

Abstract
Mast cells have diverse roles throughout the body as evidenced by their heterogeneous nature. In the heart, cardiac mast cells have been implicated in left ventricular (LV) remodeling in response to elevated myocardial stress. Accordingly, the purpose of this study was to use mast cell deficient rats (Ws/Ws) to delineate the interaction between cardiac mast cell activation and LV remodeling. LV matrix metalloproteinase (MMP) activity, fibrillar collagen, TNF-α levels, and LV diameter were compared in Ws/Ws and wild type (WT) rats subjected to 5 d (n=3/group) and 8 wks (n=4/group) of aortocaval fistula-induced volume overload. In contrast to attenuation of myocardial remodeling in the Ws/Ws group: 1) MMP-2 activity was significantly increased in the WT group at 5 d; 2) there was marked degradation of the extracellular collagen matrix in WT at 5 d and 8 wks; 3) the percent increase in LV diameter from baseline was significantly greater in WT at 2, 4, 6, and 8 wks post-fistula; and 4) myocardial TNF-α levels were markedly elevated in the WT group at 5 days post-fistula. These results underscore the importance of cardiac mast cells in mediating MMP activation, collagen degradation and LV dilatation and suggest that mast cell-derived TNF-α plays a role in early myocardial remodeling.
Keywords: Mast Cells, Collagen, matrix metalloproteinases, Cytokines, Rodent
INTRODUCTION
Mast cells are ubiquitous throughout the body, and it has become apparent that these cells have many diverse roles beyond mediating allergic responses, including counteracting bacterial and viral infections, regulating inflammatory cell recruitment and activation, as well as release of cytokines, proteases and eicosanoids [1–4]. Less well recognized is the role that cardiac mast cells play in the adverse remodeling responses of the left ventricle (LV) induced in the heart subjected to chronic elevations in myocardial stress, such as seen in heart failure [5–8], myocardial infarction [9], dilated cardiomyopathy [10] and mitral valve regurgitation [11]. Previous reports from our laboratory have provided substantial evidence supporting the role of cardiac mast cells in mediating matrix metalloproteinase (MMP) activation, which is responsible in turn for extracellular collagen matrix degradation and subsequent ventricular dilatation [5, 8, 12, 13]. These previous findings were based on pharmacological interventions aimed at preventing or producing cardiac mast cell degranulation. Accordingly, the purpose of the current study was to utilize a more direct approach to establish the causal relationship between cardiac mast cell activation and LV remodeling using mast cell-deficient rats (Ws/Ws) and their wild type (WT) controls. These studies sought to test the hypothesis that the absence of mast cells would prevent MMP-2 activation, collagen degradation and LV dilatation induced in response to increased myocardial stress. To accomplish this objective, LV diameter, myocardial MMP-2 activation, fibrillar collagen concentration and TNF-α levelswere compared in Ws/Ws and WT rats subjected to 5 days or 8 weeks of ventricular volume overload.
METHODS
This study was approved by the Institutional Animal Care and Use Committee and conformed to the principles of the National Institute of Health “Guide for the Care and Use of Laboratory Animals”. Fourteen, 3 month old male WT and Ws/Ws rats (Japan SLC, Hamamatsu, Japan) were utilized for this study. The Ws/Ws strain is the result of a mutation in the c-kit gene consisting of a 12 base pair deletion in the tyrosine kinase domain present in the WT inbred Donryu background strain [14]. Rats were housed under standard environmental conditions and were provided standard rat chow and water ad libitum.
Rats were anesthetized using pentobarbital sodium (50 mg/kg, I.P.) and volume overload was induced by creating an infrarenal aortocaval (AV) fistula as previously described [15, 16]. Briefly: 1) the aorta and caudal vena cava approximately 1.0 cm distal to the renal arteries were approached via a ventral laparotomy; 2) an 18-gauge needle was inserted into the abdominal aorta and advanced through the medial wall into the vena cava; and 3) the needle was withdrawn and the aortic puncture site sealed with cyanoacrylate. A successful AV fistula was evident by the pulsatile flow of oxygenated blood into the vena cava. Groups of rats were studied at 5 days (n = 3; Ws/Ws and WT) and 8 weeks (n = 4; Ws/Ws and WT) post-fistula. These times were chosen as they reflect the initial phase (i.e., 5 d) of the LV remodeling process when mast cell degranulation, MMP activation and extracellular matrix degradation occurs in this model, and the time point at which rats begin to transition into heart failure (i.e., 8 wks) [7, 8]. At the experimental endpoint, hearts were removed and the right ventricle (RV) and LV including septum were separated and weighed. The LV was then sectioned into apical, mid-ventricular and basal sections. The apical and basal sections were snap-frozen for biochemical analysis and the mid-ventricular section was fixed in 10% formalin for histological analysis. The lungs were also removed and weighed.
Left Ventricular Diameter
In vivo LV diameter was assessed in conscious rats by echocardiographic analysis with a P12.5 MHz transducer (ATL 5000) prior to and at 2 week intervals after creation of an AV fistula. A parasternal short axis view was used to obtain B mode 2-dimensional targeted M-mode tracings. The LV end diastolic chamber dimension was determined from the average of three consecutive M-mode tracings utilizing the leading edge method.
Collagen Volume Fraction
LV interstitial collagen volume fraction (CVF) was determined by analysis of picrosirius red stained sections. Paraffin-embedded LV cross-sections cut to 5 μm in thickness were incubated with phosphomolybdic acid (0.2%) to reduce background before staining with picrosirius red (0.1% Sirius Red F3BA in picric acid, PSR). Tissue sections were analysed in a blinded fashion using a Biorad MRC-1024 confocal laser-scanning microscope at 400× magnification. Twenty representative fields per LV cross-section were imaged. Image analysis was performed using Scion Image software (National Institutes of Health, USA). Briefly, PSR stained interstitial collagen fibers were highlighted giving a pixel count for the amount of collagen represented in each field. This was then expressed as a percentage of the overall number of pixels present in the picture giving a percentage area of interstitial collagen. Perivascular areas were excluded from the analysis.
Matrix Metalloproteinase Activity
The extent of MMP activation was determined in LV tissue extracts from the 5 day group using gelatin zymography performed by standard procedures using a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) matrix containing 1 mg/ml gelatin [5]. Each gel was run in duplicate, and in order to standardize across gels, an extract from the heart of an un-opertated Sprague-Dawley rat was used as a standard on both gels. The zymograms had two lytic bands corresponding to standards for the proenzyme and activated forms of MMP-2 (Chemicon International, Inc., Temecula, CA). The relative increase in the abundance of protein present in the 68 kDa band (i.e. the active band) was quantified by densitometry. The values thus obtained for MMP activity for each sample were first normalized for their protein concentration using a Bio-Rad Protein assay. The basal activity of the normal SD standard was set at 0% and the activities of the lytic bands in the other lanes of a gel were expressed as a percent of this standard’s activity. Once normalized in this fashion, the percent activities from hearts belonging to each group were averaged.
Quantification and Immunohistochemistry Labeling of Left Ventricular TNF-α
Levels of myocardial TNF-α were determined in LV tissue extracts from the 5 day groups using a standard ELISA analysis (Quantikine by R&D Systems, Inc., Minneapolis, USA). Immunohistochemistry for localization of TNF-α was performed with methods established and optimized in our laboratories. Briefly, hearts isolated from unoperated WT rats were fixed in methanol Carnoy’s solution (60% methanol, 30% chloroform, 10% acetic acid) and embedded in paraffin to obtain 5 μm cross sections. Serial sections were subjected to either TNF-α immunohistochemistry or Toluidine Blue staining. All sections were deparaffinized and rehydrated. Endogenous peroxidase was blocked with 1% H2O2 in methanol. Non-specific antibody binding was reduced by a 30 min incubation in 10% normal donkey serum (Fitzgerald, Concord, MA) in TBS containing 3% dry powdered milk and 0.2% BSA. Sections were then incubated with rabbit anti-TNF-α (IP-300, Genzyme, Cambridge, MA), at a 1:100 dilution for 2 hours. After incubation with biotinylated donkey anti-rabbit IgG (Fitzgerald) at 1:400 for 30 min, sections were incubated with pre-formed avidin-biotin-peroxidase complex (Vector Laboratories, Burlingame, CA) and visualized with 0.5 mg/ml 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich, St. Louis, MO) and 0.003% H2O2 in TBS. Mast cells were visualized with Toluidine Blue by incubating sections in 0.5% Toluidine Blue (Sigma-Aldrich) in 0.5 N HCl for 20 min, followed by a 10-min incubation in 0.7 N HCl. A light hematoxylin staining was used as background for both TNF-α immunohistochemistry and Toluidine Blue. Images of serial sections were taken with a 40× objective.
Statistics
All statistical analyses were performed using the SPSS 11.5 software. TNF-α analysis was performed using an unpaired sample T-test. All other grouped data comparisons were made by one-way analysis of variance (ANOVA). When a significant F ratio (p < 0.05) was obtained, intergroup comparisons were made using Fisher's protected least significant difference post-hoc testing with p < 0.05 considered significant.
RESULTS
Morphometric Parameters
Table I contains the average values for body weight as well as LV, RV and lung weights for Ws/Ws and WT groups at 8 weeks post-fistula. While the LV and RV weights tended to be slightly higher in the Ws/Ws group, these were not statistically different to the WT. Similarly, there was no statistical difference between Ws/Ws and WT for body or lung weights.
Table I.
Morphometric Parameters.
| n | Body Weight (g) | LV Weight (mg) | RV Weight (mg) | Lung Weight (mg) | |
|---|---|---|---|---|---|
| WT (8 wks) | 4 | 375 ± 8 | 1204.8 ± 77.8 | 393.3 ± 24.1 | 1636.0 ± 184.9 |
| Ws/Ws (8 wks) | 4 | 371 ± 57 | 1285.5 ± 248.0 | 437.3 ± 121.8 | 1514.0 ± 200.3 |
MMP Activity
The average values for MMP-2 activation in the Ws/Ws and WT groups are presented in Figure 1. As a result of the sustained volume overload, there was significant increase in the concentration of activated MMP-2 in the WT group at 5 days post-fistula relative to the Ws/Ws group.
Figure 1.

Left ventricular MMP-2 activity in mast cell deficient (Ws/Ws) and wild type (WT) rats at five days post fistula. * indicates p < 0.05.
Left Ventricular Structural Remodeling
Consistent with the increased MMP-2 activation, CVF was significantly decreased in the WT group when compared to the Ws/Ws group following AV fistula (Figure 2). The reduction in CVF was noted at both 5 days and 8 weeks. Furthermore, as can be seen in Figure 3, the degradation of the extracellular matrix in the WT group was concurrent with the development of significant ventricular dilatation. This was characterized by a substantial increase in the WT groups LV diameter at the initial echocardiographic evaluation 2 weeks post-fistula, followed by a progressive increase thereafter. This was in stark contrast to the Ws/Ws group, where the percent increases in LV chamber dimensions relative to baseline measurements were significantly smaller than that in the WT hearts at every time point. At 8 weeks post-fistula, the LV diameter was 80% greater than the pre-fistula value in the WT group compared to only a 37% increase in the Ws/Ws group.
Figure 2.
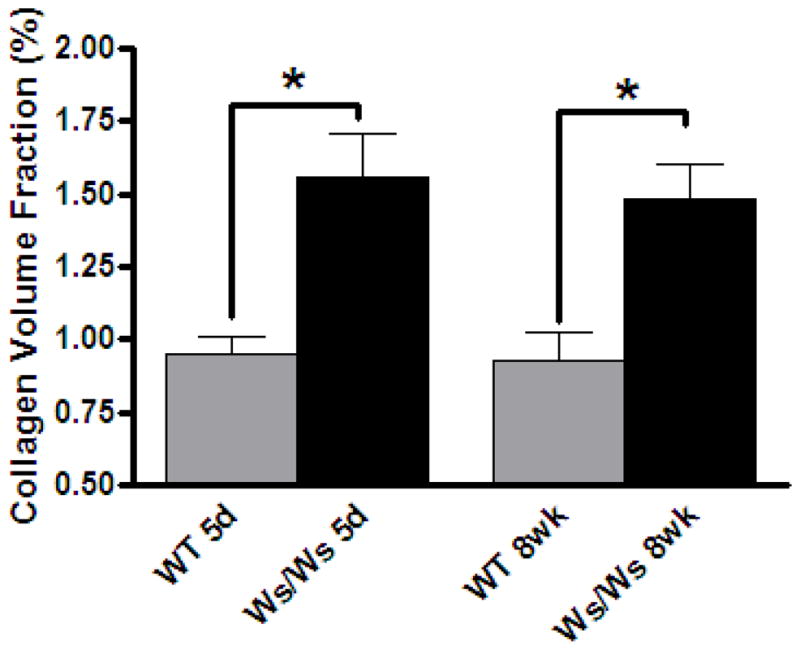
Left ventricular myocardial collagen volume fraction in mast cell deficient (Ws/Ws) and wild type (WT) rats at 5 days (n=3) and 8 weeks (n=3) post-fistula. * indicates p < 0.05.
Figure 3.

Temporal increase in left ventricular chamber diameter (post-fistula) above baseline in mast cell deficient (Ws/Ws) and wild type rats (WT). * indicates p < 0.05 to WT at corresponding time-point.
Left Ventricular TNF-α
There was a strong trend for myocardial TNF-α to be markedly elevated in the WT group relative to the Ws/Ws group following 5 days of volume overload (Figure 4), however, these values did not reach significance due to the small number of rats in each group. Consistent with the possibility of this increase being related to cardiac mast cells, histological analysis of LV sections from WT rats showed concentrated localization of TNF-α within cardiac mast cells, with additional diffuse labeling dispersed throughout the ventricle (Figure 5).
Figure 4.

Left ventricular TNF-α levels in the mast cell deficient (Ws/Ws) and wild type (WT) rats at 5 days post-fistula.
Figure 5.

Toludine blue labeling of cardiac mast cells (indicated by arrows) (A); and immuno-labeling of TNF-α (brown) (B) in LV tissue from normal WT rats. TNF-α is evident in the cardiac mast cells as well as throughout the LV.
DISCUSSION
Mast cells have a number of disparate functions throughout the body, however, until recently the role of these immune cells in the heart has been largely ignored. This changed with the advent of several studies reporting increases in cardiac mast cell density secondary to various myocardial pathologies [5, 9, 10, 17, 18]. These cardiac mast cells have been shown to be of the connective tissue type, containing a variety of biologically active mediators including cytokines and proteases that have the ability to activate MMPs. Recently we identified such a relationship between mast cell activation and MMP activity in the heart [5, 12, 13]. Treatment with the mast cell stabilizing drug, cromolyn, initiated prior to the imposition of volume overload induced by creation of an AV fistula, prevented the increases in cardiac mast cell density and MMP activation during the critical first 5 days of volume overload [5]. In a subsequent study, chronic inhibition of mast cell degranulation over an 8 wk period post-fistula effectively prevented the adverse LV structural dilatation, increased compliance and decreased contractility present in the untreated fistula group [6]. These studies used pharmacologic inhibitors of mast cell activation, however, a more direct assessment of mast cell mediated remodeling is possible in animals such as in the Ws/Ws rat that lack the normal complement of mast cells. Thus, the results presented herein, conclusively establish the direct role of cardiac mast cells in mediating the ventricular remodeling response induced secondary to chronic volume overload.
Activation of MMPs is a crucial step in producing the adverse ventricular remodeling post-fistula [12]. In the current study, a marked increase in MMP-2 activation was found in the myocardium of WT rats. This was in stark contrast to the mast cell deficient Ws/Ws rats in which MMP-2 activation following 5 days of ventricular volume overload did not occur. We have previously documented that there are no basal differences in interstitial collagen density between normal Ws/Ws and WT hearts [19]. Therefore, we would interpret the significant decrease in the concentration of fibrillar collagen in the WT fistula groups, concurrent with these differences in mast cell mediated MMP-2 activation, to be the result of collagen degradation.
Consistent with previous findings demonstrating the relationship between increased activation of MMP-2 and decreased CVF with ventricular dilatation [5, 6], we observed marked differences in the extent of ventricular dilatation between the Ws/Ws and WT groups. The relative increase in LV diameter in the WT group was substantially greater in the first 2 weeks and continued to further increase at all subsequent time points post-fistula. However, despite MMP-2 activation not being appreciable and collagen being preserved in the Ws/Ws group, a small increase in LV diameter did occur over the 8 wk period of volume overload. It is expected that this relatively small increase in ventricular diameter in the Ws/Ws could be due to either the significant increase in LV end diastolic pressure that typically occurs post-fistula [15], or could reflect compensatory adaptation due to structural dilatation and/or increased chamber compliance induced by other mechanisms possibly related to cardiomyocyte remodeling [16]. While neither of these possibilities can be discounted on the basis of an in vivo measurement of diameter, the likelihood that this relatively small increase in LV diameter observed in the Ws/Ws hearts stems from this increase in LV filling pressure, and not pathological remodeling, would be consistent with the results of Brower et al. [6] where there was no difference in the LV end diastolic pressure-volume relationship between sham-operated rats and fistula rats treated with a mast cell stabilizing drug for 8 wks of sustained ventricular volume overload. Under normal conditions, both the Ws/Ws and their WT are reported to have similar LV masses over a variety of ages. Further, given the similarity in LV weight post-fistula, there is no indication that mast cells influence the hypertrophic response. However, the smaller chamber dimension in the mast cell deficient Ws/Ws is comparable to the findings in previous studies using mast cell stabilizing compounds and ACE [20] or MMP inhibitors [12], with the corresponding increase in mass to volume ratio indicative of a compensated heart. Thus, mast cells appear essential to the pathological remodeling that occurs in response to volume overload of the heart, while alternative mechanisms are responsible for compensatory mechanisms.
Immuno-labeling of LV tissue from WT rats without an AV fistula demonstrated that concentrated TNF-α expression was localized in cardiac mast cells, in addition to being diffusely distributed throughout the myocardium. While mast cells are clearly not the only source of TNF-α in the heart under normal conditions, inhibition of the marked increase in myocardial TNF-α in the Ws/Ws rats post-fistula suggests that mast cells are essential to the increased levels of TNF-α observed following imposition of volume overload. This could be due to either direct synthesis and release by mast cells, via their stimulation of other cells to produce TNF-α, or possibly MMP mediated cleavage of pro-TNF-α present in the heart. To this end, leukocytes and viable cardiomyocytes are major producers of TNF-α following microembolism [21]. Our finding is consistent with recent ischemia-reperfusion studies showing that the initial release of TNF-α was cardiac mast cell-dependent, while subsequent induction was derived from other cell sources [22, 23]. TNF-α has been documented to exhibit multiple roles in the heart, which can be either pathological or cardioprotective [21, 24, 25]. While it is theoretically possible that the basal levels of TNF-α present in the Ws/Ws hearts reflect the attenuated LV dilatation, several lines of evidence suggest that TNF-α induces activation of MMPs in the heart. A continuous 15 day infusion of TNF-α in normal rats led to a significant progressive increase in LV diameter associated with significant reductions in interstitial collagen as assessed by CVF [26]. Further characterization at the ultrastructural level demonstrated that the fibrillar collagen weave surrounding cardiac myocytes was significantly disrupted or completely absent [26]. This was thought by the authors to be the result of TNF-α mediated MMP activation. Our laboratory subsequently tested this supposition and found myocardial MMP-2 activity to be significantly elevated in normal rats during the first week of a TNF-α infusionrelative to that in saline-infused rats [27]. As a result of this TNF-α mediated MMP activation, LV CVF was reduced. These findings together with the findings of the current study strongly suggest that mast cell-mediated increases in TNF-α play a significant role in the early stages of myocardial remodeling. This suggests the possibility that the failure of recent clinical trials examining the efficacy of TNF-α inhibition may be an issue of timing. Although TNF-α does not appear to be critical to worsening end-stage heart failure [28], our findings are suggestive of TNF-α being important in the earlier stages of myocardial remodeling prior to the transition to a decompensated state. Thus, future consideration should be given to experimentation or trials focused on intervention directed against TNF-αin the early stages of heart failure (or even diastolic dysfunction). It should be remembered, however, that several other mast cell products, including tryptase and chymase, are also capable of activating MMPs [29–31]. Furthermore, while not yet determined in cardiac mast cells, non-cardiac mast cells have been shown to produce various MMPs including MMP-1 [32], -2 [33], -9 [33, 34], and -13 [35], as well as ADAM-9, -10 and -17 [33]. It is possible that cardiac mast cells may also produce some or all of these MMPs. Therefore, although TNF-α appears to exert a prominent role in cardiac mast cell-induced myocardial remodeling, it is likely that the release of other substances, including tryptase, chymase or MMPs, from cardiac mast cells also contribute to collagen degradation in response to myocardial stress.
In summary, these results obtained in Ws/Ws rats definitively establish the central role of cardiac mast cells in initiating MMP activation, collagen degradation and subsequent myocardial dilatation and are suggestive of a role for mast cell-derived TNF-α in mediating the remodeling. These findings continue to expand the functional role of mast cells in pathologic conditions and emphasize the need to explore mast cell stabilizing compounds as a potential therapy for heart failure.
Acknowledgments
This work was supported in part by EPA grant RD831953 (GLB), Philip Morris USA Inc. and Philip Morris International (SPL) and NIH grants RO1-HL-62228 (JSJ) and HL-73990 (JSJ).
ABBREVIATIONS
- LV
left ventricle
- MMP
matrix metalloproteinase
- Ws/Ws
mast cell deficient rats
- WT
wild type
- AV
aortacaval
- CVF
collagen volume fraction
- RV
right ventricle
Footnotes
CONFLICTS OF INTEREST
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Boyce JA. Mast cells: Beyond IgE. Journal of Allergy and Clinical Immunology. 2003;111:24–32. doi: 10.1067/mai.2003.60. [DOI] [PubMed] [Google Scholar]
- 2.Metcalfe DD. Mast cells. Physiol Rev. 1997;77:1033–79. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 3.Ryan JJ, Kashyap M, Bailey D, Kennedy S, Speiran K, Brenzovich J, Barnstein B, Oskeritzian C, Gomez G. Mast cell homeostasis: a fundamental aspect of allergic disease. Crit Rev Immunol. 2007;27:15–32. doi: 10.1615/critrevimmunol.v27.i1.20. [DOI] [PubMed] [Google Scholar]
- 4.Sayed BA, Brown MA. Mast cells as modulators of T-cell responses. Immunol Rev. 2007;217:53–64. doi: 10.1111/j.1600-065X.2007.00524.x. [DOI] [PubMed] [Google Scholar]
- 5.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am J Physiol. 2002;283:H518–H525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 6.Brower GL, Janicki JS. Pharmacologic inhibition of mast cell degranulation prevents left ventricular remodeling induced by chronic volume overload in rats. J Cardiac Fail. 2005;11:548–56. doi: 10.1016/j.cardfail.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Janicki JS. The dynamic interaction between matrix metalloproteinase activity and adverse myocardial remodeling. Heart Failure Reviews. 2004;9:33–42. doi: 10.1023/B:HREV.0000011392.03037.7e. [DOI] [PubMed] [Google Scholar]
- 8.Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart J, Murray DB, Chancey AL. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovascular Research. 2006;69:657–65. doi: 10.1016/j.cardiores.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 9.Engels W, Reiters PH, Daemen MJ, Smits JF, van der Vusse GJ. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J Pathol. 1995;177:423–9. doi: 10.1002/path.1711770414. [DOI] [PubMed] [Google Scholar]
- 10.Patella V, de Crescenzo G, Lamparter-Schummert B, De Rosa G, Adt M, Marone G. Increased cardiac mast cell density and mediator release in patients with dilated cardiomyopathy. Inflamm Res. 1997;46:S31–S32. [PubMed] [Google Scholar]
- 11.Dell'italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, Machida N, Straeter-Knowlen IM, Hankes GH, Dillon R, Cartee RE, Oparil S. Volume-overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am J Physiol Heart Circ Physiol. 1997;273:H961–H970. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- 12.Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circ. 2002;105:1983–8. doi: 10.1161/01.cir.0000014686.73212.da. [DOI] [PubMed] [Google Scholar]
- 13.Murray DB, Gardner JD, Brower GL, Janicki JS. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am J Physiol Heart Circ Physiol. 2004;287:H2295–H2299. doi: 10.1152/ajpheart.00048.2004. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy RH, Hauer-Jensen M, Joseph J. Cardiac function in hearts isolated from a rat model deficient in mast cells. Am J Physiol Heart Circ Physiol. 2005;288:H632–H637. doi: 10.1152/ajpheart.00803.2004. [DOI] [PubMed] [Google Scholar]
- 15.Brower GL, Henegar JR, Janicki JS. Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am J Physiol. 1996;271:H2071–H2078. doi: 10.1152/ajpheart.1996.271.5.H2071. [DOI] [PubMed] [Google Scholar]
- 16.Brower GL, Janicki JS. Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol. 2001;280:H674–H683. doi: 10.1152/ajpheart.2001.280.2.H674. [DOI] [PubMed] [Google Scholar]
- 17.Olivetti G, Lagrasta C, Ricci R, Sonnenblick EH, Capasso JM, Anversa P. Long-term pressure-induced cardiac hypertrophy: capillary and mast cell proliferation. Am J Physiol Heart Circ Physiol. 1989;257:H1766–H1772. doi: 10.1152/ajpheart.1989.257.6.H1766. [DOI] [PubMed] [Google Scholar]
- 18.Shiota N, Rysa J, Kovanen PT, Ruskoaha H, Kokkonen JO, Lindstedt KA. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J Hypertens. 2003;21:1823–5. doi: 10.1097/00004872-200310000-00022. [DOI] [PubMed] [Google Scholar]
- 19.Joseph J, Kennedy RH, Devi S, Wang J, Joseph L, Hauer-Jensen M. Protective role of mast cells in homocysteine-induced cardiac remodeling. Am J Physiol Heart Circ Physiol. 2005;288:H2541–H2545. doi: 10.1152/ajpheart.00806.2004. [DOI] [PubMed] [Google Scholar]
- 20.Brower GL, Levick SP, Janicki JS. Inhibition of matrix metalloproteinase activity by ACE inhibitors prevents left ventricular remodeling in a rat model of heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H3057–H3064. doi: 10.1152/ajpheart.00447.2006. [DOI] [PubMed] [Google Scholar]
- 21.Dorge H, Schulz R, Belosjorow S, Post H, van de Sand A, Konietzka I, Frede S, Hartung T, Vinten-Johansen J, Youker KA, Entman ML, Erbel R, Heusch G. Coronary Microembolization: the Role of TNF- [alpha] in Contractile Dysfunction. Journal of Molecular and Cellular Cardiology. 2002;34:51–62. doi: 10.1006/jmcc.2001.1489. [DOI] [PubMed] [Google Scholar]
- 22.Reil JC, Gilles S, Zahler S, Brandl A, Drexler H, Hultner L, Matrisian LM, Welsch U, Becker BF. Insights from knock-out models concerning postischemic release of TNF[alpha] from isolated mouse hearts. Journal of Molecular and Cellular Cardiology. 2007;42:133–41. doi: 10.1016/j.yjmcc.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Gilles S, Zahler S, Welsch U, Sommerhoff CP, Becker BF. Release of TNF-[alpha] during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovascular Research. 2003;60:608–16. doi: 10.1016/j.cardiores.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 24.Skyschally A, Gres P, Hoffmann S, Haude M, Erbel R, Schulz R, Heusch G. Bidirectional Role of Tumor Necrosis Factor-{alpha} in Coronary Microembolization: Progressive Contractile Dysfunction Versus Delayed Protection Against Infarction. Circ Res. 2007;100:140–6. doi: 10.1161/01.RES.0000255031.15793.86. [DOI] [PubMed] [Google Scholar]
- 25.Thielmann M, Dorge H, Martin C, Belosjorow S, Schwanke U, van de Sand A, Konietzka I, Buchert A, Kruger A, Schulz R, Heusch G. Myocardial Dysfunction With Coronary Microembolization: Signal Transduction Through a Sequence of Nitric Oxide, Tumor Necrosis Factor-{alpha}, and Sphingosine. Circ Res. 2002;90:807–13. doi: 10.1161/01.res.0000014451.75415.36. [DOI] [PubMed] [Google Scholar]
- 26.Bozkurt B, Kribbs SB, Clubb FJ, Jr, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically Relevant Concentrations of Tumor Necrosis Factor-{alpha} Promote Progressive Left Ventricular Dysfunction and Remodeling in Rats. Circulation. 1998;97:1382–91. doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- 27.Janicki JS, Brower GL, Chancey AL, Forman MF, Jobe LJ. Cardiac mast cells as mediators of ventricular remodeling. In: Villarreal FJ, editor. Interstitial Fibrosis in the Heart. New York: Springer; 2004. pp. 197–209. [Google Scholar]
- 28.Feldman AM, Kadokami T, Higuichi Y, Ramani R, McTiernan CF. The role of anticytokine therapy in heart failure: recent lessons from preclinical and clinical trials? Med Clin North Am. 2003;87:419–40. doi: 10.1016/s0025-7125(02)00189-x. [DOI] [PubMed] [Google Scholar]
- 29.Lohi J, Harvima I, Keski-Oja J. Pericellular substrates of human mast cell tryptase: 72,000 dalton gelatinase and fibronectin. J Cell Biochem. 1992;50:337–49. doi: 10.1002/jcb.240500402. [DOI] [PubMed] [Google Scholar]
- 30.Fang KC, Raymond WW, Lazarus SC, Caughey GH. Dog mastocytoma cells secrete a 92-kD gelatinase activated extracellularly by mast cell chymase. J Clin Invest. 1996;97:1589–96. doi: 10.1172/JCI118583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lees M, Taylor DJ, Woolley DE. Mast cell proteinases activate precursor forms of collagenase and stromelysin, but not of gelatinases A and B. Eur J Biochem. 1994;223:171–7. doi: 10.1111/j.1432-1033.1994.tb18980.x. [DOI] [PubMed] [Google Scholar]
- 32.Di GN, Wakefield D. In vitro and in vivo expression of interstitial collagenase/MMP-1 by human mast cells. Dev Immunol. 2000;7:131–42. doi: 10.1155/2000/82708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards ST, Cruz AC, Donnelly S, Dazin PF, Schulman ES, Jones KD, Wolters PJ, Hoopes C, Dolganov GM, Fang KC. c-Kit immunophenotyping and metalloproteinase expression profiles of mast cells in interstitial lung diseases. J Pathol. 2005;206:279–90. doi: 10.1002/path.1780. [DOI] [PubMed] [Google Scholar]
- 34.Baram D, Vaday GG, Salamon P, Drucker I, Hershkoviz R, Mekori YA. Human Mast Cells Release Metalloproteinase-9 on Contact with Activated T Cells: Juxtacrine Regulation by TNF-{alpha} The Journal of Immunology. 2001;167:4008–16. doi: 10.4049/jimmunol.167.7.4008. [DOI] [PubMed] [Google Scholar]
- 35.Vajner L, Vytasek R, Lachmanova V, Uhlik J, Konradova V, Novotna J, Hampl V, Herget J. Acute and chronic hypoxia as well as 7-day recovery from chronic hypoxia affects the distribution of pulmonary mast cells and their MMP-13 expression in rats. Int J Exp Pathol. 2006;87:383–91. doi: 10.1111/j.1365-2613.2006.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]