

Abstract
Vascular cell adhesion molecule-1 (VCAM-1) regulates leukocyte migration from the blood into tissues. VCAM-1 expression is induced on endothelial cells during inflammatory bowel disease, atherosclerosis, allograft rejection, infection, and asthmatic responses. During these responses, VCAM-1 forms a scaffold for leukocyte migration. VCAM-1 also activates signals within endothelial cells resulting in the opening of an “endothelial cell gate” through which leukocytes migrate. Immediately following this migration, the endothelial cell–endothelial cell contact is re-established. VCAM-1 outside-in signals are mediated by NADPH oxidase production of reactive oxygen species and subsequently activation of matrix metalloproteinases. These signals are required for endothelial cell shape changes and leukocyte migration. In addition, VCAM-1-activated signals in endothelial cells are regulated by cytokines indicating that it is important to consider both endothelial cell adhesion molecule expression and function during inflammatory processes.
Keywords: VCAM-1 signals, Lymphocyte migration, Matrix metalloproteinases, Cytokine, Actin
1. Introduction
Leukocytes continuously circulate throughout the body in order to come in contact with antigens sequestered within tissues. To enter tissues, circulating leukocytes migrate from the blood, between vascular endothelial cells and into the tissue. During this migration, leukocytes initially bind to endothelial cells via low affinity adhesion receptors (Springer, 1994, 1995). The low affinity adhesion in combination with the force of the blood flow results in rolling of leukocytes on endothelial cells. Subsequently, adhesion molecule affinity is upregulated and leukocytes firmly adhere to the endothelium (Springer, 1994, 1995). Finally, bound leukocytes migrate between the endothelial cells and into the tissue. One of the endothelial cell adhesion molecules that mediate leukocyte binding is vascular cell adhesion molecule-1 (VCAM-1; Chan and Aruffo, 1993; Springer, 1994, 1995). The focus of this review is on VCAM-1, its signal transduction through reactive oxygen species (ROS), and endothelial cell function during lymphocyte migration.
VCAM-1 is involved in both disease pathogenesis and normal processes. VCAM-1 is expressed by lymph node endothelium (May et al., 1993; Cook-Mills et al., 1996) and is induced on endothelium in inflammatory sites. In atherosclerosis, VCAM-1 is expressed on endothelial cells in sites predisposed to atherosclerotic lesion formation suggesting an early role for VCAM-1 in lesions (Iiyama et al., 1999). In asthma, eosinophil migration into the lung is VCAM-1 dependent (Chin et al., 1997; Sagara et al., 1997). In experimental allergic encephalomyelitis, VCAM-1 is expressed by endothelium in the brain and is required for T cell infiltration into the brain (Baron et al., 1993). VCAM-1 also plays a role in melanoma tumor metastasis to the liver (Scherbarth and Orr, 1997; Vidal-Vanaclocha et al., 2000; Kishimoto et al., 2001). In normal processes, VCAM-1 is important during development since a VCAM-1 knockout is an embryonic lethal (Gurtner et al., 1995). In a conditional VCAM-1 knockout, B cell homing to the bone marrow is impaired (Koni et al., 2001; Leuker et al., 2001).
VCAM-1 structure and binding functions have been characterized. VCAM-1 is a member of the immunoglobulin super family and it binds to α4β1-integrin on leukocytes. There are two forms of VCAM-1 in mice and humans. In humans, VCAM-1 is a transmembrane protein containing either seven immunoglobulin-like domains or a protein with just domains 1–3 and 5–7 (Cybulsky et al., 1991; Osborn et al., 1992; Pepinsky et al., 1992; Needham et al., 1994; Chuluyan et al., 1995; Kilger et al., 1995). Domains 1 and 4 of VCAM-1 are the ligand binding domains. In mice, VCAM-1 exists as a seven domain transmembrane protein or as an alternatively spliced protein with domains 1–3 linked to glycophosphatidylinositol (GPI; Moy et al., 1993; Terry et al., 1993; Hahne et al., 1994; Kumar et al., 1994). The function of the GPI-linked form is not known. Different signals may be generated by the transmembrane and GPI-linked forms of VCAM-1. VCAM-1 signals in endothelial cells that are required for lymphocyte migration were identified by our studies on the transmembrane form of VCAM-1 (Matheny et al., 2000; Tudor et al., 2001). It has been reported that antibody crosslinking of VCAM-1 stimulates a calcium flux in endothelial cells but the relevance of this calcium flux to endothelial cell function is not known (Ricard et al., 1997). We have demonstrated that VCAM-1 activates the flavoprotein NADPH oxidase in endothelial cells for the generation of reactive oxygen species and that this function is required for VCAM-1-dependent lymphocyte migration (Matheny et al., 2000).
2. NADPH oxidase
NADPH oxidase has been primarily studied in professional phagocytes, such as neutrophils and macrophages as depicted in Fig. 1. NADPH oxidase is a complex of five proteins. In the inactive state, NADPH oxidase is composed of two membrane components (gp91 phox and p22 phox) and three cytosolic components (p47 phox, p67 phox and p40 phox; DeLeo and Quinn, 1996; Leusen et al., 1996). During activation, the three cytosolic components are recruited to the membrane. NADPH oxidase in the cell membrane catalyzes the production of extracellular superoxide which can dismutate to hydrogen peroxide. Hydrogen peroxide freely diffuses through cell membranes at a rapid diffusion rate (113 μm/s) that is just slightly more than that for water (110 μm/s; Mathai and Sitaramam, 1994). Upon diffusion into cells, many cells protect themselves from oxidative damage by scavenging hydrogen peroxide with catalase. Catalase catalyzes the dismutation of hydrogen peroxide to oxygen and water. In contrast to hydrogen peroxide, superoxide has a low diffusion rate across membranes. But if superoxide does enter cells, it can be scavenged by the enzyme superoxide dismutase which converts superoxide to hydrogen peroxide that can then be scavenged by catalases. Upon phagocytosis of a target, cell membrane containing NADPH oxidase is internalized and NADPH oxidase generates ROS within the phagosome. In a phagolysosome, a Haber–Weiss reaction occurs in which hydrogen peroxide and superoxide, in the presence of iron, react to generate hydroxyl radicals, hydroxyl ions and oxygen. The reactive hydroxyl radical and hydroxyl ions are utilized to destroy internalized pathogens. Finally, to regenerate NADPH utilized by the NADPH oxidase, NADP+ is converted back to NADPH by the hexose monophosphate shunt. In summary, hydrogen peroxide can diffuse readily through the membrane and into the cytoplasm as well as have extracellular and phagosome antimicrobial functions (Mathai and Sitaramam, 1994; Mastroeni et al., 2000; Vazquez-Torres et al., 2000, 2001; Vazquez-Torres and Fang, 2001).
Fig. 1.

NADPH oxidase generation of ROS in phagocytes.
3. A model for VCAM-1 signal transduction
Our working model for VCAM-1 signal transduction via NADPH oxidase is shown in Fig. 2. Adhesion of leukocyte α4-integrin to endothelial cell VCAM-1 activates signals for the stimulation of the endothelial cell NADPH oxidase (Matheny et al., 2000; Tudor et al., 2001). NADPH oxidase in the endothelial cell membrane generates low levels of extracellular ROS (1 μM; Tudor et al., 2001). Extracellular ROS activates local matrix metalloproteinases (MMPs; manuscript in preparation). MMPs are present on the surface of both leukocytes and endothelial cells. Hydrogen peroxide that diffuses through the membranes may modulate phosphatases (Brautigan, 1992; Hecht and Zick, 1992; Fialkow et al., 1997; Sommer et al., 2000) that potentially modulate actin binding proteins and actin structure within the endothelial cell. The degradation of extracellular matrix by metalloproteinases and changes in endothelial cell actin structure result in endothelial cell retraction at the site of leukocyte binding. This opens an “endothelial cell gate” so that leukocytes can migrate between the endothelial cells. In summary, VCAM-1 activation of endothelial cell NADPH oxidase is required for VCAM-1-dependent lymphocyte migration, indicating that the endothelial cell plays an active roll in this migration by leukocytes.
Fig. 2.

VCAM-1 signal transduction (working model).
4. VCAM-1 stimulation of endothelial cell NADPH oxidase during lymphocyte migration
VCAM-1 function was examined in endothelial cell lines as well as primary endothelial cells. The endothelial cell lines used in our studies are the lymph node-derived mouse endothelial cells, mHEVa and mHEVc (Cook-Mills et al., 1996). The important features of the endothelial cell lines for this review are that the endothelial cell lines bind resting lymphocytes, promote lymphocyte migration, express VCAM-1 and express the endothelial cell TGFβ receptor, endoglin (Cook-Mills et al., 1996). Resting lymphocytes refers to lymphocytes isolated from mice that have not been stimulated by addition of antigen. The endothelial cell lines as well as primary cultures of endothelial cells were used in order to determine whether VCAM-1 signals were consistent among endothelial cells (Matheny et al., 2000). The mHEVa cell line expresses the mouse transmembrane form and a relatively small amount of the GPI-linked form of VCAM-1, whereas the mHEVc cell line expresses only the transmembrane form (Tudor et al., 2000). We have shown that the VCAM-1 signals are similar in both of these endothelial cell lines and primary cultures of endothelial cells. Since the mHEVc cells express only the transmembrane VCAM-1, this suggests that the intracellular signals identified occur, at least, through the transmembrane form of VCAM-1 (Matheny et al., 2000).
First we characterized the interactions of endothelial cell lines with resting lymphocytes. B cells and T cells were isolated from spleens of BALB/c mice. B cells were isolated based on their preferential adhesion to plastic and T cells were isolated using nylon wool columns (Tudor et al., 2000). The lymphocytes were labeled with a fluorescent vital dye calcein AM and lymphocyte binding to endothelial cells examined in an adhesion assay (Tudor et al., 2000). B cells and T cells bound to the mHEVa and mHEVc cell lines, whereas there was very little to no adhesion to a control cell line (mouse melanoma cell line, B78H1; Tudor et al., 2000). Blocking antibodies were used to identify the receptors involved in lymphocyte adhesion to the endothelial cell lines. Anti-α4-integrin antibodies completely blocked lymphocyte adhesion to the endothelial cells and isotype control antibodies had no effect (Tudor et al., 2000). Lymphocyte α4-integrin bound to VCAM-1 on the mHEVa and mHEVc cell lines. Adhesion to the mHEVc cells was completely mediated by VCAM-1 (Tudor et al., 2000). For the mHEVa cells, 75% of the adhesion was mediated by VCAM-1 and the remainder was mediated by novel α4-integrin ligands (Tudor et al., 2000). We also examined several other adhesion molecules that may be involved in lymphocyte binding to the endothelial cell lines. The cell lines do not express PECAM-1, ICAM-1, P-selectin, E-selectin, MAdCAM-1 or the MECA antigens, indicating that these adhesion molecules on the endothelial cell would not be involved in lymphocyte adhesion (Cook-Mills et al., 1996; Tudor et al., 2000). Also, adhesion was not blocked using antibodies against the lymphocyte adhesion molecules LFA-1, ICAM-1, ICAM-2, L-selectin, activated β1-integrin and a combinatorial epitope of α4β7-integrin (Tudor et al., 2000). There is not a blocking antibody directed against resting murine β1-integrin. Therefore, for these cells, α4-integrin most likely functions in combination with non-active β1-integrin. In summary, α4-integrin on the lymphocyte binds to VCAM-1 on the endothelial cell lines. Since VCAM-1 was the only adhesion molecule on the mHEVc cells that was involved in lymphocyte binding to the endothelial cells, it provided us with a unique model to study VCAM-1 signaling in the absence of complications from binding of lymphocytes to other adhesion molecules. In addition, since VCAM-1 is constitutively expressed on the endothelial cell lines, it avoided complications of signals from cytokine induction of VCAM-1 expression on endothelial cells. Therefore, using this unique model, we examined lymphocyte migration and then VCAM-1 signals in endothelial cells that were required for VCAM-1-dependent lymphocyte migration.
Resting lymphocytes migrate across the endothelial cell lines. To examine the time for migration of individual lymphocytes, lymphocyte movement was followed by time lapse confocal microscopy. Individual lymphocytes at a site of migration passed between the endothelial cells and under a confluent endothelial cell monolayer in less than 2 min (Cook-Mills et al., 1996). However, this is an asynchronous process. Therefore, to quantify migration, a transwell migration assay was used (Matheny et al., 2000). Endothelial cells were grown on transwell polycarbonate membranes with 12 μm pores. The endothelial cell monolayers were confluent since they blocked diffusion of FITC-labeled albumin from the transwell upper chamber to the lower chamber. To examine lymphocyte migration across endothelial cell monolayers on these transwell membranes, lymphocytes from mouse spleens as well as splenic red blood cells were placed in the upper chamber and at several time points, lymphocytes from the lower chamber were collected and counted. Red blood cells were an internal control for monolayer integrity during the assay since red blood cells are not migratory and if the monolayer is disrupted, red blood cells would freely fall through the membrane. Exogeneous agents such as chemokines were not required for this migration. Lymphocyte migration was linear from 0 to 24 h and then there was a plateau between 24 and 48 h. Addition of either anti-α4-integrin or anti-VCAM-1 antibodies blocked migration, indicating that α4-integrin binding to VCAM-1 was required for the migration process (Matheny et al., 2000). Blocking antibodies against CD44 on the endothelial cells did not inhibit migration (Matheny et al., 2000). In summary, lymphocyte adhesion to VCAM-1 was necessary for migration across the endothelial cell lines.
Studies have described α4-integrin signals in lymphocytes but very little is known about VCAM-1 signals within endothelial cells or whether VCAM-1 signals in endothelial cells play a role in lymphocyte migration. VCAM-1 is a member of the immunogobulin super family and other members of this family stimulate intracellular signals. Therefore, it was determined whether some of the typical immunoglobulin superfamily signals such as PI3 kinase and tyrosine kinases function in VCAM-1 intracellular signals. To test this, monolayers of the endothelial cell lines were pretreated with irreversible inhibitors of PI3 kinase and tyrosine kinases. After washing, lymphocyte migration was examined. Lymphocyte migration was not affected when the mHEVa and mHEVc cell lines were pretreated with herbimycin A, an inhibitor of tyrosine kinases, or wortmanin, an inhibitor of PI3 kinase (Matheny et al., 2000). However, if lymphocytes were pretreated with either herbimycin A or wortmanin, washed, and then added to non-treated endothelial cells, there was a significant inhibition in migration across the mHEVa or the mHEVc cell lines (Matheny et al., 2000). This indicated that lymphocyte but not endothelial cell tyrosine kinases and PI3 kinase were required for α4-integrin/VCAM-1-dependent lymphocyte migration. In contrast, a recent report indicates that PI3 kinase is stimulated by VCAM-1 on smooth muscle cells (Lazaar et al., 2001). The function of VCAM-1-stimulated PI3 kinase in the smooth muscle cells has not been identified. Perhaps, VCAM-1-stimulated PI3 kinase is important for other signals such as increasing focal adhesions resulting in maintenance of vascular integrity during leukocyte migration.
VCAM-1 has also been shown to activate a calcium flux in endothelial cells but the function of the calcium flux has not been determined (Ricard et al., 1997). Calcium is known to modulate calmodulin as well as many other cell functions. Whether calmodulin was required for VCAM-1-dependent lymphocyte migration was examined by pretreating cells with the irreversible calmodulin inhibitors phenoxybenzamine or fluphenazine. Pretreatment of endothelial cells with phenoxybenzamine or fluphenazine had no inhibitory effect on migration, but pretreatment of lymphocytes with the calmodulin inhibitors blocked migration (Matheny et al., 2000). In summary, PI3 kinase, tyrosine kinase and calmodulin in lymphocytes were required for VCAM-1-dependent migration. However, these enzymes in the endothelial cells were not required for migration, indicating that for the migration event, VCAM-1 does not signal through tyrosine kinases, PI3 kinase or calmodulin.
Endothelial cells produce reactive oxygen species. These ROS modulate smooth muscle cell shape (Balligand and Cannon, 1997) and endothelial cells retract during leukocyte migration (Cook-Mills et al., 1996). Therefore, we determined whether endothelial cell-generated ROS were important during VCAM-1-dependent lymphocyte migration across endothelial cells. ROS production is catalyzed by flavoprotein containing enzymes. To inhibit flavoproteins, an irreversible inhibitor, diphenyliodonium (DPI), was used to pretreat either endothelial cells or the lymphocytes. There was a dose-dependent inhibition of lymphocyte migration after DPI pretreatment of endothelial cells (Fig. 3; Matheny et al., 2000). In contrast, pretreatment of lymphocytes had no effect on migration (Matheny et al., 2000). This indicates that flavoproteins in endothelial cells were required for migration, but flavoproteins in the lymphocytes were not required for VCAM-1-dependent migration. Since lymphocyte flavoproteins were not involved, specific reversible inhibitors could be added to the co-cultures and inhibition of lymphocyte flavoproteins would have no effect on migration. Inhibition of NADPH oxidase by apocynin gave a dose-dependent inhibition of migration (Fig. 3; Matheny et al., 2000), indicating that NADPH oxidase in the endothelial cells was important for the migration process.
Fig. 3.

Mouse mHEVa cell promotion of lymphocyte migration requires endothelial cell NADPH oxidase production of ROS. (A) mHEVa cells were pretreated for 30 min with DPI and washed before addition of lymphocytes. (B) Lymphocyte migration across mHEVa cells in the presence of apocynin. (C) Lymphocyte migration across mHEVa cells in the presence of superoxide dismutase (500 U/ml SOD) and/or catalase (5000 U/ml). There was no effect on cell viability. All inhibitors significantly blocked migration (P < 0.05). Similar data were obtained for mHEVc cell promotion of lymphocyte migration. (Modified and reproduced with permission from Matheny et al., 2000. J. Immunol. 164:6550–6559.)
Several other flavoprotein-containing enzymes that generate ROS were examined. For nitric oxide synthase, cells were treated with the inhibitors L-NMMA or L-NIO, which are arginine substrate analogues. Methoxsalin and troleandomycin were used to block cytochrome P450. Allopurinol was used to block xanthine oxidase. None of these flavoprotein inhibitors had an effect on lymphocyte migration (Matheny et al., 2000). Therefore, endothelial cell NADPH oxidase was required for VCAM-1-dependent lymphocyte migration, but endothelial cell nitric oxide synthase, xanthine oxidase and cytochrome P450 were not required.
To determine whether ROS were important for VCAM-1-dependent migration, ROS were scavenged. Superoxide was scavanged with superoxide dismutase, hydrogen peroxide was scavenged with catalase, or both of these ROS were scavenged by the combination of superoxide dismutase and catalase. Exogenous addition of these enzymes to the migration assay blocked migration (Fig. 3; Matheny et al., 2000), indicating that the generation of ROS was critical for VCAM-1-dependent lymphocyte migration.
It was determined whether endothelial cells produce ROS. To do this, confluent monolayers of endothelial cell lines were preloaded with an indicator, dihydrorhodamine 123 (DHR). This is a non-fluorescent dye that when oxidized becomes fluorescent. The endothelial cells were stimulated by a monolayer of lymphocytes. Accumulation of fluorescence in optical slices through the middle of the endothelial cells was examined by confocal microscopy. Fluorescence was quantified by summing the pixel intensities from the confocal micrographs. Lymphocytes stimulated an increase in ROS in the endothelial cells (Fig. 4; Matheny et al., 2000). Non-stimulated cells had a low background fluorescence caused by ROS generated during cell metabolism (Fig. 4; Matheny et al., 2000). Addition of the NAPDH oxidase inhibitor, apocynin, blocked this lymphocyte stimulation (Fig. 4; Matheny et al., 2000). Addition of soluable anti-VCAM-1 also blocked lymphocyte-stimulated endothelial cell generation of ROS (Fig. 4; Matheny et al., 2000). Addition of soluable anti-VCAM-1 in the absence of lymphocytes did not effect background fluorescence in the endothelial cells (Fig. 4; Matheny et al., 2000). This indicates that lymphocyte binding to VCAM-1 stimulates the generation of ROS in the endothelial cells and this generation of ROS is important for migration.
Fig. 4.

Lymphocytes stimulate the production of ROS by endothelial cell lines. Monolayers of mHEVc cells were preloaded with 1.5 μM dihydrorhodamine for 15 min at room temperature and not washed. Total of 2.5×106 lymphocytes/ml were added and rhodamine 123 fluorescence was examined by time lapse confocal microscopy at room temperature for 5–40 min. Shown are data from representative fields of an optical thin slice through the center of the mHEV cells at 30 min. mHEV cells incubated in the absence (A) or presence (B) of lymphocytes. The inset shows a representative phase contrast image of confluent monolayers of lymphocytes on top of confluent monolayers of the mHEVc cells. (C) Sum of the fluorescent pixel intensities/100 μm2 at the center of the mHEV cells at 30 min. L on the x-axis indicates lymphocytes. (*) P < 0.05 compared to the non-stimulated control. Similar data were obtained for mHEVa cells. (Modified and reproduced with permission from Matheny et al., 2000. J. Immunol. 164:6550–6559.)
VCAM-1-dependent lymphocyte stimulation of ROS did not demonstrate that VCAM-1 itself was signaling the production of ROS. In many lymphocyte interactions, there are multiple binding events. Therefore, it was conceivable that VCAM-1 binds and then some subsequent cell–cell interaction stimulates the generation of ROS. To determine whether VCAM-1 itself could activate the production ROS, VCAM-1 was crosslinked with 10 μm beads coated with anti VCAM-1 (Matheny et al., 2000). This did not cause what has been termed “frustrated phagocytosis” of an object too large for phagocytosis since these endothelial cells have been shown to be capable of phagocytosis of apoptotic lymphocytes which are approximately the size of the beads (Hess et al., 1997). Addition of anti-VCAM-1-coated beads stimulated the production of ROS (Fig. 5; Matheny et al., 2000). Apocynin blocked the anti-VCAM-1 bead stimulation, indicating that NADPH oxidase was required (Fig. 4C and D)(Matheny et al., 2000). Beads that were coated with another antibody (anti-CD44) bound to the endothelial cells but did not stimulate production of ROS (Fig. 5; Matheny et al., 2000). Therefore, VCAM-1 stimulates endothelial cell production of ROS.
Fig. 5.
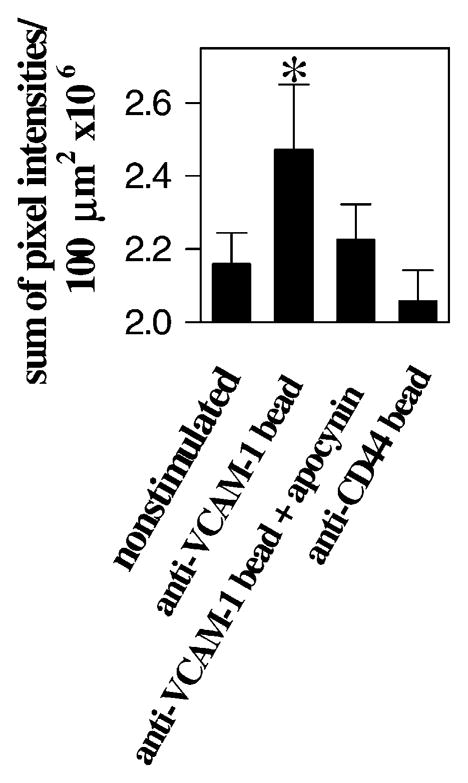
Crosslinking VCAM-1 stimulates the production of ROS by endothelial cell lines. The mHEVc cells were preloaded with 1 μM dihydrorhodamine for 15 min at room temperature and not washed. Total of 2.5 × 106 anti-VCAM-1-coated or anti-CD44-coated control beads/ml were added and rhodamine 123 fluorescence was examined by time lapse confocal microscopy at room temperature for 5–40 min. Shown are data from the sum of the fluorescent pixel intensities/100 μm2 at the center of the mHEV cells at 30 min. (*) P < 0.05 compared to the non-stimulated control. Similar data were obtained for mHEVa cells. (Modified and reproduced with permission from Matheny et al., 2000. J. Immunol. 164:6550–6559.)
Primary cultures of endothelial cells were also examined for their ability to produce ROS upon VCAM-1 stimulation. Human umbilical vein endothelial cells (HUVECs) were stimulated with TNFα overnight to induce expression of adhesion molecules including VCAM-1. VCAM-1 expression was verified by immunofluorescence and flow cytometry. To specifically activate VCAM-1 and not the other adhesion molecules on the HUVECs, VCAM-1 was crosslinked with anti-human VCAM-1-coated beads. This stimulated a significant increase in generation of ROS by HUVECs (Matheny et al., 2000). The VCAM-1 stimulation was blocked by the NADPH oxidase inhibitor apocynin (Matheny et al., 2000). Control beads coated with anti-PECAM bound to the HU-VECs but did not stimulate production of ROS (Matheny et al., 2000). Recently, van Buul et al. (2002) confirmed that crosslinking VCAM-1 activates production of ROS and they demonstrated that crosslinking ICAM-1 on human bone marrow endothelial cells does not activate the generation of ROS. In summary, VCAM-1 on endothelial cell lines and on primary endothelial cells activates NADPH oxidase for the production of ROS. These ROS are required for endothelial cell promotion of VCAM-1-dependent lymphocyte migration.
Next, we determined the level of ROS produced by VCAM-1-stimulated endothelial cells. A standard curve was generated using DHR-loaded endothelial cell lines (mHEVa and mHEVc) and exogenous hydrogen peroxide (Tudor et al., 2001). Hydrogen peroxide was used because (1) it is a metabolite of the superoxide generated by NADPH oxidase, (2) hydrogen peroxide freely diffuses through membranes (Mathai and Sitaramam, 1994), and (3) DHR within endothelial cells is oxidized by hydrogen peroxide but not by superoxide in the presence of cellular peroxidase (Henderson and Chappell, 1993). Non-stimulated cells had background levels of ROS (0.001 μM H2O2) generated from cell metabolism (Tudor et al., 2001). Addition of lymphocytes or anti-VCAM-1-coated beads stimulated endothelial cell production of approximately 1 μM hydrogen peroxide (Tudor et al., 2001). This is 50–200 times lower than that produced after stimulation of NADPH oxidase in professional phagocytes such as neutrophils and macrophages. Therefore, VCAM-1 stimulates the production of low levels of ROS for the rapid localized activation of endothelial cell function during the 2 min migration process by an individual lymphocyte.
5. Cytokine modulation of VCAM-1 signals
Cytokines are produced in most inflammatory sites with infiltrating leukocytes. Cytokines have many modulatory effects on leukocytes as well as endothelial cells. Therefore, it was determined whether cytokines alter VCAM-1-dependent lymphocyte migration and whether cytokine modulation of this migration was, at least in part, due to changes in VCAM-1 signaling. For these studies, endothelial cell lines (mHEVa and mHEVc) were treated with cytokines overnight, washed, and lymphocytes added. Transforming growth factor 1 (TGFβ1) was examined because the endothelial cell lines express the endothelial cell receptor for TGFβ, endoglin. It was first determined whether TGFβ1 pretreatment of the endothelial cell lines affected lymphocyte adhesion, VCAM-1 expression, or cell viability. TGFβ1 had no effect on adhesion of either B cells or T cells as compared to non-treated endothelial cells (Tudor et al., 2001). There was no effect of TGFβ1 on the constitutive expression of VCAM-1 by the endothelial cell lines or on endothelial cell viability (Tudor et al., 2001). In contrast, pretreatment of the endothelial cells caused a dose-dependent inhibition of B cell migration and T cell migration across the endothelial cell lines (Tudor et al., 2001).
The effect of TGFβ1 pretreatment of the endothelial cells could result from a direct effect of the cytokine on the endothelial cells. Alternatively, the cytokine may be simply bound to the surface of the endothelial cell causing a subsequent effect on lymphocytes. To test this, (1) neutralizing anti-TGFβ1 antibodies were used to block TGFβ1 function during the 18 h TGFβ1 pretreatment of the endothelial cells or (2) endothelial cells were pretreated with TGFβ1, washed and then neutralizing anti-TGFβ1 was added immediately before the addition of the lymphocytes in order to block function of TGFβ1 bound to the surface the endothelial cells. Neutralization of TGFβ1 during the TGFβ1 pretreatment of the endothelial cells blocked the inhibitory effect of TGFβ1 (Tudor et al., 2001). When the neutralizing antibody was added after TGFβ1 pretreatment, it had no blocking effect on TGFβ’s inhibition of migration (Tudor et al., 2001). Isotype control antibodies also had no affect (Tudor et al., 2001). This indicates that TGFβ1 pretreatment of endothelial cells had a direct effect on endothelial cells for the inhibition of VCAM-1-dependent lymphocyte migration.
The effect of other cytokines on VCAM-1-dependent lymphocyte migration was examined. The cytokines TNFα, IL-1, GM-CSF, and IL-6 had no effect on lymphocyte migration across the endothelial cell lines (Tudor et al., 2001). However, IFNγ pretreatment of the endothelial cell lines had a dose-dependent inhibitory effect on B cell and T cell migration across the endothelial cell line mHEVa (Tudor et al., 2001). Neutralizing antibodies blocked this effect (Tudor et al., 2001). Interferon gamma had no affect on the mHEVc cell line although both endothelial cell lines express IFNγ receptors (Tudor et al., 2001). Therefore, the mHEVc cells were a control cell line in these migration assays demonstrating a direct affect of the interferon on the mHEVa cell line. In summary, IFNγ and TGFβ1 can block VCAM-1-dependent lymphocyte migration by having a direct effect on the endothelial cells. In contrast to the inhibitory effect of IFNγ, IL-4 stimulated a two-fold increase in lymphocyte migration without affecting VCAM-1 expression (Tudor et al., 2001). Thus, IFNγ and IL-4 had opposite effects which is consistent with many opposing modulatory functions of these two cytokines.
The effect of TGFβ1 and IFNγ on VCAM-1 activation of NADPH oxidase was determined in order to define a mechanism for this cytokine inhibition of endothelial cell function during VCAM-1-dependent lymphocyte migration. TGFβ1 and IFNγ pretreatment of endothelial cells blocked activation of endothelial cell NADPH oxidase whether the endothelial cells were stimulated with lymphocytes or anti-VCAM-1 beads (Tudor et al., 2001). TGFβ1 and IFNγ in the absence of VCAM-1 stimulation had no effect on background ROS generated by endothelial cell metabolism (Tudor et al., 2001). In summary, TGFβ1 and IFNγ did not affect lymphocyte adhesion, constitutive VCAM-1 expression, or endothelial cell viability. Pretreatment of the endothelial cell lines with these cytokines did block VCAM-1-dependent lymphocyte migration. At least one mechanism for the effect of these cytokines was the inhibition of VCAM-1-stimulated endothelial cell generation of ROS. This indicates that these cytokines can have a functional effect on adhesion molecule signaling within endothelial cells.
Cytokine regulation of endothelial cell adhesion molecule function is important to consider when examining the migration of leukocytes into tissues. The adhesion molecule repertoire involved in leukocyte infiltration into a tissue is defined by the combination of functional adhesion molecules and not simply by the adhesion molecules expressed at the site. Furthermore, these studies emphasize that the endothelial cell function in this migration process is important during lymphocyte migration and that endothelial cell adhesion molecules are not simply a scaffold on which lymphocytes migrate.
6. VCAM-1-stimulated endothelial cell ROS activate matrix metalloproteinases (MMPs)
We have demonstrated that VCAM-1 stimulates endothelial cell NADPH oxidase-catalyzed production of ROS and that these ROS are required for VCAM-1-dependent lymphocyte migration (Fig. 2). Next, we determined a mechanism for ROS function in this process. Matrix metalloproteinases (MMPs) were examined because (1) purified MMPs are activated by low levels of ROS (Rajagopalan et al., 1996), (2) MMPs are expressed by both endothelial cells and leukocytes, and (3) it has been reported that T cell binding to VCAM-1 on cytokine-stimulated endothelial cells activates T cell matrix metalloproteinase 2 (MMP2; Romanic and Madri, 1994; Madri et al., 1996). Romanic and Madri (1994) also reported that inhibition of MMP2 with tissue inhibitor of metalloproteinase (TIMP-2) blocks VCAM-1-mediated T cell migration. However, they did not show the mechanism for activation of T cell MMP2. It was possible that VCAM-1-stimulated production of low levels of ROS could activate MMPs on both the endothelial cell and the leukocyte at localized interactions. This MMP activation and degradation of extracellular matrix could then cause localized retraction of the endothelial cells to allow leukocytes to migrate out of the vasculature. We have determined that the VCAM-1-stimulated endothelial cell ROS were required for activation of MMP2 and MMP9 during lymphocyte migration (manuscript in preparation). In summary, lymphocyte α4-integrin interaction with VCAM-1 activates endothelial cell NADPH oxidase for the production of ROS. These ROS activate MMPs (Fig. 2).
7. VCAM-1-stimulated generation of ROS causes localized endothelial cell actin changes
During VCAM-1-dependent lymphocyte migration, the endothelial cell retracts at the localized site of leukocyte binding within 2 min and then closes again (Cook-Mills et al., 1996). An endothelial cell shape change likely involves a cytoskeletal change. Therefore, we examined endothelial cell actin structure after VCAM-1 stimulation. Endothelial cell monolayers were stimulated with lymphocytes for 5 min and fixed. Actin was labeled with TRITC-phalloidin. Fluorescence was examined in optical slices of the cells by confocal microscopy. There was a coalescence of endothelial cell actin just below the site of lymphocyte binding (Fig. 6C; Tudor et al., 2001). There was no change in actin structure in the center of the endothelial cell (Tudor et al., 2001). Lymphocyte binding stimulated endothelial cell actin coalescence for about 35% of the bound lymphocytes (Fig. 6A; Tudor et al., 2001). Addition of the NADPH oxidase inhibitor apocynin blocked this activation (Fig. 6A; Tudor et al., 2001). Addition of the actin polymerization inhibitor, cytochalasin D, also blocked this activation (Fig. 6A; Tudor et al., 2001). Since it was possible that lymphocytes bound to VCAM-1 and then some subsequent interaction stimulated the actin structural change, we determined whether crosslinking VCAM-1 stimulated the localized endothelial cell actin structural change. Crosslinking VCAM-1 with anti-VCAM-1-coated beads activated endothelial cell actin coalescence immediately under the location of the beads (Fig. 6D; Tudor et al., 2001). Sixty-five percent of the bound beads activated actin coalescence (Fig. 6B; Tudor et al., 2001). Inhibition of NADPH oxidase with apocynin blocked VCAM-1 activation of actin coalescence to the same extent as the actin polymerization inhibitor cytochalasin D (Fig. 6B; Tudor et al., 2001). Therefore, leukocyte α4-integrin binds to VCAM-1. VCAM-1 activates endothelial cell NADPH oxidase for the production of ROS (1 μM). These low levels of ROS activate endothelial cell MMPs and localized endothelial cell cytoskeletal changes. Recent preliminary data also indicate that these VCAM-1-stimulated ROS modulate intracellular endothelial cell enzyme activities which may regulate endothelial cell actin structure (Fig. 2).
Fig. 6.

Lymphocytes and anti-VCAM-1-coated beads stimulate apocynin-inhibitable actin coalescence in endothelial cells at the site of contact. Lymphocytes and anti-VCAM-1-coated beads (2.5 × 106/ml) were incubated in the presence or absence of 2.5 mM apocynin or 1 μM cytochalasin D with confluent monolayers of mHEVc cells for 5 min. The cells were fixed, permeabilized, labeled with TRITC-phalloidin, and examined by confocal microscopy. (A) Percentage of lymphocytes or (C) % of anti-VCAM-1-coated beads with endothelial cell actin coalescence at the site of contact with the lymphocytes or beads, respectively. L indicates lymphocytes. (*) P < 0.05 compared to controls (lymphocyte-stimulated or bead-stimulated mHEV cells). Micrographs shown are representative optical thin slices of TRITC-phalloidin labeled actin in the endothelial cell at the site of lymphocyte (C) or anti-VCAM-1-coated bead (D) contact. (*) in panel C, indicates the location of the center of a lymphocyte, above the thin slice shown, as determined by labeling with FITC-conjugated anti-CD45 (data not shown). There was no effect of bead or lymphocyte binding on the actin structure at the center of the mHEV cell (data not shown). Similar data were obtained for mHEVa cells. (Modified and reproduced with permission from Matheny et al., 2000. J. Immunol. 164:6550–6559.)
8. Conclusions and future directions
In summary, the endothelial cell plays an important role in VCAM-1-dependent lymphocyte migration and endothelial cells are not simply a scaffold on which leukocytes crawl. VCAM-1 stimulates endothelial cell NADPH oxidase activity that is required for this lymphocyte migration (Matheny et al., 2000). The cytokines TGFβ1 and IFNγ block VCAM-1-dependent lymphocyte migration at least by inhibiting VCAM-1 activation of endothelial cell NADPH oxidase (Tudor et al., 2001). ROS from the VCAM-1 signaling pathway activate endothelial cell MMPs that can degrade ECM and membrane proteins (manuscript in preparation). The ROS from the VCAM-1 signaling pathway also activate localized endothelial cell actin restructuring (Matheny et al., 2000). Therefore, the endothelial cells retract, opening an “endothelial cell gate” and allowing lymphocytes to migrate from the blood and into tissues. Future studies will delineate the function of the cytoplasmic domain of VCAM-1, intermediates in the VCAM-1 signaling pathway and VCAM-1 signals in vivo.
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01 HL68171, R01AI40640, R29AI34585). We thank Tracy Deem and Dr. E. Charles Snow for review of this manuscript.
References
- Balligand JL, Cannon PJ. Nitric oxide synthases and cardiac muscle: autocrine and paracrine influences. Arterioscler Thrombo Vascular Biol. 1997;17:1846–1858. doi: 10.1161/01.atv.17.10.1846. [DOI] [PubMed] [Google Scholar]
- Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigan DL. Great expectations: protein tyrosine phosphatases in cell regulation. Biochim Biophys Acta. 1992;1114:63–77. doi: 10.1016/0304-419x(92)90007-l. [DOI] [PubMed] [Google Scholar]
- Chan PY, Aruffo A. VLA-4 integrin mediates lymphocyte migration on the inducible endothelial cell ligand VCAM-1 and the extracellular matrix ligand fibronectin. J Biol Chem. 1993;268:24655–24664. [PubMed] [Google Scholar]
- Chin JE, Hatfield CA, Winterrowd GE, Brashler JR, Vonderfecht SL, Fidler SF, Griffin RL, Kolbasa KP, Krzesicki RF, Sly LM, Staite ND, Richards IM. Airway recruitment of leukocytes in mice is dependent on alpha4-integrins and vascular cell adhesion molecule-1. Am J Physiol. 1997;272:L219–L229. doi: 10.1152/ajplung.1997.272.2.L219. [DOI] [PubMed] [Google Scholar]
- Chuluyan HE, Osborn L, Lobb R, Issekutz AC. Domains 1 and 4 of vascular cell adhesion molecule-1 (CD106) both support very late activation antigen-4 (CD49d/CD29)-dependent monocyte transendothelial migration. J Immunol. 1995;155:3135–3144. [PubMed] [Google Scholar]
- Cook-Mills JM, Gallagher JS, Feldbush TL. Isolation and characterization of high endothelial cell lines derived from mouse lymph nodes. In Vitro Cell Dev Biol. 1996;32:167–177. doi: 10.1007/BF02723682. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI, Fries JWU, Williams AJ, Sultan P, Eddy R, Byers M, Shows T, Gimbrone MA, Jr, Collins T. Gene structure, chromosomal location, and basis for alterative mRNA splicing of the human VCAM1 gene. Proc Natl Acad Sci USA. 1991;88:7859–7863. doi: 10.1073/pnas.88.17.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leuko Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- Fialkow L, Chan CK, Downey GP. Inhibition of CD45 during neutrophil activation. J Immunol. 1997;158:5409–5417. [PubMed] [Google Scholar]
- Gurtner GC, Davis V, Li H, McCoy MJ, Sharpe A, Cybulsky MI. Targeted disruption of the murine VCAM1 gene: essential role of VCAM-1 in chorioallantoic fusion and placentation. Genes Develop. 1995;9:1–14. doi: 10.1101/gad.9.1.1. [DOI] [PubMed] [Google Scholar]
- Hahne M, Lenter M, Jäger U, Vestweber D. A novel soluble form of mouse VCAM-1 is generated from a glycolipid-anchored splicing variant. Eur J Immunol. 1994;24:421–428. doi: 10.1002/eji.1830240223. [DOI] [PubMed] [Google Scholar]
- Hecht D, Zick Y. Selective inhibition of protein tyrosine phosphatase activities by H2O2 and vanadate in vitro. Biochem Biophys Res Commun. 1992;188:773–779. doi: 10.1016/0006-291x(92)91123-8. [DOI] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB. Dihydrorhodamine 123: a fluorescent probe for superoxide generation? Eur J Biochem. 1993;217:973–980. doi: 10.1111/j.1432-1033.1993.tb18328.x. [DOI] [PubMed] [Google Scholar]
- Hess KL, Tudor KSRS, Johnson JD, Osati-Ashtiani F, Askew DS, Cook-Mills JM. Human and murine high endothelial venule cells phagocytose apoptotic leukocytes. Exp Cell Res. 1997;236:404–411. doi: 10.1006/excr.1997.3745. [DOI] [PubMed] [Google Scholar]
- Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, Cybulsky MI. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199–207. doi: 10.1161/01.res.85.2.199. [DOI] [PubMed] [Google Scholar]
- Kilger G, Needham LA, Nielsen PJ, Clements J, Vestweber D, Holzmann B. Differential regulation of α4 integrin-dependent binding to domains 1 and 4 of vascular cell adhesion molecule-1. J Biol Chem. 1995;270:5979–5984. doi: 10.1074/jbc.270.11.5979. [DOI] [PubMed] [Google Scholar]
- Kishimoto H, Sasahara K, Yamazaki K, Nagata T, Uotani H, Yamashita I, Tauchi K, Tsukada K. Obstructive jaundice facilitates hepatic metastasis of B16F1 mouse melanoma cells: participation of increased VCAM-1 expression in the liver. Oncol Rep. 2001;8:575–578. doi: 10.3892/or.8.3.575. [DOI] [PubMed] [Google Scholar]
- Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar AG, Dai XY, Kozak CA, Mims MP, Gotto AM, Ballantyne CM. Murine VCAM-1: molecular cloning, mapping, and analysis of a truncated form. J Immunol. 1994;153:4088–4098. [PubMed] [Google Scholar]
- Lazaar AL, Krymskaya VP, Das SKP. VCAM-1 activates phosphatidylinosital 3-kinase and induces p120cbl phosphorylation in human airway smooth muscle cells. J Immunol. 2001;166:155–161. doi: 10.4049/jimmunol.166.1.155. [DOI] [PubMed] [Google Scholar]
- Leuker CE, Labow M, Muller W, Wagner N. Neonatally induced inactivation of the vascular cell adhesion molecule 1 gene impairs B cell localization and T cell-dependent humoral immune response. J Exp Med. 2001;193:755–768. doi: 10.1084/jem.193.6.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leusen JH, Verhoeven AJ, Roos D. Interactions between the components of the human NADPH oxidase: intrigues in the phox family. J Lab Clin Med. 1996;128:461–476. doi: 10.1016/s0022-2143(96)90043-8. [DOI] [PubMed] [Google Scholar]
- Madri JA, Graesser D, Haas T. The roles of adhesion molecules and proteinases in lymphocyte transendothelial migration. Biochem Cell Biol. 1996;74:749–757. doi: 10.1139/o96-082. [DOI] [PubMed] [Google Scholar]
- Mastroeni P, Vazquez-Torres A, Fang FC, Xu Y, Khan S, Hormaeche CE, Dougan G. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II Effects on microbial proliferation and host survival in vivo. J Exp Med. 2000;192:237–248. doi: 10.1084/jem.192.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathai JC, Sitaramam V. Stretch sensitivity of transmembrane mobility of hydrogen peroxide through voids in the bilayer: role of cardiolipin. J Biol Chem. 1994;269:17784–17793. [PubMed] [Google Scholar]
- Matheny HE, Deem TL, Cook-Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J Immunol. 2000;164:6550–6559. doi: 10.4049/jimmunol.164.12.6550. [DOI] [PubMed] [Google Scholar]
- May MJ, Entwistle G, Humphries MJ, Ager A. VCAM-1 is a CS1 peptide-inhibitable adhesion molecule expressed by lymph node high endothelium. J Cell Sci. 1993;106:109–119. doi: 10.1242/jcs.106.1.109. [DOI] [PubMed] [Google Scholar]
- Moy P, Lobb R, Tizard R, Olson D, Hession C. Cloning of an inflammation-specific phosphatidyl inositol-linked form of murine vascular cell adhesion molecule-1. J Biol Chem. 1993;268:8835–8841. [PubMed] [Google Scholar]
- Needham LA, Van DS, Pigott R, Edwards RM, Shepherd M, Hemingway I, Jack L, Clements JM. Activation dependent and independent VLA-4 binding sites on vascular cell adhesion molecule-1. Cell Adhesion Commun. 1994;2:87–99. doi: 10.3109/15419069409004429. [DOI] [PubMed] [Google Scholar]
- Osborn L, Vassallo C, Benjamin CD. Activated endothelium binds lymphocytes through a novel binding site in the alternately spliced domain of vascular cell adhesion molecule-1. J Exp Med. 1992;176:99–107. doi: 10.1084/jem.176.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepinsky B, Hession C, Chen LL, Moy P, Burkly L, Jakubowski A, Chow EP, Benjamin C, Chi-Rosso G, Luhowskyj S, Lobb R. Structure/function studies on vascular cell adhesion molecule-1. J Biol Chem. 1992;267:17820–17826. [PubMed] [Google Scholar]
- Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard I, Payet MD, Dupuis G. Clustering the adhesion molecules VLA-4 (CD49d/CD29) in Jurkat T cells or VCAM-1 (CD106) in endothelial (ECV 304) cells activates the phosphoinositide pathway and triggers Ca2+ mobilization. Eur J Immunol. 1997;27:1530–1538. doi: 10.1002/eji.1830270632. [DOI] [PubMed] [Google Scholar]
- Romanic AM, Madri JA. The induction of 72-kD gelatinase in T cells upon adhesion to endothelial cells is VCAM-1 dependent. J Cell Biol. 1994;125:1165–1178. doi: 10.1083/jcb.125.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara H, Matsuda H, Wada N, Yagita H, Fukuda T, Okumura K, Makino S, Ra C. A monoclonal antibody against very late activation antigen-4 inhibits eosinophil accumulation and late asthmatic response in a guinea pig model of asthma. Intl Arch Allergy Immunol. 1997;112:287–294. doi: 10.1159/000237468. [DOI] [PubMed] [Google Scholar]
- Scherbarth S, Orr FW. Intravital videomicroscopic evidence for regulation of metastasis by the hepatic microvasculature: effects of interleukin-1 alpha on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res. 1997;57:4105–4110. [PubMed] [Google Scholar]
- Sommer D, Fakata KL, Swanson SA, Stemmer PM. Modulation of the phosphatase activity of calcineurin by oxidants and antioxidants in vitro. Eur J Biochem. 2000;267:2312–2322. doi: 10.1046/j.1432-1327.2000.01240.x. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Ann Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- Terry RW, Kwee L, Levine JF, Labow MA. Cytokine induction of an alternatively spliced murine vascular cell adhesion molecule (VCAM) mRNA encoding a glycosylphosphatidylinositol-anchored VCAM protein. Proc Natl Acad Sci USA. 1993;90:5919–5923. doi: 10.1073/pnas.90.13.5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor KSRS, Deem TL, Cook-Mills JM. Novel α4-integrin ligands on an endothelial cell line. Biochem Cell Biol. 2000;78:99–113. [PubMed] [Google Scholar]
- Tudor KSRS, Hess KL, Cook-Mills JM. Cytokines modulate endothelial cell intracellular signal transduction required for VCAM-1-dependent lymphocyte transendothelial migration. Cytokine. 2001;15:196–211. doi: 10.1006/cyto.2001.0922. [DOI] [PubMed] [Google Scholar]
- van Buul JD, Voermans C, van den Berg V, Anthony EC, Mul FPJ, van Wetering S, van der Schoot EC, Hordijk PL. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J Immunol. 2002;168:588–596. doi: 10.4049/jimmunol.168.2.588. [DOI] [PubMed] [Google Scholar]
- Vazquez-Torres A, Fang FC. Oxygen-dependent anti-Salmonella activity of macrophages. Trends Microbiol. 2001;9:29–33. doi: 10.1016/s0966-842x(00)01897-7. [DOI] [PubMed] [Google Scholar]
- Vazquez-Torres A, Jones-Carson J, Mastroeni P, Ischiropoulos H, Fang FC. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I Effects on microbial killing by activated peritoneal macrophages in vitro. J Exp Med. 2000;192:227–236. doi: 10.1084/jem.192.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Torres A, Fantuzzi G, Edwards CK, III, Dinarello CA, Fang FC. Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in tumor necrosis factor p55 receptor-deficient macrophages. Proc Natl Acad Sci USA. 2001;98:2561–2565. doi: 10.1073/pnas.041618998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Vanaclocha F, Fantuzzi G, Mendoza L, Fuentes AM, Anasagasti MJ, Martin J, Carrascal T, Walsh P, Reznikov LL, Kim SH, Novick D, Rubinstein M, Dinarello CA. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci USA. 2000;97:734–739. doi: 10.1073/pnas.97.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]