 mm. The lesion exerts mass effect with compression of the right lateral ventricle and displacement of the midline structures (Figure 1D, 1E). Total surgical excision was undertaken, but Post operatoire CT scan showed the persistence of a tumor residue in the inner surface of the cavity (Figure 1F).
Histopathological examination showed a cerebral parenchyma infiltrated by a biphasic tumoral tissue pattern with alternating areas displaying glial and mesenchymal differentiation (Figure 2b). The gliomatous component strongly GFAP positive (Figure 2a) was intermingled with the sarcomatous tumour cells that demonstrated a vimentine expression (Figure 2c). A diagnosis of GS was made. The patient received chemotherapy with a basis of bevacizumab 10mg/kg every 2 weeks and irinotecan 125 mg/m2 every 2 weeks. The patient died from tumor progression after two cycles, approximately 27 months after the diagnosis of GB was made.
Fig. 2.
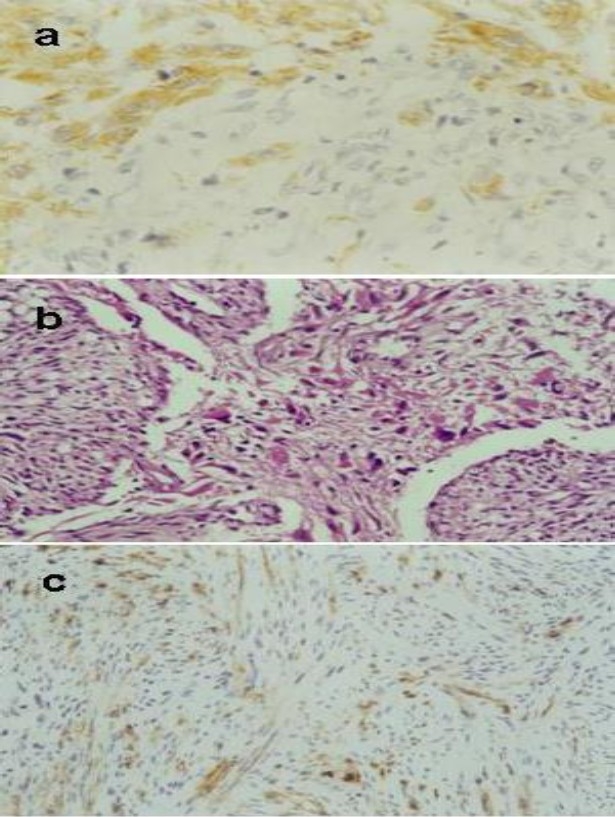
Photomicrographs of the tumor. a: The gliomatous component GFAP positive (×100). b: Tumour characterised by a biphasic tissue pattern with alternating areas displaying glial and mesenchymal differentiation (HE×40). c: the sarcomatous component vimentine positive (×10).
Discussion
GS is currently defined as amorphological variant of GBM, which accounts for between 1.8 and 8% of cases. It typically affects older men, with onset between the fourth and sixth decades of life and a male/female ratio of 1.8/1[1].
Secondary gliosarcoma after treatment of primary glioblastoma multiforme and radiation-induced gliosarcoma are exceedingly rare. Only 54 cases were reported in the literature. Most GS are the novo, and are hence termed primary GS, whereas those detected at subsequent surgery for previously resected and irradiated GBM are termed SGS. The causal relationship between therapeutic irradiation and the delayed induction of neoplastic processes is well established for meningioma, cerebral fibrosarcoma, other sarcomatous variants, and more rarely GBM. The criteria for radiation-induced tumors require that: 1) the tumor appear in the area of irradiation, with a significant latency period between irradiation and the appearance of the tumor, Most post-irradiation soft tissue sarcomas develop no sooner than two to three years after the administration of radiotherapy. The latency period is shortened, and the risk of post-irradiation sarcoma is increased, with increased radiation dose and the administration of chemotherapy. 2) The tumor is absent prior to irradiation, and 3) The new tumor is of a histologically distinct type from the first tumor[4,5].
In our observation, the latency period of 16 months appears to be perhaps sufficient to incriminate radiotherapy in the appearance of gliosarcoma. But, it may be a lack of sampling primary tumor that has enabled to see only the component glial.
GS are usually located in the cerebrum, involving the temporal, parietal, frontal, and occipital lobes in decreasing order of frequency[6]. Our patient presented a temporal lobe lesion.
GS corresponds to a WHO grade IV tumor. It is a bimorphic tumor, containing both gliomatous and mesenchymal regions. The sarcomatous region consists of neoplastic mesenchymal cells with associated reticulin formation. These cells are spindle shaped, and demonstrate nuclear atypia, increased mitotic activity and necrosis. The glial regions in gliosarcoma are typical of glioblastoma, with a varying degree of anaplasia and GFAP expression. GFAP immunostaining is most important in distinguishing between GS and GB, with GFAP being present in glial regions, but found in only very low quantities in sarcomatous regions. Vimentin is a marker of mesenchymal cells, with strong vimentin staining in sarcomatous areas but almost no staining in glial regions[5].
Multiple reports in the literature describe the higher tendency of GS to metastasise to extracranial and intraaxial sites than that of GB. This tendency is reported to be as high as 15% in some studies[7].
The histogenesis of GS has been the subject of debate. Feigin and Gross, who first described GS, suggest that the cell of origin would arise from neoplastic transformation of blood vessels in a pre-existing GB. However, immunohistochemical studies have failed to detect endothelial markers in the sarcomatous component. Some studies have shown expression of monohistiocytic markers, suggesting that GS develop from histiocytes, whereas others suggest that the origin is from fibroblasts, pluripotent mesenchymal cells of the perivascular adventitia, or perivascular spaces. The presence of identical p53 mutations and similar chromosomal imbalances and cytogenetic alterations in both gliomatous and sarcomatous components strongly supports the concept of a monoclonal origin of GS[8].
Several authors reported significant biological similarities in the behavior of GS and GBM, suggesting that the same treatment should be applied for these two kinds of tumor[6]. Treatment modalities described for GS include tumor resection, postoperative radiation therapy, and chemotherapy with nitrosureas, misonidazole, dacarbazine, mithramycin, ametophterin, thalidomide, temozolomide, irinotecan, vincristine, cisplatin, or doxorubicin[9]. The majority of information on GS therapy is derived from published case series. The total dose delivered in radiotherapy ranged from 45 to 81 Gy in these reports. The mean overall survival of patients receiving radiotherapy is longer (10.6 months) than for those treated only with surgery (6.2 months).[10].GS is a chemoresistant tumor, but the literature suggests that the use of temozolomide at the same time as radiotherapy ,and after the radiotherapy treatment is concluded, as an adjuvant treatment slightly increases survival[11].
Currently, there is very little data regarding the response of GS to novel therapies that are being developed and studied for malignant gliomas, such as immunotherapy and cancer vaccine therapies. Most trials with malignant glioma include GS as a variant of GBM, and roles of novel therapies in management of GS becomes difficult to determine[12]. Current evidence suggests that angiogenesis inhibitors may have clinical utility for GBM patients. The FDA recently approved bevacizumab for use in recurrent GBM patients, based on the clinical benefits observed in two recent phase II trials[13,14].
The prognosis for GS is generally poor, with a mean survival of 10.5 months since time of GS diagnosis (range 4.5-21 months)[4,15].In our case the patient survived for 5 months after gliosarcoma diagnosis. The brain edema with increase in intracranial pressure and herniation of the temporal lobe are the immediate causes of death[1].
Conclusion
The data underscore the difficulty associated with management of this disease. The poor survival of patients with SGS who had previously received combined radiation and temozolomide chemotherapy for GBM may reflect a unique molecular profile of GBM that eventually recurs as SGS.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgments
K A S was the principal physician who managed our patient, performed the literature research, and wrote the manuscript. O M made the histological diagnosis. M E helped write the manuscript and performed the literature Review. H S performed and approved the radiotherapy part of our patient's treatment. K H contributed to the treatment of the patient and has been responsible for his follow-up examinations. H M helped with modifications and revisions to the manuscript, also in the final conception of the article, principally in the redaction of the manuscript. All authors read and approved the final manuscript.
References
- 1.Pardo J, Murcia Mauricio, García Felip, Alvarado Arnaldo. Gliosarcoma: A rare primary CNS tumor.Presentation of two cases. Rep Prac Onco Radiother. 2010;15(4):98–102. doi: 10.1016/j.rpor.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stroebe H. Ueber Entstehung und Bau der Gehirngliome. Beitr Pathol Anat. 1895;18:405–486. [Google Scholar]
- 3.Cheong J-H, Kim C-H, Kimand J-M, Oh Y-M. Transformation of intracranial anaplastic astrocytoma associated with neurofibromatosis type I into gliosarcoma: Case report. Clin Neurol Neuroserg. 2010;112(8):701–706. doi: 10.1016/j.clineuro.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Han SJ, Yang I, Tihan T, Chang SM, Parsa AT. Secondary gliosarcoma: a review of clinical features and pathologic diagnosis. J Neurosurg. 2010;112:26–32. doi: 10.3171/2009.3.JNS081081. [DOI] [PubMed] [Google Scholar]
- 5.Alatakis S, Stuckey S, Siu K, McLean C. Gliosarcoma with osteosarcomatous differentiation: review of radiological and pathological features. J Clin Neurosci. 2004;11(6):650–656. doi: 10.1016/j.jocn.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Lutterbacha J, Guttenberger R, Pagenstecher A. Gliosarcoma: a clinical study. Radiother Oncol. 2001;61:57–64. doi: 10.1016/s0167-8140(01)00415-7. [DOI] [PubMed] [Google Scholar]
- 7.Salvati M, Caroli E, Raco A, Giangaspero F, Delfini R, Ferrante L. Gliosarcomas: analysis of 11 cases do two subtypes exist? J Neuro Oncol. 2005;74:59–63. doi: 10.1007/s11060-004-5949-8. [DOI] [PubMed] [Google Scholar]
- 8.Cincu R, Juan F, Lázaro M, Luis J, Liesa C, Eiras J. Gliosarcoma: An Uncommon Brain Tumor Pak J Neurol Sci. 2009;4(1):20–22. [Google Scholar]
- 9.Han SJ, Yang I, Tihan T, Prados MD, Parsa AT. Primary gliosarcoma: key clinical and pathologic distinctions from glioblastoma with implications as a unique oncologic entity. J Neurooncol. 2010;96:313–320. doi: 10.1007/s11060-009-9973-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CH, Horton J, Schoenfeld D, et al. Comparison of post-operative radiotherapy and combined post-operative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas.A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Int J Radiat Oncol Biol Phys. 1983;52:997–1007. doi: 10.1002/1097-0142(19830915)52:6<997::aid-cncr2820520612>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Mason WP, Maestro RD, Eisenstat D, et al. Canadian recommendations for the treatment of glioblastoma multiforme. Curr Oncol. 2007;14(3):110–117. doi: 10.3747/co.2007.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prados MD, Chang SM, Butowski N, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009;27:579–584. doi: 10.1200/JCO.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cloughesy TF, Prados MD, Wen PY, et al. A phase II, randomized, non-comparative clinical trial of the effect of bevacizumab (BV) alone or in combination with irinotecan (CPT) on 6-month progression free survival (PFS6) in recurrent, treatment-refractory glioblastoma (GBM) J Clin Oncol. 2008;26:15S. [Google Scholar]
- 14.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–745. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han SJ, Yang I, Otero JJ, et al. Secondary gliosarcoma after diagnosis of glioblastoma: clinical experience with 30 consecutive patients. J Neurosurg. 2010;112:990–996. doi: 10.3171/2009.9.JNS09931. [DOI] [PubMed] [Google Scholar]