

Abstract
Small molecules experience diffusion retardation when transferred from phosphate-buffered saline into a peptide hydrogel of the same pH and ionic strength. The extent of retardation increases linearly with logPoct, their lipophilicity.
Peptide hydrogels are finding increasing applications as cell growth and differentiation media1-4 with commercial products on the market (e.g., PuraMatrix™). Compared with collagens extracted from animal sources, synthetic peptides have the benefits of lot-to-lot consistency and the absence of antigens and pathogens. We previously developed a mixing-induced approach to the assembly of peptide hydrogels.5-7 In this approach, a hydrogel is assembled by mixing two oppositely charged oligopeptides. As potential media for cell growth and differentiation, this type of hydrogels has several suitable features: the two peptides can be pre-dissolved in physiological buffers with gelation induced by mixing the two peptide solutions at the desired temperature;5 the hydrogels can recover rapidly from shear-induced breakdowns and thus are injectable;6 proteins inside the hydrogels retain their native conformation,7 which is important for biocompatibility.
Aside from biocompatibility, another requirement for cell growth and differentiation media is transport properties: the matrix should allow an efficient diffusion of nutrients and metabolites. Extensive work has been done on the diffusion of proteins in peptide hydrogels for controlled release.8-10 In this project, the diffusion of small molecules inside a mixing-induced peptide hydrogel is evaluated. The focus is on how molecular characteristics, such as charge and lipophilicity, affect diffusion.
The hydrogel was assembled from a pair of oppositely charged undecapeptides, K11 and E11, whose sequences are shown in Table 1. The sequence of the positive peptide module, K11, alternates between positively charged and neutral amino acids, and the sequence of the negative peptide module, E11, alternates between negatively charged and neutral amino acids.
Table 1.
Sequences, molecular weight of undecapeptides K11 and E11
| peptides | sequences | MW (Da) |
|---|---|---|
| K11 | acetyl-KWKAKAKAKWK-amide | 1,413 |
| E11 | acetyl-EWEAEAEAEWE-amide | 1,419 |
A, alanine; E, glutamic acid; K, lysine; W, tryptophan. The N- and C-termini of each peptide are acetylated (acetyl-) and amidated (-amide), respectively. Each peptide contains two tryptophans for concentration determination through UV spectroscopy.11
Gelation in phosphate-buffered saline (PBS) and in H2O, both of pH 7.4, was monitored using dynamic rheometry at 25°C. Gelation in PBS was slower but reached a higher elastic modulus G’ value (Fig. 1 and Fig. S21). The PBS gel also has a higher strain yield value (~3%) than the H2O gel (~1%) (Fig. S22).
Fig. 1.
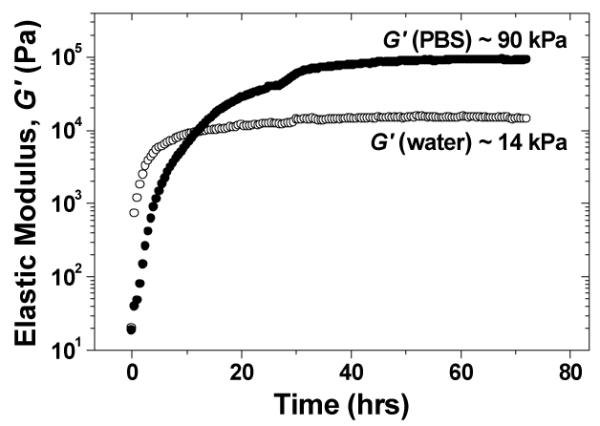
Elastic modulus vs. time for the hydrogel assembled by mixing K11 and E11 in PBS (●) and H2O (○).
To investigate small molecule diffusion inside this hydrogel, a set of four phenylalanine analogues, 1, 2, 3 and 4 (Scheme 1), was used as diffusants. Each compound contains a −CF3 group so that it can be traced by 19F NMR without any interference from the hydrogel. 1 was purchased from Chem-Impex, Inc (Wood Dale, IL, USA). C-terminal amidation of 1 gives 2; N-terminal formylation of 1 gives 3; N-terminal formylation of 2 gives 4. The purity and molecular weight of each compound were verified respectively using analytical HPLC and mass spectrometry (ESI).
Scheme 1.
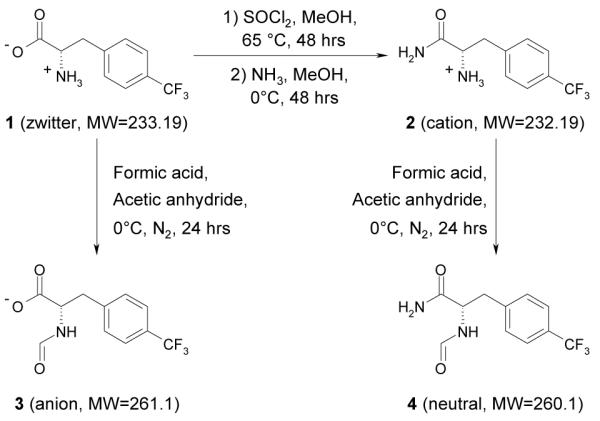
Synthesis of fluorinated phenylalanine analogues.
In terms of charge status, 4 is analogous to the solvent (water) as both are neutral; 3 is analogous to E11 as both contain negative charge carried by the carboxylic group (−COO−); 2 is analogous to K11 as both contain positive charge carried by the amino group (−NH3+); 1 is analogous to the hydrogel as both contain equal amount of positive and negative charges from −NH3+ and −COO−.
Diffusion of each compound was investigated in four types of media: solvent, solution of 8 mM K11 peptide, solution of 8 mM E11 peptide, and the hydrogel which contains 8 mM K11 and 8 mM E11. Pairing of diffusants and media is shown in Scheme 2.
Scheme 2.
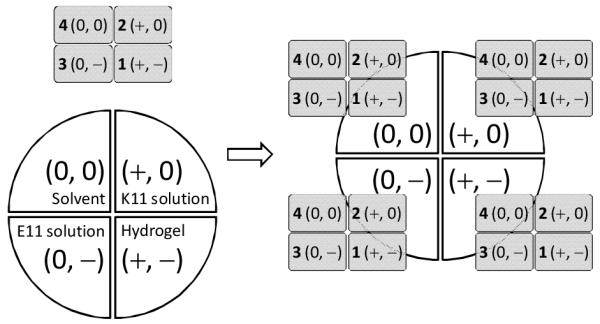
Pairing of four diffusants with four media. + denotes positive charge carried by −NH3+; - denotes negative charge carried by −COO−; 0 denotes a molecule or a medium devoid of −NH3+ or −COO−, e.g., solvent and 4 are denoted as (0, 0) as they contain neither −NH3+ nor −COO−.
Diffusion coefficient (D) was measured at 25°C using pulsed field gradient (PFG) NMR spectroscopy (bipolar pulse longitudinal eddy current delay, BPP-LED).12 D of 1, 2, 3 and 4 was measured using the 19F signal. D of H2O was measured as a reference point.
D values of four diffusants in four types of media are plotted in Fig. 2, along with D of H2O. There are two series: a PBS series and a D2O series. As each compound transfers from solvents to peptide solutions, there is a diffusion retardation, i.e., D(X in peptide solution) < D(X in solvent) (X = 1, 2, 3, 4 and H2O); as each compound transfers from solvents to hydrogels, there is a diffusion retardation, i.e., D(X in hydrogel) < D(X in solvent); and as each compound transfers from D2O to PBS, there is a diffusion retardation, i.e., D(X in PBS) < D(X in D2O). Clearly, diffusion retardation is caused by the interaction between diffusants and non-solvent components of the media (peptides, hydrogel matrix and salts). The question is, in a given medium, does diffusion retardation correlate with molecular characteristics of the diffusants? This is answered by comparing diffusion retardation of the four closely related diffusants, 1, 2, 3 and 4.
Fig. 2.
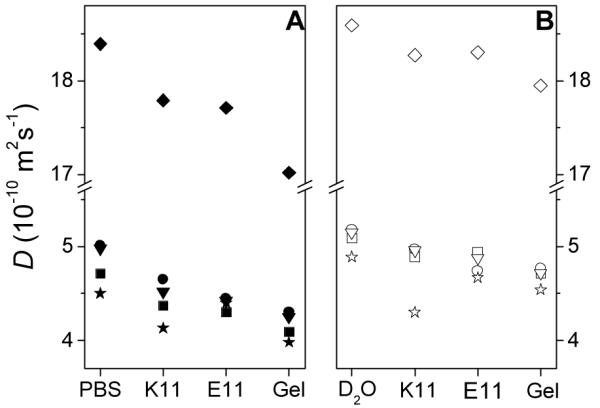
Diffusion coefficients of compounds in various media. (A) In four PBS-based media; (B) In four D2O-based media. The pH of each medium was 7.4. ◆, H2O; ■, 1; ●, 2; ★, 3; ▼, 4. Solid symbols: in PBS media; hollow symbols: in D2O media. Standard deviation of D(H2O) in solvent is 0.08 × 10−10 m2s−1, based on 4 repeated measurements.
Even in PBS and D2O, 1, 2, 3 and 4 have slightly different D values, probably due to difference in hydrodynamic radii as a result of difference in hydration and counter-ion paring. Thus, a meaningful comparison of their diffusion retardation in a given medium should be based on normalized diffusion retardation, Re, defined as:
| (1) |
with X = 1, 2, 3, 4 and H2O; and medium = solvent, K11 solution, E11 solution and hydrogel. Table 2 gives Re values for 1, 2, 3, 4 and H2O in different media. In PBS or D2O, Re is 0 by definition.
Table 2.
Re (in %) of 1, 2, 3, 4 and H2O in various media. The charge status symbols are the same as in Scheme 2
| diffusants | 1 | 2 | 3 | 4 | H2O | |
|---|---|---|---|---|---|---|
| media | (+, −) | (+, 0) | (0, −) | (0, 0) | (0, 0) | |
| PBS (0, 0) |
0 | 0 | 0 | 0 | 0 | |
| PBS series (pH 7.4) |
K11 sol (+, 0) |
7.2 | 7.2 | 8.2 | 9.2 | 3.3 |
| E11 sol (0, −) |
8.7 | 11.2 | 2.2 | 11.2 | 3.7 | |
| Hydrogel (+, −) |
13.2 | 14.2 | 11.6 | 14.7 | 7.5 | |
|
| ||||||
| D2O (0, 0) |
0 | 0 | 0 | 0 | 0 | |
| D2O series (pH 7.4) |
K11 sol (+, 0) |
3.9 | 4.1 | 12.0 | 3.7 | 1.7 |
| E11 sol (0, −) |
3.0 | 8.5 | 4.5 | 5.2 | 1.6 | |
| Hydrogel (+, −) |
7.7 | 7.9 | 7.2 | 8.4 | 3.4 | |
With two exceptions, hydrogels cause larger diffusion retardation than peptide solutions, i.e.
| (2) |
Considering that a hydrogel contains twice as much peptide (8 mM K11 + 8 mM E11) as a peptide solution (8 mM K11 or 8 mM E11), this is to be expected. In fact, for H2O, Re satisfies the following additive relationship in both PBS and D2O series:
| (3) |
However, such additivity does not hold for the non-solvent diffusants 1, 2, 3 and 4. The implication is that the interactions between these diffusants and peptides are quite different in hydrogels and in solutions.
As 1, 2, 3 and 4 differ in their charge status and the two peptides carry multiple charges, one might expect 2 and 3 to have larger Re than 1 and 4, because 2 and 3 carry net charge. However, this is true only in the D2O solution of E11 (anion) in which 2 (cation) has the largest Re; and in the D2O solution of K11 (cation) in which 3 (anion) has the largest Re. In all other media, Re of 2 and 3 is no higher than Re of 1 and 4. This result suggests that only in D2O solutions, do electrostatic attraction between diffusants and peptides dominate diffusion retardation. In other media, non-electrostatic interactions become important.
In addition to charge status, another difference among the four diffusants is their lipophilicity, which was quantified by Poct, the 1-octanol/water partition coefficient. Poct values of the four diffusants, measured in both PBS and H2O, are listed in Table 3.
Table 3.
Poct of 1, 2, 3 and 4 at pH 7.4.
| compounds | Poct (1-octanol/PBS) | Poct (1-octanol/water) |
|---|---|---|
| 1 (zwitter ion) | 0.84 | 0.76 |
| 2 (cation) | 4.87 | 1.92 |
| 3 (anion) | 0.02 | 0.01 |
| 4 (neutral) | 13.01 | 13.65 |
When Re is plotted against logPoct (Fig. 3), a clear pattern emerges: Re increases monotonously with logPoct in hydrogels but not in solutions. Further, in the PBS hydrogel, Re has a nearly perfect linear dependency on logPoct (R2 = 0.996).
Fig. 3.

Re vs. logPoct. R2 is the goodness of linear fitting. ■, 1; ●, 2; ★, 3; ▼, 4. Solid symbols: PBS media; Hollow symbols: D2O media.
It thus appears that both electrostatic attraction and hydrophobic interaction can retard the diffusion of a small molecule in a medium. As to which one dominates, it depends on whether other electrolytes (salts and peptides) are present. We have three scenarios in this work.
In D2O solutions, electrostatic attraction dominates due to the absence of other electrolytes. Thus, in the D2O solution of K11 (cation), 3 (anion) has the largest Re; in the D2O solution of E11 (anion), 2 (cation) has the largest Re. Further, these two cases are the only two exceptions to Eqn. (2), attesting to the prominence of electrostatic attraction in the absence of other electrolytes.
Salt is known screen electrostatic attraction and enhance hydrophobic interaction.13 Due to the added salts in PBS solutions, Re of 3 (charged, lowest Pcot) decreases, attesting the decline of electrostatic attraction; Re of 4 (neutral, highest Pcot) increases, attesting rise of hydrophobic interaction; Re of 2 (charged, 2nd highest Pcot) increases, attesting that hydrophobic interaction has become more important than electrostatic attraction. However, hydrophobic interaction does not fully dominate diffusion retardation and there is no monotonous increase of Re with logPoct in PBS solutions.
In hydrogels, electrostatic attraction is primarily between the two oppositely charged peptides as they both carry multiple charges. As a result, electrostatic attraction between peptides and diffusants is diminished to such an extent that hydrophobic interaction between peptides and diffusants dominates, leading to Re increasing monotonously with logPoct. Any residual electrostatic attraction between peptides and diffusants in the D2O hydrogel is further abolished by the addition of salts. Consequently, in the PBS hydrogel, Re has a linear dependency on logPoct.
This above analysis also sheds light on the gelation process shown in Fig. 1. As a longer range interaction, electrostatic attraction drives peptide association. In D2O, electrostatic attraction is unscreened, leading to faster gelation. In PBS, electrostatic interaction is screened, leading to slower gelation.
Finally, we wish to point out that the close structural similarity of 1, 2, 3 and 4 is likely crucial in revealing the linear relationship between Re and logPoct, as many confounding factors, such as size and conformation of the diffusants, are eliminated.
Conclusion
When small molecules of similar size are transferred from phosphate-buffered saline to a peptide hydrogel of the same pH and ionic strength, their diffusion is retarded by 10-15%. The extent of retardation has a linear dependency on logPoct. Overall, this type of mixing-induced peptide hydrogels has excellent transport properties for small molecules.
Supplementary Material
Acknowledgments
We thank the NIH for financial support (EB004416).
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/b000000x/
Notes and references
- 1.Gelain F, Bottai D, Vescovi A, Zhuang S. PLoS ONE. 2006;1:e119. doi: 10.1371/journal.pone.0000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmes TC, de Lacalle S, Su X, Liu G, Rich A, Zhang S. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6728. doi: 10.1073/pnas.97.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 4.Kretsinger JK, Haines LA, Ozbas B, Pochan DJ, Schneider JP. Biomaterials. 2005;26:5177. doi: 10.1016/j.biomaterials.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran S, Taraban MB, Trewhella J, Gryczynski I, Gryczynski Z, Yu YB. Biomacromolecules. 2010;11:1502. doi: 10.1021/bm100138m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramachandran S, Tseng Y, Yu YB. Biomacromolecules. 2005;6:1316. doi: 10.1021/bm049284w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramachandran S, Flynn P, Tseng Y, Yu YB. Chem. Mater. 2005;17:6583. [Google Scholar]
- 8.Koutsopoulos S, Unsworth LD, Nagai Y, Zhang S. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4623. doi: 10.1073/pnas.0807506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Branco MC, Pochan DJ, Wagner NJ, Schneider JP. Biomaterials. 2009;30:1339. doi: 10.1016/j.biomaterials.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branco MC, Pochan DJ, Wagner NJ, Schneider JP. Biomaterials. 2010;31:9527. doi: 10.1016/j.biomaterials.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill SC, Vonhippel PH. Anal. Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 12.Wu DH, Chen AD, Johnson CS. J. Magn. Reson., A. 1995;115:260. [Google Scholar]
- 13.Selb J, Biggs S, Renoux D, Cadau F. Adv. Chem. 1996;248:251. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.