

Abstract
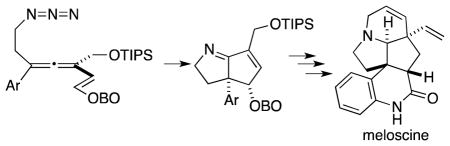
The pentacyclic alkaloid (±)-meloscine was prepared in 19 steps through a reaction sequence that features a putative azatrimethylenemethane intermediate, generated through cascade cyclization of an allenyl azide substrate, to deliver the core azabicyclo[3.3.0]octadiene substructure. Subsequent manipulation of the peripheral functionality then delivered (±)-meloscine.
Diradical combination holds great promise for the construction of C–C bonds in sterically hindered environments. For example, joining two t-butyl radicals to form the exceedingly hindered hexamethylethane is only 30 times slower than similar combination of two methyl radicals.1 When spanning the α,ω positions of a chain, this bond formation constitutes an effective ring forming strategy, provided that efficient means of generating the singlet diyl is available. The recent development of allenyl azide cascade cyclizations (Scheme 1) represents one such emerging methodology, wherein strain-driven release of N2 from cycloadduct 2 promotes formation of an azatrimethylenemethane 3a/3b en route to the bicyclic product 4.2 If high levels of stereochemical control upon formation of bond a in 4 can be achieved, then this methodology might find use in the synthesis of cognate alkaloid structures. One such target is the Melodinus alkaloid meloscine (5),3 which features an azabicyclo[3.3.0]octane core embedded in a complex pentacyclic framework. In this report, we describe the application of the allenyl azide cyclization cascade to the efficient synthesis of this alkaloid. The synthesis strategy unsurprisingly focuses on ring formation, with the following key components: (a) ultimate ring E closure via a diastereoselective ring closing metathesis,3f (b) ring B closure via an intramolecular amidation of an aryl bromide, and (c) formation of the bicyclic C/D core via allenyl azide chemistry.
Scheme 1.

Allenyl azide cyclization cascade; application to the synthesis of meloscine
The synthesis plan commenced with formation of the allene precursor carbonate 13 from the known azido ketone 12, itself accessible in three steps from 2-bromobenzaldehyde (Scheme 2).2d Addition of lithiated O-TIPS-protected propargyl alcohol4 to 12 followed by treatment of the crude alcoholate with methyl chloroformate afforded the desired carbonate.
Scheme 2.

Synthesis and cyclization of the allenyl azide substrate.
Treatment of propargyl carbonate 13 with the 4-methyl-2,6,7-trioxabicyclo[2.2.2]octane (OBO) functionalized vinyl zincate 145 under palladium-mediated allene synthesis conditions furnished tetrasubstituted allene 10.6 Unfortunately, this molecule did not tolerate even mild SiO2 chromatographic conditions, with significant decomposition attending isolation of only trace impure product. Earlier work had shown that allenyl azide cyclization cascades were insensitive to contaminants derived from the zincate addition chemistry, and so the thermal cyclization was performed on the crude allene mixture to eliminate the problematic purification step. Thermolysis of a dilute (0.003 M) solution of crude allene 10 in deoxygenated toluene gave the desired bicycle 8 in moderate yield. The structure and relative stereochemistry of unsaturated imine 8 was ascertained by single crystal X-ray analysis (see Supporting Information for details).7
Elaboration of the core continued with a two-stage reduction of the α,β-unsaturated imine moiety in 8 (Scheme 3). High-pressure hydrogenation of 8 over Pt/C in the presence of triethylamine gave imine 15 as an inconsequential mixture of diastereomers in 80% yield. Super-hydride reduction of the imine gave the free amine after ammonium chloride workup to cleave the intermediate amine-borane complex. This crude amine was protected as its t-butyl carbamate to facilitate subsequent oxidation chemistry. The low nucleophilicity of the pyrrolidine nitrogen necessitated the use of the highly reactive BOC-ONH2 generated in situ from a mixture of BOC2O, hydroxylamine hydrochloride and triethylamine.8 It was necessary to employ an excess of reagents to drive the BOC protection to completion, and that exigency made purification of the fully protected core problematic and inefficient. Instead, a hexanes/MeCN extraction was utilized in order to separate the polar contaminants from excess BOC2O and the protected bicycle. Then, Bu4NF-mediated removal of the TIPS group permitted uneventful isolation of alcohol 16 in a 59% yield over the three-step sequence. Rapid installation of the 1,3-diol was achieved by a 2 step oxidation/Tollens-type aldol sequence.9 In this procedure, a buffered, water promoted Dess-Martin oxidation10 of 16 afforded the expected aldehyde, which was then treated with KOH and formaldehyde in ethylene glycol/water/CH2Cl2 to give diol 17 in excellent yield.
Scheme 3.

Functionalizing the upper periphery of the bicyclic core in anticipation of E-ring installation.
Hydrolysis of orthoester 17 to afford an ester diol proceeded smoothly under mildly acidic conditions, but this ester proved to be recalcitrant to aminolysis (Scheme 4). Treatment of the derived ester with 7N methanolic ammonia led to incomplete and inconsistent conversion of the ester to the primary amide. Switching ammonia sources to gaseous ammonia and elevating the temperature led to complete consumption of the starting ester, but significant byproduct formation was observed; analysis of the crude reaction mixture suggested that significant methyl ester formation occurred even after extended reaction times, implying that this methyl ester may be unreactive to the aminolysis conditions. Switching solvents from methanol to the less nucleophilic isopropanol led to rapid and clean conversion of the ester derived from 17 into primary amide 18. After several unsuccessful attempts to perform an oxidation/olefination sequence on the diol function of 18, a rerouting that featured initial closure of the lactam ring was pursued. Thus, microwave assisted Goldberg-type coupling was employed to form the B ring of 19.11
Scheme 4.

Formation of the B-ring.
The last transformation of note focused on installation of the 1,3-divinyl moiety that would be used to close the final (E) ring of meloscine (Scheme 5). The initial approach to this goal involved a Swern-type oxidation of the diols in 19. Unfortunately, formation of variable mixtures of the mono-, di- and unoxidized compounds plagued these initial studies, plausibly due to steric congestion-induced retroaldol reaction of an intermediate β-hydroxyaldehyde or perhaps by nucleophile-induced decarbonylation of the product dialdehyde.
Scheme 5.

Completion of the synthesis of (±)-meloscine.
The solution to this problem proved to be a water-promoted Dess-Martin oxidation effected through microwave heating. In the absence of water, only mono-oxidation of the substrate was observed and in the absence of microwave heating, the dialdehyde decomposed before the reaction had gone to completion. In tandem, however, these conditions allowed for the rapid and clean conversion of the diol to the desired dialdehyde. Wittig olefination of this intermediate dialdehyde gave the desired divinyl compound 20. Again, the byproducts of the reaction sequence impeded purification, and so the crude 20 mixture was carried through the N-deprotection/allylation sequence to give trialkenyl tetracycle 6 in 57% overall yield. Finally, treatment of this triene with 5% Hoveyda-Grubb’s second-generation catalyst as per the procedure described by Mukai3f gave (±)-meloscine (5) as a single diastereomer. No signals for a C(3) epimer were evident in the 1H NMR spectrum of the crude metathesis reaction product, an observation consistent with Mukai’s findings in his meloscine synthesis.3f
In summary, the Melodinus alkaloid (±)-meloscine was synthesized in 19 steps from 2-bromobenzaldehyde. The azabicyclo[3.3.0]octadiene core was constructed efficiently, in moderate yield, and with excellent stereochemical control using an allenyl azide cycloaddition/diradical cyclization cascade strategy. Robust and efficient methods for the installation and subsequent oxidation of an intermediate 1,3-diol moiety were key to the completion of this synthesis.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health, General Medical Sciences (GM 72572) for support of this work. The X-ray facility is supported by NSF CHE 0131112.
Footnotes
Supporting Information Available. Full experimental procedures and copies of the 1H NMR and 13C NMR spectra for 13, 8, 15–19, 6, and 5; X-ray data for 8. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Tsang W, Hampson RF. J Phys Chem Ref Data. 1986;15:1087–1222. (see pg. 1095) [Google Scholar]; (b) Parkes DA, Quinn CP. J Chem Soc Farad Soc I. 1976;72:1952–1970. [Google Scholar]; (c) Tsang W. J Am Chem Soc. 1985;107:2872–2880. [Google Scholar]; (d) Choo KY, Beadle PC, Piszkiewicz W, Golden DM. Int J Chem Kinet. 1976;8:45–58. [Google Scholar]
- 2.(a) Feldman KS, Iyer MR. J Am Chem Soc. 2006;127:4590–4591. doi: 10.1021/ja050757w. [DOI] [PubMed] [Google Scholar]; (b) Feldman KS, Iyer MR, Hester DK., II Org Lett. 2006;8:3113–3116. doi: 10.1021/ol061216u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) López CS, Faza ON, Feldman KS, Iyer MR, II, DKH J Am Chem Soc. 2007;129:7638–7646. doi: 10.1021/ja070818l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Feldman KS, Iyer MR, López CS, Faza ON. J Org Chem. 2008;73:5090–5099. doi: 10.1021/jo8008066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Feldman KS, Hester DK, López CS, Faza ON. Org Lett. 2008;10:1665–1668. doi: 10.1021/ol800429j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Feldman KS, Hester DK, Iyer MR, Munson PJ, López CS, Faza ON. J Org Chem. 2009;74:4958–4974. doi: 10.1021/jo900659w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Faza ON, Feldman KS, López CS. Curr Org Chem. 2010;14:1646–1657. doi: 10.2174/138527210793563305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isolation: Bernauer K, Englert G, Vetter W, Weis E. Helv Chim Acta. 1969;52:1886–1905.Synthesis studies: Lévy J, Hugel G. J Org Chem. 1986;51:1594–1595.Schultz AG, Dai M. Tetrahedron Lett. 1999;40:645–648.Total syntheses: Overman LE, Robertson GM, Robichaud AJ. J Am Chem Soc. 1991;113:2598–2610.Selig P, Herdtweck E, Bach T. Chem Eur J. 2009;15:3509–3525. doi: 10.1002/chem.200802383.Hayashi Y, Inagaki F, Mukai C. Org Lett. 2011;13:1778–1780. doi: 10.1021/ol200311y.Zhang H, Curran DP. J Am Chem Soc. 2011;133:10376–10378. doi: 10.1021/ja2042854.
- 4.Keck D, Vanderheiden S, Bräse S. Eur J Org Chem. 2006;21:4916–4923. [Google Scholar]
- 5.Su Y, Jung Y, Seo S, Min K, Shin D, Lee Y, Kim S, Park H. J Org Chem. 2002;67:4127–4137. doi: 10.1021/jo0110855. [DOI] [PubMed] [Google Scholar]
- 6.Mandai T, Ogawa M, Yamaoki H, Nakata T, Murayama H, Kawada M, Tsuji J. Tetrahedron Lett. 1991;32:3397–3398. [Google Scholar]
- 7.Cambridge Crystallographic Data Centre deposition number for 8: CCDC 844410. The data can be obtained free from Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
- 8.(a) Harris RB, Wilson IB. Tetrahedron Lett. 1983;24:231–232. [Google Scholar]; (b) Harris RB, Wilson IB. Int J Peptide Protein Res. 1984;23:55–60. doi: 10.1111/j.1399-3011.1984.tb02692.x. [DOI] [PubMed] [Google Scholar]
- 9.Gunic E, Girardet J, Pietrzkowski Z, Esler C, Wang G. Bioorg Med Chem. 2001;9:163.170. doi: 10.1016/s0968-0896(00)00221-2. [DOI] [PubMed] [Google Scholar]
- 10.Meyer SD, Schreiber SL. J Org Chem. 1994;24:7549–7552. [Google Scholar]
- 11.(a) Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2008;130:9971–9983. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang H, Yan X, Zhu W, Liu H, Jiang H, Chen K. J Comb Chem. 2008;10:617–619. doi: 10.1021/cc800048p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.