

Abstract
The construction of two libraries of triazole-containing isothiazolidine 1,1-dioxides is reported utilizing either a one-pot click/aza-Michael or click/OACC esterification protocol. One core dihydroisothiazole 1,1-dioxide scaffold was prepared rapidly on multi-gram scale via RCM and was subjected to a one-pot multi-component click/aza-Michael protocol with an array of amines and azides for the generation of a 180-member triazole-containing isothiazolidine 1,1-dioxide library. Alternatively, three daughter scaffolds were generated via the aza-Michael of three amino alcohols, followed by a one-pot, multi-component click/esterification protocol utilizing a ROMP-derived coupling reagent, oligomeric alkyl carbodiimide (OACC) to generate a 41-member library of triazole-containing isothiazole 1,1-dioxides.
Keywords: Triazole-containing isothiazolidine 1, 1-dioxide, Sultams, One-pot, aza-Michael, RCM, ROMP
1. Introduction
The need for discovery of new pharmaceutical leads and small molecule probes has led to efforts focusing on the advancement of current high-throughput screening and the corresponding small molecule screening collections. This effort has been driven by the development and emergence of methods, protocols and technologies to access diverse collections of small molecules in a rapid fashion.1 Sultams (cyclic sulfonamide analogues) have surfaced in recent years as important targets in drug discovery due to their extensive chemical and biological profiles.2 In this regard, β-amino sultams and their corresponding sulfonate analogues are a relatively new chemotype that has shown interesting biological properties. Such reports include the inhibition of HIV-1 replication and antibacterial activity (Figure 1).3
Figure 1.

Biologically active β-amino sultams and sulfonates.
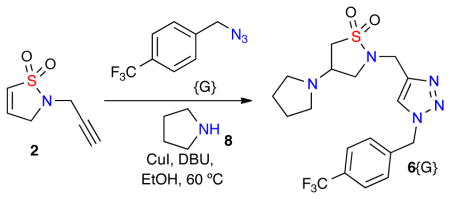
Building on these aforementioned reports, it was envisioned that a library of β-amino sultams, specifically triazole-containing isothiazolidine 1,1-dioxides, could be rapidly generated via a facile one-pot click/aza-Michael diversification protocol of RCM-derived dihydroisothiazole 1,1-dioxide core scaffold 2 with a series of amines, azides and acids (Figure 2).
Figure 2.

One-Pot, multi-component synthesis of triazole-containing isothiazole 1,1-dioxides via orthogonal reactions.
Utilizing RCM,4 simple 5-, 6- and 7-membered sultam derivatives from the corresponding allyl and vinyl sulfonamides were readily accessed.5 In the cases of RCM with vinyl sulfonamides, the corresponding sultam retains the α,β-unsaturated functionality incorporated within the cycle for diversification utilizing an aza-Michael protocol. Aza-Michael reactions are efficient pathways that, historically, have been broadly utilized to access a variety of heterocycles.6 Specifically, the hetero-Michael reaction has been utilized as an efficient cyclization protocol to access a variety of sultam motifs leading to the proposed triazole-containing isothiazolidine 1,1-dioxide library.7
2. Results and Discussion
The construction of isothiazolidine 1,1-dioxides libraries was envisioned by the diversification of core dihydroisothiazole 1,1-dioxide core scaffolds via a one-pot, multi-component protocol pairing an aza-Michael diversification reaction with other orthogonal reaction pathways. To this effect, the corresponding core scaffold 2-(prop-2-yn-1-yl)-2,3-dihydroisothiazole 1,1-dioxide 2 was rapidly generated on multi-gram scale via a 3-step sulfonylation, RCM, propargylation protocol (Scheme 1).9,10 Notably, the addition of metathesis catalyst [(IMesH2)(PCy3)(Cl)2Ru=CHPh; cat-B],9,10 in 5 equal portions of 0.5 mol% (total 2.5 mol%) every 30 minutes was key to maintaining the observed high conversation of the RCM cyclization.
Scheme 1.

Gram synthesis of core 2-(prop-2-yn-1-yl)-2,3-dihydroisothiazole 1,1-dioxide 2 via RCM.
With the desired core sultam 2 in hand, initial efforts focused on the diversification of the core via solely an aza-Michael or click reaction.8 After successful diversification of 2 utilizing either reactions in high yield, the combination of both reactions in a one-pot protocol was investigated. In this regard, the one-pot click/aza-Michael protocol with azide {G} and pyrrolidine 6 was investigated (Table 1). Initial efforts combined both reaction conditions into the same pot (Table 1, entry 1) yielding the desired product in 62% yield. This yield was improved to 96% after increasing the CuI catalyst load to 30 mol% (Table 1, entry 2), while additional optimization led to the use of lower equivalents of amine without affecting yield (Table 1, entry 5).
Table 1.
Optimization of one-pot click/aza-Michael reaction conditions
 | |||||
|---|---|---|---|---|---|
| entrya | azide {G} (equiv.) | amine 6 (equiv.) | CuI (mol %) | DBU (mol%) | yield (%) |
| 1 | 2 | 2 | 10 mol% | 10 mol% | 62 % |
| 2 | 2 | 2 | 30 mol% | 10 mol% | 96 % |
| 3 | 1 | 1 | 30 mol% | 10 mol% | 78 % |
| 4 | 2 | 1.2 | 30 mol % | 10 mol% | 95 % |
| 5 | 2 | 1.2 | 30 mol% | 10 mol% | 96 % |
Reactions carried out utilizing 2 (50 mg, 0.32 mmol, 1 equiv.) in 0.5 M EtOH at 60 °C for 12 hrs.
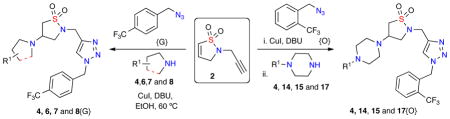
With these optimized conditions in hand, a validation library was investigated for the diversification of dihydroisothiazole 1,1-dioxide 2 with a variety of 2° amine nucleophiles (Table 2). Reactions were carried out in 1-dram vials using reaction blocks, whereby crude reaction mixtures were diluted in EtOAc, filtered through a SiO2 SPE and subjected to a QC check, followed by purification via automated mass-directed LCMS.
Table 2.
Library prototype utilizing dihydroisothiazole 1,1-dioxide 2.
 | |||||||
|---|---|---|---|---|---|---|---|
| entrya | yield | final purityc | Mass | entryb | yield | final purityc | mass |
| 4{G} | 52 % | 99 % | 89.3 mg | 4{O} | 43 % | 100 % | 22.9 mg |
| 6{G} | 45 % | 99 % | 75.4 mg | 14{O} | 41 % | 98 % | 22.3 mg |
| 7{G} | 42 % | 92 % | 56.2 mg | 15{O} | 40 % | 97 % | 19.4 mg |
| 8{G} | 47 % | 99 % | 63.9 mg | 16{O} | 43 % | 98 % | 22.3 mg |
Reaction conditions: Dihydroisothiazole 1,1-dioxide 2 (50 mg, 1 equiv.), azide (2 equiv.), amine (1.2 equiv.), CuI (30 mol%), DBU (10 mol%), dry EtOH (0.5 M), 60 °C, 12 hrs.
Reaction conditions: Dihydroisothiazole 1,1-dioxide 2 (20 mg, 1 equiv.), azide (2 equiv.), amine (1.2 equiv.), CuI (30 mol%), DBU (10 mol%), dry EtOH (0.5 M), 60 °C, 12 hrs.
Purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy). Purity was determined by HPLC with peak area (UV) at 214 nm and % rounded up to nearest 1%.
With the successful synthesis of the 8-member validation library, two libraries A and B were proposed for the synthesis of 180-triazole-containing isothiazole 1,1-dioxides derivatives via the diversification of dihydroisothiazole 1,1-dioxide 2 utilizing amines (3–17) and azides {A–R} (Figure 2).
Figure 2.

Amine (3–17) and Azide {A–R} library building blocks.
Library Design
For all three libraries A, B and C, a full matrix library was designed using in-silico analysis, literature precedence, and observed synthetic results.11 A virtual library incorporating all possible building block combinations of azides, 2° amines and acids (library C) was constructed for each scaffold (2, 18, 19 and 20) (Figure 3). Physico-chemical property filters were applied, guiding the elimination of undesirable building blocks that led to products with undesirable in-silico properties.12 These metric filters included standard Lipinski Rule of 5 parameters (molecular weight <500, ClogP <5.0, number of H-acceptors <10, and number of H-donors <5), in addition to consideration of the number of rotatable bonds (<5) and polar surface area. Absorption, distribution, metabolism and excretion (ADME) properties were calculated along with diversity analysis using standard H-aware 3D BCUT descriptors comparing against the MLSMR screening set (ca. 7/2010; ~330,000 unique chemical structures). Guided by this library design analysis, the corresponding amines 3–17, azides {A–R} and acids 21–25 and sultams (2, 18–20) [Figure 2 & 3] were chosen to generate 234 proposed sultams (libraries A, B and C}.
Figure 3.

Library C: Core Scaffolds 18, 19 and 20, azide building blocks M–R and acid building blocks {21–25}.
Library A and B
Utilizing the optimized conditions, library A (132-member) was designed and generated utilizing azides A–L with amines 3–13 via a one-pot click/aza-Michael transformation. Library B (48-member) was also generated utilizing azides A–L with piperazines 14–17 via a sequential 2-step click/aza-Michael protocol, instead of the previously used one-pot method. This 2-step sequence was necessary to efficiently remove the Cu catalyst from the crude material without the need of an aqueous work-up due to increased affinity for the SiO2 SPE of the corresponding final compounds bearing both a triazole and piperazine moiety. Overall, all 180 triazole-containing isothiazole-1,1-dioxide members of library A and B were successfully generated, with 167 out of the 180 compounds possessing >90% final purity after purification by automated mass-directed LCMS (Graph 1).13
Graph 1.


Library A and B: Representative library members demonstrating final mass, purity and overall yield.
Within the 132-member library A is a unique set of 12 bis-triazole-containing isothiazole 1,1-dioxides 13{A–L} which were generated through a bis-click/aza-Michael due to the use of N-methyl propargyl amine 13 as the Michael component in the presence of 2 equivalents of the corresponding azide (Scheme 2).
Scheme 2.

One-Pot bis-click/aza-Michael to produce compounds 13{A–L}.
Library C
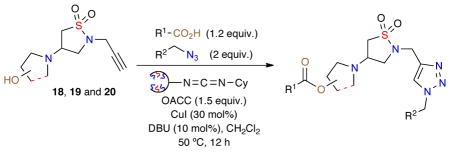
Building on the synthesis of 180-member triazole-containing isothiazole 1,1-dioxides, library C, a 90-member compound set of derivatives was investigated. This involved the synthesis of three daughter scaffolds (18, 19 and 20, Figure 3) synthesized from dihydroisothiazole 1,1-dioxide 2, which underwent diversification via a one-pot procedure click/oligomeric alkyl cyclohexylcarbodiimide (OACC) esterification to generate the desired 90-membered library (Scheme 3).
Scheme 3.

Library C; One-pot, click/OACC esterification of isothiazole 1,1-dioxides 18, 19 and 20.
Key to the combination of these orthogonal reaction pathways is the utilization of an immobilized, soluble ring-opening metathesis polymerization (ROMP)-derived coupling reagent OACC.14 This high load reagent allows for efficient coupling of a variety of acids with 18, 19 or 20, followed by facile removal via a simple precipitation/filtration protocol.
Initially, the three daughter isothiazole 1,1-dioxide scaffolds 18, 19 and 20 were generated via aza-Michael of the corresponding amino alcohols with dihydroisothiazole 1,1-dioxide 2 in an efficient multi-gram synthesis. With these core isothiazole-1,1-dioxide (18, 19 and 20) scaffolds prepared, a 90-member library was designed utilizing a combination of 18, 19 and 20 with azides M–P and acids {21–25} (Figure 3).
Using the aforementioned method, a one-pot click/OACC esterification protocol for the synthesis of library C was achieved utilizing 1.2 equivalents of acid {21–25}, azide (2 equiv.), OACC (1.5 equiv.), CuI (30 mol%), DBU (10 mol%) and anhydrous CH2Cl2 (Table 3). Reactions were carried out at 50 °C for 12 h, whereby upon completion the crude reactions were diluted, filtered and concentrated. Utilizing these conditions an initial validation set of isothiazolidine-1,1-dioxides was investigated to evaluate the reaction conditions (Table 3).
Table 3.
One-Pot, click/OACC esterification.
 | ||||
|---|---|---|---|---|
| entry | scaffold | azide | acid | yield (%) |
| 1 | 18 | P | 21 | 48 |
| 2 | 18 | M | 22 | 52 |
| 3 | 18 | M | 23 | 47 |
| 4 | 18 | P | 23 | 60 |
| 5 | 19 | M | 22 | 40 |
| 6 | 19 | M | 23 | 42 |
| 7 | 19 | M | 24 | 43 |
Reaction conditions: Isothiazole 1,1-dioxide (40 mg, 1 equiv.), acid (1.2 equiv.), azide (2 equiv.), OACC (1.5 equiv.), CuI (30 mol%), DBU (10 mol%), dry CH2Cl2 (0.2 M), 50 °C, 12 h.
Isolated yields after standard column chromatography (EtOAc, Rf = 0.3–0.6).
Upon completion, the validation set was diluted in EtOAc to precipitate the spent oligomer, followed by filtration through a SiO2 SPE and concentration. All samples were purified by column chromatography, characterized and then submitted to purification by reverse-phase automated mass-directed LCMS.15 Despite their stability when isolated after standard chromatography, it was observed that esters generated with acids 21 and 23 underwent complete hydrolysis during automated mass-directed LCMS. Analysis indicated that after successful purification, hydrolysis occurred during the final step of removal of the solvent utilized (CH3CN, H2O, DMSO and NH4OH). Due to this observation, library C was redesigned removing acids 21 and 23 to propose a 54-member library (Scheme 5).
Scheme 5.

Generation of Library C via an One-pot click/OACC esterification.
Under these conditions, 41 out of 54 compounds were successfully isolated >90% purity after purification by automated mass-directed LCMS. Crude reaction analysis indicated that all 54 reactions worked, however it is proposed that the failed reactions and low yields observed were due to hydrolysis of the desired product in the final stages of purification.
Conclusion
In conclusion, three libraries of triazole-containing isothiazolidine 1,1-dioxides were prepared utilizing either a one-pot click/aza-Michael or click/esterification protocol for utilization in high throughput screening (HTS) collections. One core dihydroisothiazole 1,1-dioxide scaffold was prepared on multi-gram scale via RCM and rapidly diversified via a one-pot multi-component click/aza-Michael protocol to generate a 180-triazole-containing isothiazole 1,1-dioxide library (A and B). All 180 compounds were successfully generated, with 167 possessing >90% final purity after purification by automated mass-directed LCMS. Building on this success, a 41-member library C of triazole-containing isothiazole 1,1-dioxide library was prepared via a one-pot, click/OACC esterification utilizing the soluble oligomeric coupling reagent OACC. Taken collectively, a total of 208 sultams were successfully generated (89% success rate) with purities >90%. This screening set of sultams represent a diverse motif not currently reported in the literature and has been submitted for biological evaluation via high-throughput screening.
Experimental Section
General procedure A for the synthesis of library I via a one-pot click/aza-Michael protocol. To a 1-dram vial containing 2-(prop-2-yn-1-yl)-2,3-dihydroisothiazole 1,1-dioxide 2 (50 mg, 0.32 mmol, 1 equiv.) was added CuI (18.2 mg, 30 mol%), DBU (5 μL, 10 mol%), dry EtOH (0.64 mL, 0.5 M), amine (0.38 mmol, 1.2 equiv.) and azide (0.64 mmol, 2 equiv.). The reaction was heated at 60 °C on a reaction block for for 12 hrs, after which time the reactions were cooled, filtered through SiO2 SPE into pre-weighed bar-coded vials, washed with eluent (2 mL, EtOAc:MeOH 95:5) and concentrated under reduced pressure. The crude reaction was concentrated and QC/purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy).
Note: All reactions involving the use and heating of azides were carried out behind a safety shield taking extra precautions due to the explosive nature of these materials.
General procedure B for two-step click, aza-Michael with amines 14–17 and azides A–L. To a 1-dram vial containing 2-(prop-2-yn-1-yl)-2,3-dihydroisothiazole 1,1-dioxide 2 (50 mg, 0.32 mmol, 1 equiv) was added CuI (18.2 mg, 30 mol%), DBU (5 μL, 10 mol%), dry EtOH (0.64 mL, 0.5 M) and azide (0.64 mmol, 2 equiv.). The reaction was heated at 60 °C on a reaction block for 4 hrs, after which time the reactions were cooled, filtered through SiO2, washed with eluent (2 mL, EtOAc) and concentrated under reduced pressure. The crude was transferred to a 1-dram vial, where DBU (5 μL, 10 mol%), dry EtOH (0.64 mL, 0.5 M), amine (0.38 mmol, 1.2 equiv.) were added. The reaction was subsequently heated at 60 °C on a reaction block for 10 hrs, after which time the reactions were cooled, filtered through a SiO2 SPE and concentrated into pre-weighed barcoded vials, washed with eluent (2 mL, EtOAc:MeOH 95:5) and concentrated under reduced pressure. The crude reaction was concentrated and QC/purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy).
General procedure C for the synthesis of cores 18, 19 and 20 via aza-Michael. Into a round-bottom flask was added a solution of 2-(prop-2-yn-1-yl)-2,3-dihydroisothiazole 1,1-dioxide 2 (1 equiv.) in dry MeOH (1 M). To the stirring solution was added DBU (10 mol %) and the corresponding amino alcohol (1.5 equiv.), and the reaction mixture was heated at 60 °C for 12 hrs. After such time, the reaction was diluted in CH2Cl2:MeOH (9:1), filtered through a silica SPE and flushed with CH2Cl2:MeOH (9:1). The resulting isothiazolidine 1,1-dioxide (18, 19 or 20) was concentrated and carried forward without the need for further purification.
General procedure D for synthesis of library C via one-pot click/OACC esterification protocol. To a 1-dram vial containing sultam 18, 19, or 20 (40 mg, 1 equiv.) was added CuI (30 mol%), DBU (10 mol%), dry CH2Cl2 (0.2 M), acid (1.2 equiv.) and azide (2 equiv.). To the crude mixture was added a solution of OACC (1.5 equiv.) in dry CH2Cl2 (0.2 M) and the resulting reaction mixture was heated at 50 °C on a reaction block for 12 hrs. After which time, the reactions were cooled, diluted in EtOAc (2 mL) and the corresponding suspension filtered through a SiO2 SPE into pre-weighed bar-coded vials, washed with eluent (2 mL, EtOAc) and concentrated under reduced pressure. The crude reaction was concentrated and QC/purified by an automated preparative reverse phase HPLC (detected by mass spectroscopy).
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Science [Center in Chemical Methodologies and Library Development at the University of Kansas, (KU-CMLD), NIH P50 GM069663, NIH P41-GM076302 and the NIH-STTR R41 GM076765]. Undergraduate funding was provided by a KU Undergraduate Research Award from the KU Honors Department and the KU Center for Research (S.V.K and M.B).
Footnotes
Supporting Information Available: Experimental procedures, tabulated results for all libraries, and full characterization data for representative compounds. This material is available free of charge via the Internet at http://acs.pubs.org/
Reference and Notes
- 1.(a) Dolle RE, Bourdonnec BL, Worm K, Morales GA, Thomas CJ, Zhang W. Comprehensive Survey of Chemical Libraries for Drug Discovery and Chemical Biology: 2009. J Comb Chem. 2010;12:765–806. doi: 10.1021/cc100128w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dolle RE, Bourdonnec BL, Goodman AJ, Morales GA, Thomas CJ, Zhang W. Comprehensive Survey of Chemical Libraries for Drug Discovery and Chemical Biology: 2008. J Comb Chem. 2009;11:739–790. doi: 10.1021/cc9000828. [DOI] [PubMed] [Google Scholar]; (c) Dolle RE, Bourdonnec BL, Goodman AJ, Morales GA, Thomas CJ, Zhang W. Comprehensive Survey of Chemical Libraries for Drug Discovery and Chemical Biology: 2007. J Comb Chem. 2008;10:753–802. doi: 10.1021/cc800119z. [DOI] [PubMed] [Google Scholar]
- 2.Drews J. Drug Discovery: A Historical Perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960.Navia MA. A Chicken in Every Pot, Thanks to Sulfonamide Drugs. Science. 2000;288:2132–2133. doi: 10.1126/science.288.5474.2132.Page MI. β-Sultams Mechanism of Reactions and Use as Inhibitors of Serine Proteases. Acc Chem Res. 2004;37:297–303. doi: 10.1021/ar0200899.For an extensive list of biologically active sultams see Rolfe A, Young K, Hanson PR. Domino Heck-Aza-Michael Reactions: A One-pot, Multi-Component Approach to 1,2-Benzisothiazoline-3-acetic acid 1,1-dioxides. Eur J Org Chem. 2008:5254–5262. doi: 10.1002/ejoc.200800651.
- 3.(a) Nhien ANV, Tomassi C, Len C, Marco-Contelles JL, Balzarini J, Pannecouque C, Clerq ED, Postel D. First Synthesis and Evaluation of the Inhibitory Effects of Aza Analogues of TSAO on HIV-1 Replication. J Med Chem. 2005;48:4276–4284. doi: 10.1021/jm050091g. [DOI] [PubMed] [Google Scholar]; (b) Sluis-Cremer N, Hamamouch N, San-Felix A, Velazquez S, Balzarini J, Camarasa MJ. Structure-Activity Relationships of [2′,5′-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-3′-spiro-5″-(4″-amino-1″,2″-oxathiole-2″,2″-dioxide)thymine Derivatives as Inhibitors of HIV-1 Reverse Transcriptase Dimerization. J Med Chem. 2006;49:4834–4841. doi: 10.1021/jm0604575. [DOI] [PubMed] [Google Scholar]; (c) Chen Z, Demuth TP, Jr, Wireko FC. Stereoselective synthesis and antibacterial evaluation of 4-amido-isothiazolidinone oxides. Bioorg Med Chem Lett. 2001;11:2111–2115. doi: 10.1016/s0960-894x(01)00409-7. [DOI] [PubMed] [Google Scholar]
- 4.(a) van den Brook SAM, Meeuwissen SA, van Delft FL, Rutjes FPJT. Natural products containing medium-sized nitrogen heterocycles synthesized by ring-closing alkene metathesis. In: Cossy J, Areniyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley-VCH; Weinheim, Germany: 2010. pp. 45–85. [Google Scholar]; (b) Thomas CD, Hanson PR. Phosphorus and Sulfur Heterocycles via Ring-Closing Metathesis: Application in Natural Product Synthesis. In: Cossy J, Areniyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley-VCH; Weinheim, Germany: 2010. pp. 129–144. [Google Scholar]; (c) Deiters A, Martin SF. Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; (d) Hanson PR, Stoianova DS. Ring-closing metathesis strategy to P-heterocycles. Tetrahedron Lett. 1999;40:3297–3330. [Google Scholar]; (e) Stoianova DS, Hanson PR. A Ring-Closing Metathesis Strategy to Phosphonosugars. Org Lett. 2001;21:3285–3288. doi: 10.1021/ol016491p. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hanson PR, Probst DA, Robinson RE, Yau M. Cyclic sulfonamides via the ring-closing metathesis reaction. Tetrahedron Lett. 1999;40:4761–4764. [Google Scholar]; (b) McReynolds MD, Dougherty JM, Hanson PR. Synthesis of Phosphorus and Sulfur Heterocycles via Ring-Closing Olefin Metathesis. Chem Rev. 2004;104:2239–2258. doi: 10.1021/cr020109k. [DOI] [PubMed] [Google Scholar]; (c) Jiménez-Hopkins M, Hanson PR. An RCM Strategy to Stereodiverse d-Sultam Scaffolds. Org Lett. 2008;10:2223–2236. doi: 10.1021/ol800649n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hanessian S, Sailes H, Therrien E. Synthesis of functionally diverse bicyclic sulfonamides as constrained proline analogues and application to the design of potential thrombin inhibitors. Tetrahedron. 2003;59:7047–7056. [Google Scholar]
- 6.(a) Ying Y, Kim H, Hong J. Stereoselective Synthesis of 2,6-cis- and 2,6-trans-Piperidines through Organocatalytic Aza-Michael Reactions: A Facile Synthesis of (+)-Myrtine and (-)-Epimyrtine. Org Lett. 2011;13:796–799. doi: 10.1021/ol103064f. [DOI] [PubMed] [Google Scholar]; (b) Fustero S, Monteagudo S, Sánhez-Roselló M, Flores S, Barrio P, Pozo del C. N-Sulfinyl Amines as a Nitrogen Source in the Asymmetric Intramolecular Aza-Michael Reaction: Total Synthesis of (-)-Pinidinol. Chem Eur J. 2010;16:9835–9845. doi: 10.1002/chem.201000615. [DOI] [PubMed] [Google Scholar]; (c) Cai Q, Zheng C, You SL. Enantioselective Intramolecular Aza-Michael Additions of Indoles Catalyzed by Chiral Phosphoric Acids. Angew Chem Int Ed. 2010;49:8666–8669. doi: 10.1002/anie.201003919. [DOI] [PubMed] [Google Scholar]; (d) Ghorai MK, Halder S, Das RK. Domino Imino-Aldol-Aza-Michael Reaction: One-Pot Diastereo- and Enantioselective Synthesis of Piperidines. J Org Chem. 2010;75:7061–7072. doi: 10.1021/jo101680f. [DOI] [PubMed] [Google Scholar]; (e) Fustero S, Moscardó J, Sánchez-Roselló M, Rodríguez E, Barrio P. Tandem Nucleophilic Addition-Intramolecular Aza-Michael Reaction: Facile Synthesis of Chiral Fluorinated Isoindolines. Org Lett. 2010;12:5494–5497. doi: 10.1021/ol102341n. [DOI] [PubMed] [Google Scholar]; (f) Che C, Li S, Jiang X, Quan J, Lin S, Yang Z. One-Pot Syntheses of Chromeno[3,4-c]pyrrole-3,4-diones via Ugi-4CR and Intramolecular Michael Addition. Org Lett. 2010;12:4682–4686. doi: 10.1021/ol1020477. [DOI] [PubMed] [Google Scholar]; (g) Fustero S, Catalán S, Sánchez-Roselló M, Simón-Fuentes A, Pozo C del. Tandem Asymmetric Michael Reaction-Intramolecular Michael Addition. An Easy Entry to Chiral Fluorinated 1,4-Dihydropyridines. Org Lett. 2010;12:3484–3487. doi: 10.1021/ol101318t. [DOI] [PubMed] [Google Scholar]; (h) Priebbenow DL, Henderson LC, Pfeffer FM, Stewart SG. Domino Heck-Aza-Michael Reactions: Efficient Access to 1-Substituted Tetrahydro-β-carbolines. J Org Chem. 2010;75:1787–1790. doi: 10.1021/jo902652h. [DOI] [PubMed] [Google Scholar]; (i) Liu SW, Hsu HC, Chang CH, Tsai HHG, Hou DR. Asymmetric Synthesis of (–)-Lentiginosine by Double Aza-Michael Reaction. Eur J Org Chem. 2010:4771–4773. [Google Scholar]; (j) Comesse S, Sanselme M, Daïch A. New and Expeditious Tandem Sequence Aza-Michael/Intramolecular Nucleophilic Substitution Route to Substituted -Lactams: Synthesis of the Tricyclic Core of (±)-Martinellines. J Org Chem. 2008;73:5566–5569. doi: 10.1021/jo702752w. [DOI] [PubMed] [Google Scholar]; (k) Enders D, Narine AA, Toulgoat F, Biscchops T. Asymmetric Brønsted Acid Catalyzed Isoindoline Synthesis: Enhancement of Enantiomeric Ratio by Stereoablative Kinetic Resolution. Angew Chem Int Ed. 2008;47:5661–5665. doi: 10.1002/anie.200801354. [DOI] [PubMed] [Google Scholar]; (l) Carlson EC, Rathbone LK, Yang H, Collett ND, Carter RG. Improved Protocol for Asymmetric, Intramolecular Heteroatom Michael Addition Using Organocatalysis: Enantioselective Syntheses of Homoproline, Pelletierine, and Homopipecolic Acid. J Org Chem. 2008;73:5155–5158. doi: 10.1021/jo800749t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Takasu K, Nishida N, Tomimura A, Ihara M. Convenient Synthesis of Substituted Piperidinones from α,β-Unsaturated Amides: Formal Synthesis of Deplancheine, Tacamonine, and Paroxetine. J Org Chem. 2005;70:3957–3962. doi: 10.1021/jo050261x. [DOI] [PubMed] [Google Scholar]
- 7.(a) Zhou A, Rayabarapu D, Hanson PR. “Click, Click, Cyclize”: A DOS Approach to Sultams Utilizing Vinyl Sulfonamide Linchpins. Org Lett. 2009;11:531–534. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou A, Hanson PR. Synthesis of Sultam Scaffolds via Intramolecular Oxa-Michael and Diastereoselective Baylis-Hillman Reactions. Org Lett. 2008;10:2951–2954. doi: 10.1021/ol8009072. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zang Q, Javed S, Ullah F, Zhou A, Knudtson CA, Bi D, Bashac FZ, Organ MG, Hanson PR. Application of a Double aza-Michael Reaction in a “Click, Click, Cy-Click” Strategy: From Bench to Flow. Synthesis. 2011 doi: 10.1055/s-0030-1260112. Manuscript in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scholl M, Ding Lee CW, Grubbs RH. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 10.A variety of metathesis catalyst were investigated, including [(PCy3)2(Cl)2Ru=CHPh; cat-A], [(IMesH2)(PCy3)(Cl)2Ru=CHPh; cat-B], and the Hoveyda-Grubbs 2nd-generation catalyst.
- 11.Akella LB, Marcaurelle LA. Application of a Sparse Matrix Design Strategy to the Synthesis of DOS Libraries. ACS Comb Sci. 2011 doi: 10.1021/co200020j. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Full in-silico data and detailed calculation information is provided in the supplementary information.
- 13.Representative compounds with full data set available in supplementary information.
- 14.Zhang M, Vedantham P, Flynn DL, Hanson PR. High-Load, Soluble Oligomeric Carbodiimide: Synthesis and Application in Coupling Reactions. J Org Chem. 2004;69:8340–8344. doi: 10.1021/jo048870c. [DOI] [PubMed] [Google Scholar]
- 15.Despite purification via standard column chromatography, all compounds generated as part of the validation library are submitted to reverse-phase automated mass-directed LCMS to validate the stability of these compounds under this process along with purity analysis required by the NIH MLPCN for HTS screening.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.