

Abstract
Atherosclerosis is characterized by hyperplastic neointima and an inflammatory response with cytokines such as TNFα. TNFα is a pleiotropic cytokine that mediates inflammatory, proliferative, cytostatic and cytotoxic effects in a variety of cell types, including endothelial cells and vascular smooth muscle cells (VSMCs). Interestingly, TNFα has been shown to play two very opposing roles in these cell types; it inhibits proliferation and induces apoptosis in endothelial cells, while it enhances the proliferation and migration of VSMCs. Here we show that TNFα is capable of stimulating proliferation of rat VSMCs as well as human VSMCs in a Raf-1/MAPK-dependent manner. TNFα could increase the expression of E2F-regulated proliferative cdc6, Thymidylate synthase (TS) and cdc25A genes in Aortic smooth muscle cells (AoSMC), as seen by real time PCR assays. There is an activation of the stress-induced kinase, JNK1, in VSMCs upon TNFα stimulation. TNFα was capable of inducing binding of the Raf-1 kinase to Rb, and treatment with the Rb-Raf-1 inhibitor, RRD-251, could prevent TNFα-induced S-phase entry in AoSMCs. In addition, inhibition of Raf-1 or Src kinases using pharmacologic inhibitors could also prevent S-phase entry, while inhibition of JNK was not as effective. These results suggest that inhibiting the Rb-Raf-1 interaction is a potential avenue to prevent VSMC proliferation associated with atherosclerosis.
Key words: cell cycle, RRD-251, Rb-Raf interaction, MAP kinase cascade, E2F1 transcription factor
Introduction
Atherosclerosis is the hardening of the arteries that leads to stroke or heart attack and is characterized by many complex changes to the arteries.1 Development of atherosclerosis is a stringently regulated and complex process that results from aberrations in endothelial cell and vascular smooth muscle cell (VSMCs) function.2,3 Endothelial cells form the lining of the blood vessels and the heart, functioning as a barrier by regulating permeability, thrombogenicity and production of growth inhibitory molecules.4 Endothelial cells also respond to mechanical forces. Endothelial cells are contact-inhibited under normal conditions, but when endothelial cells sense an injury such as abrasion of a vessel, they proliferate and migrate, leading to re-endothelialization at sites of injury.4 At the same time, vascular smooth muscle cells proliferate and migrate from the injured arterial wall into the vessel lumen, leading to vessel thickening and occlusion, called restenosis.5,6 Intimal hyperplasia characterized by VSMC proliferation and extracellular matrix (ECM) deposition is a major process contributing to restenosis.7 Atherosclerotic lesions can be blocked if inhibition of VSMCs is effective.8 Several growth factors and cytokines are capable of stimulating VSMC migration and proliferation, such as platelet-derived growth factor (PDGF), which plays a vital role in the development of restenosis.9–11
PDGF can stimulate VSMC proliferation and migration at sites of stress.10,12 It has been shown that suppression of PDGFR activation can inhibit VSMC proliferation by decreasing activation of its downstream signaling molecules.13–15 PDGF is a potent mitogen that mediates arterial response and stimulates proliferation and matrix production.16 PDGF signaling leads to downstream activation of proliferative genes that contribute to atherosclerosis and restenosis.17
Tumor necrosis factor α (TNFα) is a pleiotropic inflammatory cytokine known to play a pathophysiological role in vascular atherosclerosis.18–20 It is known to be expressed by vascular smooth muscle cells in atheromatous plaques.21 By signaling through cell membrane receptors, TNFα can induce mitogenic and proapoptotic effects, activating monocytes, endothelial cells and macrophages.22 Accelerated atherosclerosis is characterized by hyperplastic neointima and an inflammatory response with cytokines such as TNFα.23–25 TNFα has been shown to play two opposing roles in cardiac pathology, including inhibition of endothelial cell proliferation and enhancement of apoptosis, while stimulating vascular smooth muscle cell proliferation and migration.8,26,27 Although there are conflicting reports on the ability of TNFα to stimulate VSMC proliferation, there is compelling evidence defining the migration stimulating activity of this cytokine.28,29 TNFα, like other chemoattractants, such as PDGF, stimulates VSMC migration through the MAPK pathway.30
The apoptosis induced by TNF superfamily requires binding of a ligand to its receptor, leading to oligotrimerization of receptors.31–34 This results in aggregation of death domain-containing proteins allowing recruitment of TRADD (TNF receptor 1-associated death domain protein). TRADD binds FADD (Fas-associated death domain-containing protein) and TRAF-2 (TNF receptor 1-associated protein 2) proteins, which, in turn, leads to activation of procaspase-8 and apoptosis signal-regulating kinase 1 (ASK1), respectively.32,35–37 TNFα treatment leads to simultaneous activation of the ASK1-JNK/p38 death signal.36–39 Reports on the effects of TNFα on apoptosis or proliferation in VSMCs are conflicting.26 Several investigations report that TNFα itself does not induce VSMC proliferation, while other studies suggest TNFα induces proliferation of VSMCs through NFκB-mediated transcription mechanisms.26,40 Regarding apoptosis, there are also inconsistent reports. Certain studies have shown that TNFα could induce apoptosis in VSMCs via caspase-3 activation, while others found no pro-apoptotic activity for TNFα in these cells.28 Further investigations revealed that effects of TNFα in VSMCs can vary depending on the phenotype of the cells.26
Although PDGF and TNFα have very different signaling intermediates, their downstream functions require MAPK activation. It is therefore important to identify the upstream mechanisms contributing to increased proliferation by these two stimuli. Stimulation of VSMCs with PDGF leads to downstream activation of Erk1/2 via the Ras/Raf/MAPK (mitogen-activated protein kinase/extracellular-signal-regulated-kinase) pathway. Activated ERK1/2 rapidly translocates to the nucleus where they target transcription of genes that regulate cell cycle progression, such as cyclin D1.41 Cyclin D1 binds cdk4/6, and together they facilitate S-phase entry through phosphorylation of the retinoblastoma (Rb) protein.42 Stimulation of VSMCs with TNFα has been shown to enhance proliferation through ERK1/2,43 although the exact mechanism is not known.
The retinoblastoma tumor suppressor protein, Rb is a vital regulator of the mammalian cell cylcle, and its inactivation facilitates S-phase entry.44,45 During cell cycle progression, Rb is inactivated through multiple waves of phosphorylation mediated by kinases associated with D- and E-type cyclins in the G1 phase. Rb controls the G1-S boundary by repressing the transcriptional activity of the E2F family of transcription factors.46 E2Fs transcriptionally regulate the expression of many genes necessary for DNA synthesis and cell cycle progression, such as cyclins A and E, cdc2, thymidylate synthase (TS), dihydrofolate reductase, cdc6 and cdc25A.47–49 However, Rb-E2F interaction also has a pivotal role in regulating apoptosis and the intricacies of Rb-E2F interaction, and its regulation of cell fate is context dependent.50,51 Cyclins and cyclin-dependent kinases phosphorylate Rb in mid- to late G1 phase releasing transcriptionally active E2F1, whereas Raf-1 kinase binds to Rb and phosphorylates Rb in the early G1 phase. This priming phosphorylation by Raf-1 is necessary for subsequent phosphorylation and inactivation of Rb.52 Our previous studies demonstrated that disruption of Rb/Raf-1 interaction significantly inhibits proliferation, angiogenesis as well as tumor growth in vivo in an Rb-dependent manner.53 The Rb-Raf-1 pathway has been shown to play a proliferative role in response to growth factor signaling as well as nicotine stimulation of a variety of cell lines,53–55 but the contribution of this pathway in VSMC proliferation is not known. Here, we show that TNFα stimulates proliferation of VSMCs by activating Raf-1/MEK/ERK pathway and facilitating Rb-Raf-1 interaction, leading to the expression of E2F-regulated proliferative genes. These studies were conducted on primary human aortic smooth muscle cells (AoSMCs) as well as rat A10 cells, which are immortalized vascular smooth muscle cells. Further, disruption of the Rb-Raf-1 interaction by the small molecule inhibitor RRD-251 prevented TNFα-induced proliferation of AoSMCs, raising the possibility that this could be a viable mechanism to combat certain steps of atherosclerosis.
Results
TNFα stimulates proliferation of vascular smooth muscle cells.
Since there are conflicting reports on the effects of TNFα-induced proliferation of VSMCs, we examined the effects of serum, TNFα and PDGF on VSMCs by BrdU incorporation assays. Rat A10 cells, which are vascular smooth muscle cells derived from thoracic aorta of embryonic rat, were serum starved for 24 h and subsequently re-stimulated with serum, TNFα (100 ng/ml) or PDGF (100 ng/ml) for 18 h and S-phase entry was measured using standard BrdU incorporation assays. TNFα could stimulate proliferation in vascular smooth muscle cells to a certain extent (Fig. 1A). In the same manner, we examined the effects of TNFα in primary human aortic vascular smooth muscle cells (AoSMCs). As shown in Figure 1B, TNFα could stimulate S-phase entry comparable to PDGF in human VSMCs; serum was used as the positive control.
Figure 1.

TNFα stimulates proliferation in vascular smooth muscle cells. (A) Rat A10 VSMCs were serum starved and subsequently stimulated with serum, TNFα or PDGF for 18 h, and BrdU incorporation was measured. (B) A similar assay was done using human AoSMCs.
TNFα activates Raf/MAPK pathway in vascular smooth muscle cells.
TNFα has been shown to induce migration of aortic smooth muscle cells through ERK1/2 activation.30 We wanted to examine if TNFα treatment had any effects on Raf-1 kinase in these cells. Treatment of aortic smooth muscle cells with TNFα for 10 min, 30 min, 1 h and 2 h led to a shift in Raf-1 migration, indicative of Raf-1 phosphorylation, as seen by western blotting (Fig. 2A). Indeed, phosphorylated Raf-1 can be seen by western blotting for serine 338 on Raf-1; activation was highest at 1 h of TNFα treatment (Fig. 2A). ERK1/2 activation was seen in response to TNFα and was highest at 30 min of treatment (Fig. 2A). The stress-activated protein kinase/Jun-N-terminal kinase (SAPK/JNK) is a member of the MAPK family that is potently and preferentially activated by stresses such as UV irradiation, ceramides and cytokines like TNFα.34 In certain instances, JNK can be activated by growth factors.56–58 We next examined if TNFα was capable of activating stress kinases in a proliferative scenario. To this end, AoSMCs were serum starved and stimulated with TNFα or PDGF for 30 min (time point when Raf-1 activation and ERK1/2 activation was present). Western blotting for JNK activation using an antibody that recognizes phosphorylated Thr183/Tyr185 residues revealed TNFα- and PDGF-activated JNK in aortic smooth muscle cells (Fig. 2B). It was next examined whether TNFα and PDGF activate downstream kinases following similar kinetics. AoSMCs were rendered quiescent by serum starvation and stimulated with TNFα or PDGF for 10 min, 30 min or 2 h. Western blotting using antibodies to pERK1/2 as well as pJNK showed that these kinases were activated at 10 min of stimulation with either agent. The levels of total JNK or ERK1/2 remained mostly unchanged (Fig. 2C). It thus appears that TNFα and PDGF induce ERK1/2 as well as JNK activation following similar kinetics.
Figure 2.

TNFα activates the Raf/MAPK pathway in VSMCs. (A) Time-course stimulation of AoSMCs results in Raf-1 activation, highest at 1 h, and ERK1/2 activation peaks at 30 min. (B) Activation of ERK1/2 coincides with JNK1 activation from 30 min of TNFα treatment. (C) PDGF and TNFα time-course stimulation shows ERK1/2 and JNK activation occurs simultaneously from the different stimuli.
TNFα-induced AoSMC proliferation is abrogated by targeting upstream activators of Raf-1.
Since TNFα led to activation of Raf-1 in vascular smooth muscle cells, and since Raf-1 plays a vital role in cell proliferation, we examined if vascular smooth muscle cell proliferation could be inhibited via targeting Raf-1 or kinases that activate Raf-1. Both Src and PKC kinases are known to activate Raf-1 in response to growth factor signaling.59 To evaluate the importance of these kinases in TNFα-induced proliferation, the Src inhibitor PP2 and the PKC inhibitor Ro-31-8220 were used in BrdU incorporation assays. AoSMCs were serum starved and subsequently stimulated with PDGF or TNFα in the presence or absence of the aforementioned inhibitors for 18 h. BrdU incorporation assays revealed that both inhibition of Src and PKC could efficiently block TNFα- or PDGF-induced S-phase entry (Fig. 3A). Targeting upstream of Raf-1 or Raf-1 itself using the multikinase inhibitor BAY-43-9006 could completely inhibit S-phase entry induced by PDGF or TNFα (Fig. 3B). Next we examined if inhibition of downstream activation of JNK could prevent S-phase entry induced by PDGF or TNFα. Inhibition of downstream JNK activation did not significantly block proliferation, suggesting that inhibition of Raf-1 function is a viable way to block TNFα-induced proliferation of AoSMCs (Fig. 3B). Further, we examined whether MEK inhibitor PD98059 could block PDGF- and TNFα-induced proliferation of AoSMCs. BrdU incorporation assays revealed that inhibition of MEK could block TNFα- and PDGF-induced cell proliferation (Fig. 3C).
Figure 3.

Targeting Raf-1 activation blocks AoSMC proliferation. (A) Src inhibitor (PP2) and PKC inhibitor (Ro-31-8220) block TNFα- and PDGF-induced proliferation. (B) Multi-kinase inhibitor BAY-43-9006, which targets Raf-1, can completely inhibit PDGF- and TNFα-induced proliferation, while the JNK inhibitor (SP600125) does not significantly affect TNFα-induced proliferation. (C) MEK inhibitor PD98059 could block TNFα- and PDGF-induced S-phase entry as seen by BrdU incorporation assays. (D) Raf-1 depletion using Raf-1 siRNAs could inhibit TNFα- and PDGF-induced AoSMC proliferation.
To confirm the results obtained by targeting Raf-1 by chemical inhibitors, we employed siRNA transfections. AoSMCs were transfected with siRNAs targeting Raf-1, subsequently serum starved and induced with TNFα or PDGF for 18 h. BrdU incorporation assays showed that Raf-1 depletion could abrogate TNFα- and PDGF-induced AoSMC proliferation compared with a non-targeting control siRNA (Fig. 3D and Sup. Fig. 1).
TNFα treatment induces E2F regulated genes involved in proliferation.
Since TNFα was functioning similar to a growth factor in stimulating the proliferation of AoSMCs, we examined whether TNFα-induced proliferation was an E2F-dependent mechanism. To this end, real-time PCR was performed on three E2F-responsive genes from AoSMCs that were serum starved and subsequently stimulated with either TNFα or PDGF for 18 h. TNFα could induce cdc25A, cdc6 and TS (thymidylate synthase) gene expression 3.5-, 3- and 1.8-fold respectively (Fig. 4A–C). These results were confirmed by western blotting, which showed a reduction in cdc 6, cdc 25A and TS protein levels when Raf-1 as well as E2F1 were depleted by siRNA transfection, confirming their role in the normal expression of cdc 6, cdc 25A and TS (Fig. 4E and F). Next, we examined if E2F1 was, in fact, present on these proliferative promoters in response to TNFα. Chromatin immunoprecipitation assays were conducted for this purpose. Treatment of quiescent AoSMCs with TNFα or PDGF for 18 h led to an increase in the association of E2F1 on the cdc25A, cdc6 and TS promoters with a concomitant dissociation of Rb (Fig. 4D). In quiescent cells, we consistently observed a faint band for E2F1, whereas a significantly higher amount of Rb was found to be associated with these promoters in serum-starved cells. The c-fos promoter was used as a negative control. These results suggest that TNFα can induce changes in the promoter association of E2F1 as well as Rb in AoSMCs comparable to a growth factor.
Figure 4.

TNFα and PDGF induce E2F regulated genes in AoSMCs. (A) Treatment with TNFα and PDGF for 18 h led to 3.5- and 4-fold increase, respectively, in cdc25A gene expression in real-time PCR assays. (B) Treatment with TNFα and PDGF for 18 h led to 3.5- and 7-fold increase, respectively in cdc6 gene expression in real-time PCR assays. (C) Treatment with TNFα and PDGF for 18 h led to 1.8 and 3.8 fold increase, respectively in TS gene expression in real time PCR assays. (D) Treatment with TNFα or PDGF led to an increase in E2F1 and dissociation of Rb on the proliferative promoters cdc25A, cdc6 and TS in ChIP assays; c-fos was used as the negative control. (E) Western blots for cdc 6, cdc 25A and TS after Raf-1 and E2F1 depletion. (F) Densitometric analysis of the bands to quantify the western blot data.
TNFα-induced AoSMC proliferation involves Rb-Raf-1 interaction.
Our previous studies have shown the importance of the Rb-Raf-1 interaction in mediating proliferation in a wide array of cell lines.53,60 Since Raf-1 activation is evident in response to TNFα-induced proliferation in AoSMCs, we examined if Raf.1-Rb interaction is involved in mediating these effects. Treatment with the Rb-Raf-1 inhibitor RRD-251 in the presence of TNFα or PDGF for 2 h could efficiently reduce Raf-1 levels in both AoSMCs and rat A10 cells (Fig. 5A and B). Next, we examined if TNFα stimulation of AoSMCs could induce the Rb-Raf-1 interaction; this was done by IP-WB analysis. Treatment with TNFα and PDGF for 2 h led to an increase in Raf-1 bound to Rb; in addition, there was less E2F1 associated in the TNFα- and PDGF-stimulated complexes (Fig. 5C). The Rb-Raf-1 inhibitor RRD-251 could effectively inhibit TNFα-induced Rb-Raf-1 interaction as revealed by immunoprecipitation followed by western blot assays (Fig. 5D). These results suggest that treatment of AoSMCs with TNFα leads to an increased association of the Raf-1 kinase with Rb, comparable to growth factor stimulation of cancer cell lines, and that RRD-251 can effectively inhibit this interaction in smooth muscle cells.
Figure 5.

RRD-251 inhibits Rb-Raf-1 interaction in AoSMCs. (A and B) Treatment with TNFα or PDGF in the presence of RRD-251 inhibits Raf-1 levels in AoSMCs (A) and A10s (B). (C) TNFα and PDGF treatment induced Rb-Raf-1 binding in AoSMCs. (D) RRD-251 inhibits TNFα-induced Rb-Raf-1 interaction.
We next examined if RRD-251 could prevent serum-, TNFα- or PDGF-induced proliferation in AoSMCs. AoSMCs were serum starved and subsequently stimulated with serum, TNFα or PDGF in the presence or absence of 20 µM RRD-251. In response to all three stimuli, RRD-251 was capable of inhibiting S-phase entry in AoSMCs (Fig. 6A). It is known that binding of Raf-1 to Rb facilitates the complete inactivation of Rb by phosphorylation, resulting in its dissociation from E2F and proliferative promoters, leading to their expression; disrupting this interaction in epithelial cells led to the retention of Rb on E2F1-regulated proliferative promoters, leading to their repression and cell cycle arrest. Given this background, we examined the role of proliferative E2F family members E2F1, E2F2 and E2F3 in TNFα- or PDGF- induced proliferation of AoSMCs. AoSMCs were transfected with E2F1, E2F2, E2F3 or control siRNAs and subsequently induced with TNFα or PDGF. We observed that E2F1 and E2F3 depletion resulted in a significant reduction in S-phase entry as seen by BrdU incorporation assays, whereas E2F2 had a marginal effect (Fig. 6D and Fig. S2). We further examined the effect of RRD-251 in regulating the expression of E2F target genes, such as cdc6, TS and cdc 25A in response to TNFα and PDGF treatment of AoSMCs. Treatment of AoSMC cells with RRD-251 in the presence of TNFα or PDGF for 18 h could efficiently inhibit the expression of cdc 6, TS and cdc 25A as seen by RT-PCR (Fig. 6B and C). These results suggest that inhibiting Rb-Raf-1 interaction leads to the repression of E2F1-regulated proliferative promoters, as in the case of growth factor stimulation of epithelial cell lines, and this is a possible mechanism by which RRD-251 is bringing about the growth arrest of AoSMCs. These observations also raise the possibility that targeting this interaction might be a viable alternative to prevent atherosclerosis.
Figure 6.

Inhibition of Rb-Raf interaction prevents proliferation and expression of E2F1 target proliferative genes in AoSMC. (A) Treatment with RRD-251 inhibits AoSMC proliferation induced by serum, TNFα and PDGF. (B and C) Treatment with RRD-251 inhibits expression of E2F target genes cdc 6, TS and cdc 25A, as seen by RT-PCR. (D) Depletion of E2F1 and E2F3 using siRNAs resulted in significant inhibition of TNFα and PDGF induced AoSMC proliferation, whereas E2F2 depletion had only a marginal effect.
Discussion
The dynamics of endothelial and vascular smooth muscle cells play a predominant role in the progression of atherosclerosis and restenosis.5,10 Migration, proliferation and differentiation of endothelial cells as well as VSMCs are important pathological responses that contribute to the development of vascular lesions.61 The “switch” of VSMCs from the quiescent phenotype to the proliferative and migratory phenotype is a vital event in the pathogenesis of atherosclerosis and restenosis post-angioplasty.10,17 Therefore, VSMC proliferation and migration both serve as suitable targets for drug therapy in vascular proliferative disorders.1,2
This study provides evidence that TNFα and PDGF evoke similar signaling mechanisms that contribute to VSMC proliferation. Although they are not equally efficacious in activating these pathways, TNFα is capable of activating proliferative signaling pathways in the smooth muscle cells, leading to S-phase entry and cell cycle progression. The proliferative response also increased the expression of E2F-regulated genes cdc6, TS and cdc25A; further, TNFα stimulation led to an increase in the association of E2F1 on these proliferative promoters, with a dissociation of Rb. Interestingly, in response to TNFα stimulation, Raf/MAPK activation occurred, and this coincided with an activation of the stress kinase JNK1. Inhibition of TNFα- or PDGF-induced cell proliferation was observed when pharmacologic inhibitors targeting Raf-1 or upstream regulators of Raf-1 like Src or PKC were used; inhibiting JNK had no effect. This suggests that JNK activation most likely is not responsible for the proliferative responses seen with TNFα, even though it is activated in these cells.
Studies from our lab have shown that upon TNFα treatment, the kinase ASK1 mediates Rb inactivation as an initial signaling event in Ramos and Jurkat cells.54 These studies had shown that inactivation of Rb by ASK1 kinase was necessary for TNFα to induce apoptosis; further, a mutant ASK1 kinase that was unable to bind to Rb was impaired in its ability to facilitate TNFα-mediated apoptosis in Jurkat and 3T3 cells.
In a similar vein, our recent studies show that TNFα stimulation of human aortic endothelial cells led to significant induction of apoptosis.62 This coincided with Rb phosphorylation, which was mediated by ASK1 kinase. A peptide competitor, which could effectively prevent the binding of ASK1 to Rb could inhibit TNFα-induced apoptosis of endothelial cells. These studies showed that p53 had a limited role in the process. Indeed, the role of p53 in TNFα-induced apoptosis has been controversial.63,64 We also find that TNFα has no effect on p53 expression; however, p73α levels were found to be upregulated, implicating p73α as the major contributing factor to apoptosis induced by TNFα in endothelial cells. This induction of p73α by TNFα was dependent on E2F1 function. Supporting this contention, depletion of E2F1 or ASK1 significantly reduced the ability of TNFα to induce apoptosis in endothelial cells. Taken together, studies from our lab show that Rb interacts with ASK1 upon apoptotic stimuli, and ASK1 has to overcome Rb function to execute its pro-apoptotic functions, suggesting that Rb acts as a critical connector between apoptotic and proliferative pathways by interacting with the functionally distinct kinases like Raf-1 and ASK1.54 Thus, the role of Rb phosphorylation by specific kinases is pertinent for directed signaling for apoptotic or proliferative pathways.65
The contrasting observation in AoSMCs, where TNFα treatment resulted in a lack of apoptotic response and an increase in proliferation suggests that TNFα is involved in multiple pathways depending on the cellular context (Fig. 7). We observed activation of Raf-1 and ERK in response to TNFα stimulation, indicative of a proliferative response. It has been shown that a colocalization of TNFα and ERK1/2 occurs, and that ERK 1/2 activation induces the expression of Ets-1, Egr-1 and c-fos in neointimal lesions from rat aortae two weeks post balloon injury.66 The ChIP and RT-PCR experiments showed recruitment of E2F1 to proliferative promoters, suggesting that E2F1 is a key mediator in the TNFα-induced proliferative or apoptotic pathways in VSMCs or endothelial cells, respectively.
Figure 7.
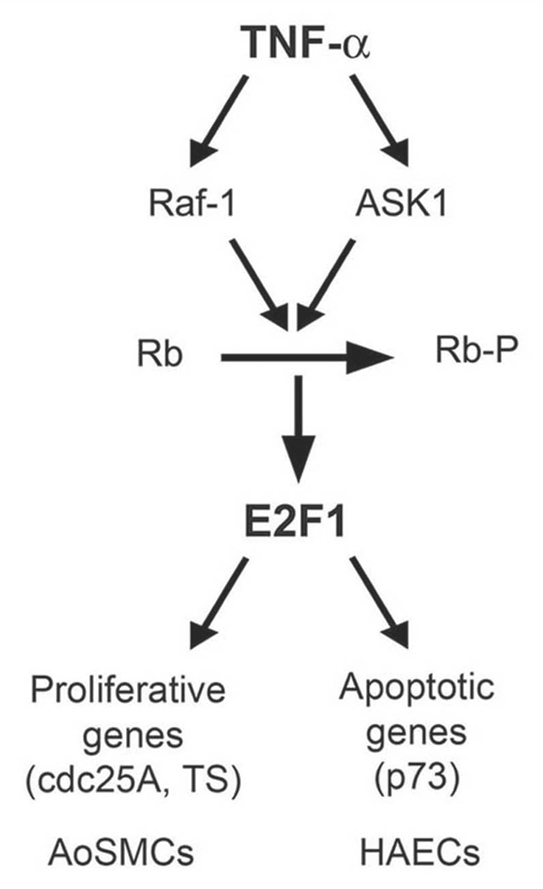
Schematic showing the contrasting roles of TNFα in AoSMCs and HAECs. In HAECs, TNFα stimulation resulted in Rb inactivation by ASK1 leading to elevated E2F1 activation and upregulation of proapoptotic protein p73. resulting in apoptosis.62 Whereas in AoSMCs, TNFα stimulation led to Raf-1-dependent Rb inactivation, elevated E2F1 activity, leading to the expression of proliferative promoters, resulting in increased cell proliferation.
Rb-Raf-1 interaction was found to play a vital role in serum-, PDGF- and TNFα-induced proliferation of VSMCs. Targeting Rb-Raf-1 interaction using RRD-251 could completely inhibit S-phase entry in these cells. Our lab has previously shown that disrupting the Rb-Raf-1 interaction can prevent endothelial cell adhesion, migration and proliferation.53,54,60 Raf-1 is known to play an essential role in normal wound healing in vivo and migration of keratinocytes and fibroblasts in vitro.67 This novel function of Raf-1 is independent of its kinase activity and is brought about by the spatial regulation of Rho signaling during migration.68 Apart from its role in cell cycle regulation, emerging evidences also indicate the involvement of Rb-E2F pathway in cell migration and invasion.69–71 Taken together, the importance of this interaction in both endothelial as well as vascular smooth muscle cell physiology in atherosclerotic lesions needs further evaluation and may provide useful tools in development of therapies for heart disease. The divergent responses of AoSMCs and HAECs (human aortic endothelial cells) to TNFα thus provide unique therapeutic possibilities: simultaneously targeting the cell cycle of two different cell types within same tissue microenvironment, resulting in opposite and biologically complimentary effects.
Materials and Methods
Cell culture.
Human aortic smooth muscle cells were obtained from Lonza (CC-2571) and cultured in smooth muscle basal medium (SmBM) supplemented with growth factors and 5% FBS, according to the manufacturer's instructions (CC-3182). TNFα (Promega) was added at 100 ng/ml. PDGF (Biosource) was added at 100 ng/ml concentration.
Lysate preparation, immunoprecipitation and western blotting.
Lysates from cells treated with different agents were prepared by NP-40 lysis as described earlier in reference 52. Polyclonal E2F1, c-Raf, ASK1, cyclin D and E and monoclonal antibodies for cdc 25A, cdc 6 and TS were obtained from Santa Cruz Biotechnology. Monoclonal Rb and Raf-1 were supplied by BD Transduction Laboratories. Polyclonal antibodies to phospho-Rb (807, 811), phospho-Raf-(338), phospho-JNK, phospho ERK 1/2 and phospho Mek1/2 were supplied by Cell Signaling.
Chromatin immunoprecipitation (ChIP) assay.
ChIP lysates were prepared, and immunoprecipitations were conducted using antibodies against E2F1, Rb and the association with specific promoters detected by PCR as previously described in reference 72. Rabbit anti-mouse secondary antibody was used as the control for all reactions. The sequences of cdc25A, TS, cdc6 and c-fos primers are described in reference 52.
Real-time PCR.
AoSMC cells were subjected to serum starvation for 24 h, followed by treatment with PDGF or TNFα in presence or absence of RRD-251. Unstimulated serum-starved cells were used as a control. Total RNA was isolated by an RNeasy miniprep kit from QIAGEN following the manufacturer's protocol. One microgram of RNA was DNase treated using RQ1 DNase (Promega), followed by first-strand cDNA synthesis using the iScript cDNA synthesis kit (Bio-Rad). A fraction (1/20) of the final cDNA reaction volume was used in each PCR.73 Primer sequences are as follows: 5′-CTG CCA GCT GTA CCA GAG AT-3′ (TS forward primer), 5′-ATG TGC ATC TCC CAA AGT GT-3′ (TS reverse primer), 5′-CCC CAT GAT TGT GTT GGT AT-3′ (Cdc6 forward primer), 5′-TTC AAC AGC TGT GGC TTA CA-3′ (Cdc6 reverse primer), 5′-CTC AAC ACG GGA AAC CTC AC-3′ (18S forward primer), and 5′-AAA TCG CTC CAC CAA CTA AGA A-3′ (18S reverse primer). Real-time PCR was performed using a Bio-Rad iCycler.
siRNA transfections.
siRNA oligonucleotides targeting human E2F1, E2F2, E2F3 and Raf-1 were purchased from Santa Cruz biotechnology. Non-target siRNA was used as the control. AoSMCs were transfected either with control siRNA, E2F1, E2F2, E2F3 or Raf-1 siRNA (100 pmol) using oligofectamine (Invitrogen) in accordance with the manufacturer's recommendations. For western blots, cells were harvested 48 h after transfection, and lysates were made. For BrdU incorporation asays, cells were serum starved for 24 h after transfection and induced with TNFα or PDGF for 18 h.
Proliferation assays.
Bromodeoxyuridine (BrdU) labeling kits were obtained from Roche Biochemicals. Cells were plated in poly-d-lysine coated chamber slides at a density of 10,000 cells per well and rendered quiescent by serum starvation for 24 h. Cells were then re-stimulated with serum, TNFα or PDGF in the presence or absence of the indicated drugs for 18 h. S-phase cells were visualized by microscopy and quantified by counting three fields in quadruplicate.
Acknowledgments
These studies were supported by the grant CA118210 from the NCI. R.D. is a recipient of a Predoctoral Fellowship from the American Heart Association (grant number 0615168B).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/S0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 2.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 3.Soufi M, Sattler AM, Maisch B, Schaefer JR. Molecular mechanisms involved in atherosclerosis. Herz. 2002;27:637–648. doi: 10.1007/s00059-002-2431-2. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 5.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abedi H, Zachary I. Signalling mechanisms in the regulation of vascular cell migration. Cardiovasc Res. 1995;30:544–556. [PubMed] [Google Scholar]
- 7.Schwartz RS, Huber KC, Murphy JG, Edwards WD, Camrud AR, Vlietstra RE, et al. Restenosis and the proportional neointimal response to coronary artery injury: results in a porcine model. J Am Coll Cardiol. 1992;19:267–274. doi: 10.1016/0735-1097(92)90476-4. [DOI] [PubMed] [Google Scholar]
- 8.Losordo DW, Isner JM, Diaz-Sandoval LJ. Endothelial recovery: the next target in restenosis prevention. Circulation. 2003;107:2635–2637. doi: 10.1161/01.CIR.0000071083.31270.C3. [DOI] [PubMed] [Google Scholar]
- 9.Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 10.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 11.Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. 1991;253:1129–1132. doi: 10.1126/science.1653454. [DOI] [PubMed] [Google Scholar]
- 12.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 13.Bilder G, Amin D, Morgan L, McVey M, Needle S, Galczenski H, et al. Stent-induced restenosis in the swine coronary artery is inhibited by a platelet-derived growth factor receptor tyrosine kinase inhibitor, TKI963. J Cardiovasc Pharmacol. 2003;41:817–829. doi: 10.1097/00005344-200306000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimura H, Nariai Y, Terashima M, Mitani T, Tanigawa Y. Taurine suppresses platelet-derived growth factor (PDGF) BB-induced PDGFbeta receptor phosphorylation by protein tyrosine phosphatase-mediated dephosphorylation in vascular smooth muscle cells. Biochim Biophys Acta. 2005;1745:350–360. doi: 10.1016/j.bbamcr.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Bilder G, Wentz T, Leadley R, Amin D, Byan L, O'Conner B, et al. Restenosis following angioplasty in the swine coronary artery is inhibited by an orally active PDGF-receptor tyrosine kinase inhibitor, RPR101511A. Circulation. 1999;99:3292–3299. doi: 10.1161/01.cir.99.25.3292. [DOI] [PubMed] [Google Scholar]
- 17.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 18.Beg AA, Baltimore D. An essential role for NFkappaB in preventing TNFalpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNFalpha and IL-1beta mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- 20.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 21.Prins JB, Ledgerwood EC, Ameloot P, Vandenabeele P, Faraco PR, Bright NA, et al. Tumor necrosis factor-induced cytotoxicity is not related to rates of mitochondrial morphological abnormalities or autophagy-changes that can be mediated by TNFR-I or TNFR-II. Biosci Rep. 1998;18:329–340. doi: 10.1023/A:1020261316486. [DOI] [PubMed] [Google Scholar]
- 22.Rakesh K, Agrawal DK. Cytokines and growth factors involved in apoptosis and proliferation of vascular smooth muscle cells. Int Immunopharmacol. 2005;5:1487–1506. doi: 10.1016/j.intimp.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 23.McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. 2009;6:410–417. doi: 10.1038/nrcardio.2009.57. [DOI] [PubMed] [Google Scholar]
- 24.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol Ther. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Selzman CH, Shames BD, McIntyre RC, Jr, Banerjee A, Harken AH. The NFkappaB inhibitory peptide, IkappaBalpha, prevents human vascular smooth muscle proliferation. Ann Thorac Surg. 1999;67:1227–1231. doi: 10.1016/S0003-4975(99)00252-0. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, Park Y, Wu J, Chen X, Lee S, Yang J, et al. Role of TNFalpha in vascular dysfunction. Clin Sci (Lond) 2009;116:219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factoralpha and interleukin-1beta. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.ATV.16.1.19. [DOI] [PubMed] [Google Scholar]
- 29.Sawada H, Kan M, McKeehan WL. Opposite effects of monokines (interleukin-1 and tumor necrosis factor) on proliferation and heparin-binding (fibroblast) growth factor binding to human aortic endothelial and smooth muscle cells. In Vitro Cell Dev Biol. 1990;26:213–216. doi: 10.1007/BF02624115. [DOI] [PubMed] [Google Scholar]
- 30.Goetze S, Xi XP, Kawano Y, Kawano H, Fleck E, Hsueh WA, et al. TNFalpha-induced migration of vascular smooth muscle cells is MAPK dependent. Hypertension. 1999;33:183–189. doi: 10.1161/01.hyp.33.1.183. [DOI] [PubMed] [Google Scholar]
- 31.O'Donnell MA, Ting AT. Chronicles of a death foretold: dual sequential cell death checkpoints in TNF signaling. Cell Cycle. 2010;9:1065–1071. doi: 10.4161/cc.9.6.10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 33.Choy JC, Granville DJ, Hunt DW, McManus BM. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol. 2001;33:1673–1690. doi: 10.1006/jmcc.2001.1419. [DOI] [PubMed] [Google Scholar]
- 34.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 35.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 36.Ichijo H. Molecular mechanisms for cell life and cell death. Kokubyo Gakkai Zasshi. 1998;65:155–163. doi: 10.5357/koubyou.65.155. [DOI] [PubMed] [Google Scholar]
- 37.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J Biochem. 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct Funct. 2003;28:23–29. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- 40.Selzman CH, Shames BD, Reznikov LL, Miller SA, Meng X, Barton HA, et al. Liposomal delivery of purified inhibitory-kappaBalpha inhibits tumor necrosis factor-alpha-induced human vascular smooth muscle proliferation. Circ Res. 1999;84:867–875. doi: 10.1161/01.res.84.8.867. [DOI] [PubMed] [Google Scholar]
- 41.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, et al. Ras signalling linked to the cell cycle machinery by the retinoblastoma protein. Nature. 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- 43.Ouyang P, Peng LS, Yang H, Peng WL, Wu WY, Xu AL. Recombinant human interleukin-10 inhibits proliferation of vascular smooth muscle cells stimulated by advanced glycation end products and neointima hyperplasia after carotid injury in the rat. Sheng Li Xue Bao. 2003;55:128–134. [PubMed] [Google Scholar]
- 44.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 45.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 47.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 48.DeGregori J. The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim Biophys Acta. 2002;1602:131–150. doi: 10.1016/s0304-419x(02)00051-3. [DOI] [PubMed] [Google Scholar]
- 49.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 50.Sun B, Wingate H, Swisher SG, Keyomarsi K, Hunt KK. Absence of pRb facilitates E2F1-induced apoptosis in breast cancer cells. Cell Cycle. 2010;9:1122–1130. doi: 10.4161/cc.9.6.10990. [DOI] [PubMed] [Google Scholar]
- 51.Rogoff HA, Kowalik TF. Life, death and E2F: linking proliferation control and DNA damage signaling via E2F1. Cell Cycle. 2004;3:845–846. doi: 10.4161/cc.3.7.975. [DOI] [PubMed] [Google Scholar]
- 52.Dasgupta P, Sun J, Wang S, Fusaro G, Betts V, Padmanabhan J, et al. Disruption of the Rb-Raf-1 interaction inhibits tumor growth and angiogenesis. Mol Cell Biol. 2004;24:9527–9541. doi: 10.1128/MCB.24.21.9527-41.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kinkade R, Dasgupta P, Carie A, Pernazza D, Carless M, Pillai S, et al. A small molecule disruptor of Rb/Raf-1 interaction inhibits cell proliferation, angiogenesis and growth of human tumor xenografts in nude mice. Cancer Res. 2008;68:3810–3818. doi: 10.1158/0008-5472.CAN-07-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dasgupta P, Betts V, Rastogi S, Joshi B, Morris M, Brennan B, et al. Direct binding of apoptosis signal-regulating kinase 1 to retinoblastoma protein: novel links between apoptotic signaling and cell cycle machinery. J Biol Chem. 2004;279:38762–38769. doi: 10.1074/jbc.M312273200. [DOI] [PubMed] [Google Scholar]
- 55.Dasgupta P, Chellappan SP. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5:2324–2328. doi: 10.4161/cc.5.20.3366. [DOI] [PubMed] [Google Scholar]
- 56.Kyriakis JM. Activation of the AP-1 transcription factor by inflammatory cytokines of the TNF family. Gene Expr. 1999;7:217–231. [PMC free article] [PubMed] [Google Scholar]
- 57.Kyriakis JM. Making the connection: coupling of stress-activated ERK/MAPK (extracellular-signal-regulated kinase/mitogen-activated protein kinase) core signalling modules to extracellular stimuli and biological responses. Biochem Soc Symp. 1999;64:29–48. [PubMed] [Google Scholar]
- 58.Leppä S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- 59.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351:289–305. doi: 10.1042/0264-6021:3510289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kinkade R, Dasgupta P, Chellappan S. The ABC's of Targeting Raf: Novel Approaches to Cancer Therapy. Curr Can Ther Rev. 2006;2:305–314. doi: 10.2174/157339406778699204. [DOI] [Google Scholar]
- 61.Libby P. Vascular biology of atherosclerosis: overview and state of the art. Am J Cardiol. 2003;91:3–6. doi: 10.1016/S0002-9149(02)03143-0. [DOI] [PubMed] [Google Scholar]
- 62.Rastogi S, Rizwani W, Joshi B, Kunigal S, Chellappan SP. TNFalpha response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.93. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chau BN, Chen TT, Wan YY, DeGregori J, Wang JY. Tumor necrosis factor alpha-induced apoptosis requires p73 and c-ABL activation downstream of RB degradation. Mol Cell Biol. 2004;24:4438–4447. doi: 10.1128/MCB.24.10.4438-47.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C. TNF-induced enterocyte apoptosis in mice is mediated by the TNF receptor 1 and does not require p53. Eur J Immunol. 1998;28:3499–3505. doi: 10.1002/(SICI)1521-4141(199811)28:11<3499::AIDIMMU3499>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 65.Wang S, Nath N, Minden A, Chellappan S. Regulation of Rb and E2F by signal transduction cascades: divergent effects of JNK1 and p38 kinases. EMBO J. 1999;18:1559–1570. doi: 10.1093/emboj/18.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goetze S, Kintscher U, Kaneshiro K, Meehan WP, Collins A, Fleck E, et al. TNFalpha induces expression of transcription factors c-fos, Egr-1 and Ets-1 in vascular lesions through extracellular signal-regulated kinases 1/2. Atherosclerosis. 2001;159:93–101. doi: 10.1016/S0021-9150(01)00497-X. [DOI] [PubMed] [Google Scholar]
- 67.Ehrenreiter K, Piazzolla D, Velamoor V, Sobczak I, Small JV, Takeda J, et al. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168:955–964. doi: 10.1083/jcb.200409162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matallanas D, Birtwistle M, Romano D, Zebisch A, Rauch J, von Kriegsheim A, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClellan KA, Ruzhynsky VA, Douda DN, Vanderluit JL, Ferguson KL, Chen D, et al. Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Mol Cell Biol. 2007;27:4825–4843. doi: 10.1128/MCB.02100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li J, Hu SX, Perng GS, Zhou Y, Xu K, Zhang C, et al. Expression of the retinoblastoma (RB) tumor suppressor gene inhibits tumor cell invasion in vitro. Oncogene. 1996;13:2379–2386. [PubMed] [Google Scholar]
- 71.Andrusiak MG, McClellan KA, Dugal-Tessier D, Julian LM, Rodrigues SP, Park DS, et al. Rb/E2F regulates expression of neogenin during neuronal migration. Mol Cell Biol. 2011;31:238–247. doi: 10.1128/MCB.00378-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pillai S, Dasgupta P, Chellappan SP. Chromatin immunoprecipitation assays: analyzing transcription factor binding and histone modifications in vivo. Methods Mol Biol. 2009;523:323–339. doi: 10.1007/978-1-59745-190-1_22. [DOI] [PubMed] [Google Scholar]
- 73.Rastogi S, Joshi B, Dasgupta P, Morris M, Wright K, Chellappan S. Prohibitin facilitates cellular senescence by recruiting specific corepressors to inhibit E2F target genes. Mol Cell Biol. 2006;26:4161–4171. doi: 10.1128/MCB.02142-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.